

Abstract
Severe infections like sepsis lead frequently to cardiomyopathy. The mechanisms are unclear and an optimal therapy for septic cardiomyopathy still lacks. The aim of this study is to establish an endotoxin-induced inflammatory model using human induced pluripotent stem cell (hiPSC) derived cardiomyocytes (hiPSC-CMs) for mechanistic and therapeutic studies. hiPSC-CMs were treated by lipopolysaccharide (LPS) in different concentrations for different times. ELISA, FACS, qPCR, and patch-clamp techniques were used for the study. TLR4 (Toll-like receptor 4) and its associated proteins, CD14, LBP (lipopolysaccharide binding protein), TIRAP (toll-interleukin 1 receptor domain containing adaptor protein), Ly96 (lymphocyte antigen 96) and nuclear factor kappa B as well as some pro-and anti-inflammatory factors are expressed in hiPSC-CMs. LPS-treatment for 6 hours increased the expression levels of pro-inflammatory and chemotactic cytokines (TNF-a, IL-1ß, IL-6, CCL2, CCL5, IL-8), whereas 48 hour-treatment elevated the expression of anti-inflammatory factors (IL-10 and IL-6). LPS led to cell injury resulting from exaggerated cell apoptosis and necrosis. Finally, LPS inhibited small conductance Ca2+-activated K+ channel currents, enhanced Na+/Ca2+-exchanger currents, prolonged action potential duration, suggesting cellular electrical dysfunctions. Our data demonstrate that hiPSC-CMs possess the functional reaction system involved in endotoxin-induced inflammation and can model some bacterium-induced inflammatory responses in cardiac myocytes.
Introduction
A severe infection by Gram-negative bacteria, like sepsis, can lead to multi organ dysfunctions including heart failure and arrhythmias1, 2. Lipopolysaccharide (LPS), the major component of the outer membrane of Gram-negative bacteria, is known to be a key pathogenic stimulator for the dysfunctions. Under septic circumstances circulating LPS as a pathogen associated molecular pattern (PAMP) can stimulate the innate immune system, which mediates a local or systemic inflammatory response. LPS can also stimulate non-immune cells and initiate the inflammatory process. Exaggerated inflammatory responses are usually detrimental. The innate LPS-pattern recognition receptor, the Toll-like receptor 4 (TLR4) is widely expressed in the body including cardiomyocytes3, 4. Therefore, the innate inflammatory response can be evoked in cardiomyocytes by LPS irrespective of the involvement of immune cells. This could be a reason for the facts that cardiac dysfunctions were frequently observed in patients with sepsis and also in animals injected with LPS5–7. The underlying mechanisms, however, remain largely unclear.
Two hypotheses have been proposed to explain sepsis-induced cardiac dysfunctions, i.e., inadequate coronary blood flow and chemical mediators were supposed to be causative for the dysfunctions. The former was based on studies in animals8, 9, showing that coronary blood flow was reduced by infusion of endotoxin. This theory, however, was refuted after other reports showing a marked coronary vasodilation and even higher coronary flow in patients with sepsis10, 11. The later was supported by increasing evidences. Administration of endotoxin depressed cardiac function12, 13. Cardiomyocytes exposed to the serum of patients with sepsis displayed impaired cell shortening and the causative factor has been proved to be the tumor necrosis factor alpha (TNF-a)14–16. Later, interleukin-1ß, nitric oxide, and reactive oxygen species (ROS), have been implicated in pathogenesis17–19. Despite the improvements in our current understanding of cardiac dysfunction in sepsis, the exact mechanisms have till now not been clarified. Therefore optimal therapy for septic cardiomyopathy still lacks and the mortality of patients with septic cardiomyopathy stays still high19.
To explore the signaling involved in the pathogenesis of septic cardiomyopathy, animals or animal cells have been frequently employed for different studies. Due to the differences between man and animals, animals or animal cells are not ideal for mimicking the human cardiac diseases. Because of limited availability and difficulty in long-time culture of human cardiomyocytes, some human cell lines, e.g., HEK293 and HeLa cells have been used for some studies investigating some inflammation-related signaling20–22. These non-cardiac cells are not ideal for functional studies because they cannot generate action potential and contraction. Since the successful reprogramming of adult somatic cells to induced pluripotent stem (iPS) cells and generation of functional cardiomyocytes from human iPS cells (hiPSC-CM)23–26, hiPSC-CMs have been demonstrated to have the electrophysiological and pharmacological properties including action potentials and responses to antiarrhythmic drugs which are similar to those of native cardiomyocytes27, 28. Therefore hiPSC-CMs can be a good alternative for modeling cardiac diseases. This has been confirmed by emerging evidences showing that the hiPSC-CMs derived from patients with genetic heart diseases recapitulated the phenotype of the disease29–31. Recently, it has been reported that hiPSC-CMs can model the coxsackievirus B3-induced myocarditis32. Here, we report that hiPSC-CMs can recapitulate LPS-induced inflammatory responses with electrophysiological dysfunctions and can serve as an in vitro model for mechanistic and drug-screening studies for some bacterium-induced inflammatory cardiomyopathy.
Results
LPS induced inflammatory responses in hiPSC-CMs
To check whether LPS-challenge can induce inflammatory responses in hiPSC-CMs, we first investigated the mRNA expression of LPS-receptor, the Toll-like receptor 4 (TLR4), and its associated signaling proteins in cells during the cardiac differentiation process. The qPCR-analysis showed that TLR4 and its associated proteins, CD14, LBP (lipopolysaccharide binding protein), TIRAP (toll-interleukin 1 receptor domain containing adaptor protein) and Ly96 (lymphocyte antigen 96, also known as MD2) are expressed in hiPSC-CMs. In addition, the expression of the gene RelA and NF-κB1, two subunits of nuclear factor-κB (NF-κB), an important downstream signaling factor of TLR4, was also detected (Fig. 1). Notably, on day 35 and 50 after differentiation, during which cells were used for the following studies, the mRNA expression levels of the receptor-associated proteins except MD2 on day 35 and TLR4 on day 50 are higher in hiPSC-CMs than that in hiPS cells.
Figure 1.
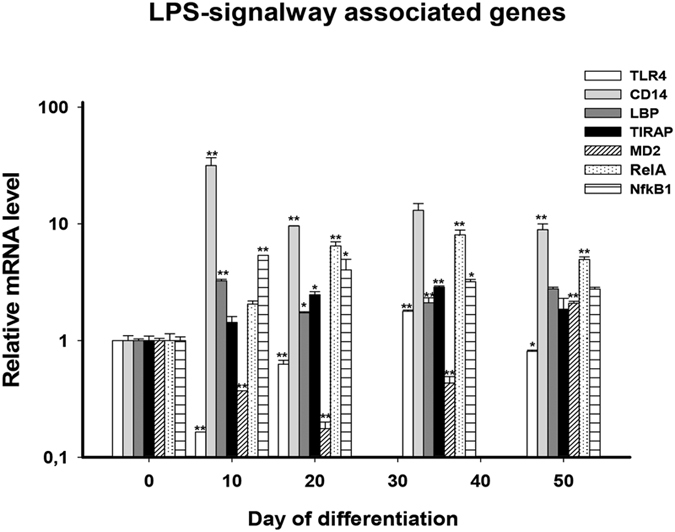
Expression of TLR4-associated signaling genes. Shown are the relative mRNA expression levels of LPS-signaling associated genes at different times of cardiac differentiation process (day 10 to day 50). The hiPSC-status at day 0 was used as relative control. *p < 0.05, **p < 0.01, ***p < 0.001 vs. day 0.
Then we analyzed the mRNA expression of inflammatory factors in hiPSC-CMs treated by different concentrations of LPS for different incubation times. The qPCR-analysis was performed at 6 and 48 hours after LPS-challenge to check whether there are differences of pro- and anti-inflammatory response in acute and prolonged LPS-exposure. The results displayed that the mRNA expression levels of interleukin-1ß (IL-1ß) was increased by LPS treatment for 6 hours (Fig. 2A), reflecting its roles in the pro-inflammatory responses, whereas interleukin-10 (IL-10) was increased by LPS only at 48 hour-treatment (Fig. 2B), consistent with its anti-inflammatory effects. TNF-α, an important mediator of some acute and chronic inflammation, and interleukin-6 (IL-6), a well-known cytokine with pro- and anti-inflammatory properties during acute phase response, was enhanced by LPS-treatment at both 6 and 48 hours (Fig. 2C,D). In addition, the mRNA expression of some chemotactic cytokines, e.g., interleukin-8 (IL-8), CCL2 and CCL5, also increased at 6 hours after LPS-challenge (Fig. 2E–G). Furthermore, TLR4 mRNA expression levels were elevated by LPS irrespective of treating time (Fig. 2H).
Figure 2.
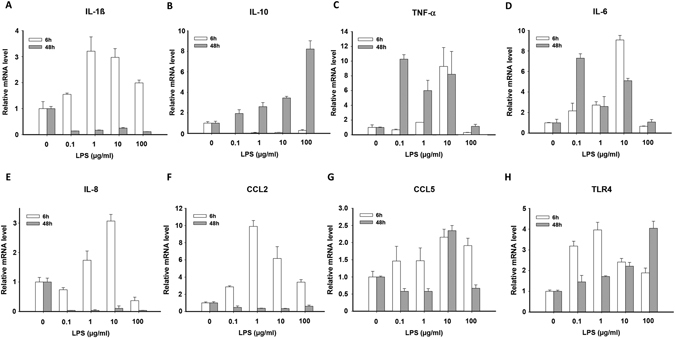
LPS-induced changes in the expression of inflammatory cytokines. Shown are the relative mRNA expression levels of different cytokines after LPS treatment in increasing concentrations for 6 h (white bar) and 48 h (gray bar). 6 hours treatment increased mRNA expression of pro-inflammatory factors IL1 (A), IL8 (E), CCL2 (F), and CCL5 (G), while prolonged treatment (48 h) enhanced expression of anti-inflammatory genes of IL10 (B). TNFα (C) and IL6 (D) were increased by LPS-treatment for both 6 h and 48 h. Main LPS-receptor TLR4 (H) was time-independently raised by LPS-treatment. p-values (One-way ANOVA) for 6 h- and 48 h-treatment (6 h/48 h): CCL2: 0.008/0.0156; CCL5: 0.0855/0.001; IL-1b: 0.0038/ < 0.0001; IL-6: <0.0001/0.0002; IL-8: 0.0008/0.005; TLR4: 0.0021/0.0011; TNF-α: 0.0012/0.0171; IL-10: 0.0010/0.0072.
To check whether the results on mRNA level could be confirmed on protein level we exemplarily applied an ELISA analysis for human IL-6. We found that the detectable IL-6 protein in the supernatants of hiPSC-CMs was also increased after LPS-treatment for 6 h and 48 h (Figure S1A). Then we used fluorescence-activated cell sorting (FACS) to check the receptors of IL-6 in LPS-treated hiPSC-CMs. We could demonstrate that CD 126, a major receptor of IL-6, was undetectable in hiPSC-CMs either with or without LPS-challenging (Figure S1B, p3 gate), whereas cardiac troponin T (TNNT) was detected in 50–90% of the cells (Figure S1B, p2 gate). These data suggest that CD126 does not exist in hiPSC-CMs. Further, we analyzed soluble CD130 (sCD130), a well-known IL-6 signal transducer33, 34. We observed a significant increase of sCD130 in hiPSC-CMs supernatants after prolonged LPS-treatment (48 h) with higher concentrations (Figure S1C).
For optical verifying of our results we further established an immunohistochemistry protocol. First we stained our hiPSC-CMs monolayer with cardiac TNNT. Nuclear staining was performed with DAPI. We could show striated cardiac muscle cells (Figure S2A,B). In the next step we supplemented a titin antibody to the first protocol, where we could detect the structural proximity of both proteins in cardiac muscle cells (Figure S2C). Finally we applied 10 µg/ml LPS on hiPSC-CMs monolayer and stained after 6 h treatment with a NF-κB antibody, which resulted in nuclear-near fluorescence signals (Figure S2D).
LPS induced cell injury
Cell injury happens frequently in inflammatory responses. Troponin measurement for sepsis-induced cardiac damage was previously described35. Therefore we assessed the cell injury in hiPSC-CMs using different techniques. First we used ELISA to measure the concentration of TNNT in culture medium with the same amount of cells treated either with vehicle or different concentrations of LPS for 6 and 48 hours. We found that the TNNT level in the culture medium was not significantly changed by LPS-treatment for 6 hours but increased by LPS-treatment for 48 hours in a dose-dependent manner (p < 0.05, one-way ANOVA, Fig. 3A).
Figure 3.
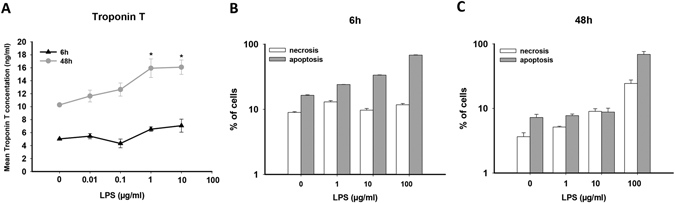
LPS induced cell impairment. (A) ELISA analysis showing that 48 h-but not 6 h-treatment with LPS increased concentrations of human Troponin T (TNNT) in supernatants of LPS-treated hiPSC-CMs. p-values (test for linear trend): 6 h > 0.05, 48 h < 0.05. (B,C) FACS analysis for necrosis (7AAD positive) and apoptosis (Annexin-V positive) ratio of hiPS-CMs treated by LPS in different concentrations for 6 and 48 hours. 6 h-treatment showed a dose dependent increase of apoptotic cells ratio, but no influence on necrotic ratio (B). 48 h-treatment showed a dose dependent increase of necrotic cells ratio (C). The increase in apoptotic ratio was observed only when LPS concentration was raised to 100 µg/ml (p < 0.001 vs. 0 µg/ml). Test for linear trend: 6 h, necrosis, p > 0.05, apoptosis, p < 0.001; 48 h, necrosis, p = 0.003.
Then we examined apoptosis- and necrosis- rate of hiPSC-CMs challenged by LPS. For this aim, FACS with a Annexin-V-APC/7-AAD conjugated antibody was employed to analyze the rate of apoptotic and necrotic cells treated with 1, 10 and 100 µg/ml of LPS for 6 or 48 hours (Fig. 3B,C). In 6 hour-treatment, the rate of apoptosis but not necrosis was enhanced by LPS with increasing concentrations (Fig. 3B). In 48 hour-treatment, the dose-dependent enhancement of necrosis was clearly displayed, whereas the apoptosis rate was not elevated until 100 µg/ml LPS (Fig. 3C).
LPS changed expression level of ion channels
The clinic fact that patients with severe infections have frequently heart dysfunctions including arrhythmias led us to check whether ion channel expression levels are also changed in hiPSC-CMs treated by LPS. To this end, hiPSC-CMs were treated for 48 hours by 3 concentrations of LPS (0.1, 1, and 10 µg/ml). Expression levels of mRNA of ion channels were analyzed by qPCR. LPS of 0.1 and 1 µg/ml reduced the expression level of SCN5A (Na+ channel, Nav1.5, Fig. 4A) but increased the expression of SCN10A (Na+ channel, Nav1.8, Fig. 4B), CACNA1C (L-type Ca2+ channel, Fig. 4C), KCNH2 (IKr, Kv11.1, Fig. 4D), KCND3 (Ito, Kv4.3, Fig. 4E), ABCC8 (KATP, beta-subunit SUR1, Fig. 4I), KCNN2 (SK2, Fig. 4J). No significant changes in expression of KCNQ1 (IKs, Kv7.1, Fig. 4F), KCNK3 (TASK-1, Fig. 4G), KCNJ11 (KATP, alpha-subunit, Fig. 4H), KCNN3 (SK3, Fig. 4K), and SLC8A1 (NCX1, Fig. 4L) were detected. Remarkably, 10 µg/ml LPS reduced consistently the channel expression either significantly or by tendency except for KCNH2 and SLC8A1, probably due to cell injury by high concentration of LPS. Therefore, only the concentrations of 0.1 and 1 µg/ml were used for the following studies.
Figure 4.
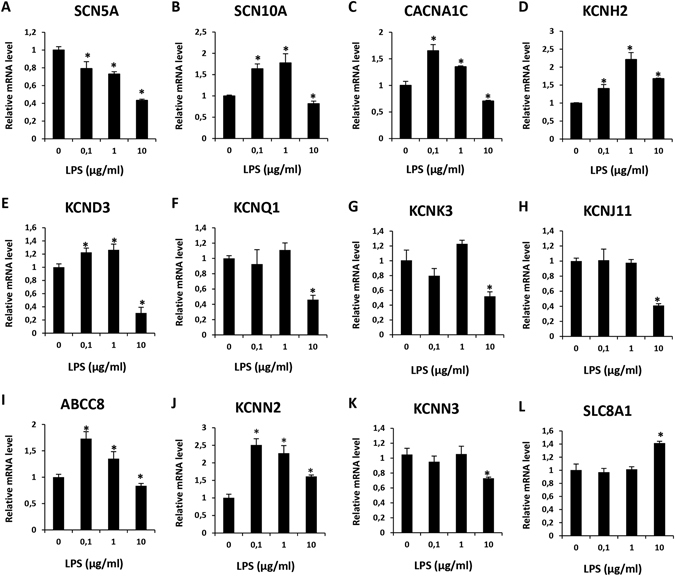
LPS-induced changes in mRNA expression of ion channels. Relative mRNA levels of different ion channels were analyzed by qPCR in hiPSC-CMs after treatment with LPS in different concentrations for 48 h, Values given are mean ± SEM. *p < 0.05 vs control (0 µg/ml).
LPS prolonged action potential duration (APD)
To investigate the effects of LPS on the electrophysiological properties of cardiomyocytes, hiPSC-CMs were treated by 0.1 and 1 µg/ml LPS for 48 hours and then the membrane potentials and currents were measured. LPS-treatment did not change the RP (resting potential) and APA (amplitude of action potential), but enhanced Vmax (maximal velocity of depolarization) and prolonged both APD50 (repolarization at 50%) and APD90 (repolarization at 90%) significantly (Fig. 5). Vmax was enhanced from 36. ± 1 4.5 V/s to 46.6 ± 1.8 V/s (p = 0.054). APD50 was prolonged from 62.6 ± 11.2 ms to 116.8 ± 18.6 ms (p < 0.05) and APD90 from 219.1 ± 35.4 ms to 377.4 ± 33.5 (p < 0.05).
Figure 5.
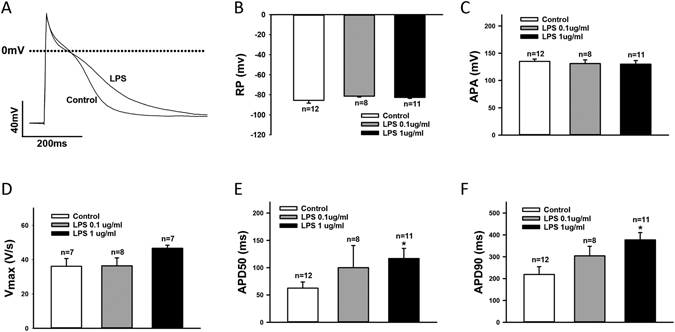
LPS-treatment prolonged APD. (A) Representative traces of action potentials (AP) in control and LPS-treated hiPSC-CMs. (B) Mean values of resting potentials (RP). (C) Mean values of action potential amplitude (APA). (D) Mean values of maximal upstroke velocity of AP (Vmax). (E) Mean values of APD at 50% repolarization (APD50). (F) Mean values of APD at 90% repolarization (APD90).Values given are mean ± SEM. n, number of cells. *p < 0.05.
Effects of LPS on sodium channel currents
To assess the ion channel currents that may contribute to the observed changes of APs by LPS, sodium channel currents (INa) were checked in LPS-treated hiPSC-CMs (Fig. 6). LPS (1 µg/ml) increased the peak INa but reduced the late INa (Fig. 6B,C). Although the changes did not reach statistical significance, the test for linear trend showed a tendency of significance (p = 0.08). The peak INa was changed by 1 µg/ml LPS from 41.2 ± 10.7 to 131.7 ± 50.7 pA/pF (p > 0.05,) and the late INa from 620.6 ± 172.2 to 531.1 ± 64.8 pA*mV/pF (p > 0.05), respectively. No changes in the activation of INa were detected (Fig. 6D and G). A shift to more negative potential of the inactivation curve was observed in cells treated by 0.1 µg/ml LPS (Fig. 6E and H), i.e, V0.5 (the half maximal voltage of inactivation) was shifted from −74.4 ± 1.9 to −83.7 ± 3.4 mV (p < 0.05). In addition, the recovery from inactivation of INa was speeded up by 0.1 µg/ml LPS (Tau was reduced from 31.0 ± 1.3 to 20.8 ± 0.5 ms, p < 0.05, Fig. 6F and I).
Figure 6.
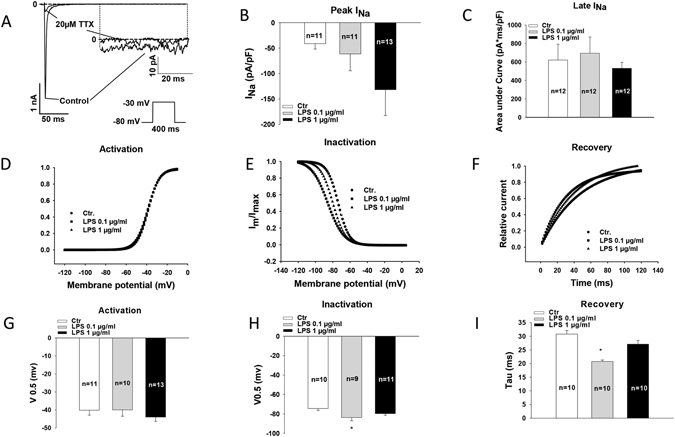
Effects of LPS on INa. (A) Representative traces of INa at −30 mV in absence and presence of 20 µM TTX. (B) Averaged peak INa at −30 mV in control and LPS-treated hiPSC-CMs. (C) Averaged values of late INa at −30 mV. (D) Representative activation curves of peak INa. (E) Representative inactivation curves of peak INa. (F) Representative curves of recovery from inactivation of peak INa. (G) Mean values of voltages at 50% of steady-state activation of peak INa (V0.5). (H) Mean values of voltages at 50% of steady-state inactivation of peak INa. (I) Mean values of time constants (Tau) of recovery from inactivation of peak INa. Values given are mean ± SEM. n, number of cells. *p < 0.05.
LPS reduced small conductance calcium-activated K+ channel currents (ISK1–3)
The detected effects of LPS on INa cannot explain the APD-prolongation. Therefore we further assessed the ion channel currents possible for prolonging APD. We found that ISK1–3 was significantly reduced by LPS-treatment (Fig. 7 and S3,S4). To separate ISK1–3 from other ion channel currents, two SK1–3 channel blockers, apamin (100 nM) and NS8593 (10 µM), were used. The blocker-sensitive currents were analyzed as ISK1-3. In the presence of 1 µg/ml LPS, apamin- and NS8593-sensitive currents (at 40 mV) were reduced from 1.3 ± 0.6 pA/pF to 0.1 ± 0.1 pA/pF (p < 0.05) and from 1.2 ± 0.3 pA/pF to 0.4 ± 0.1 pA/pF (p < 0.05), respectively.
Figure 7.
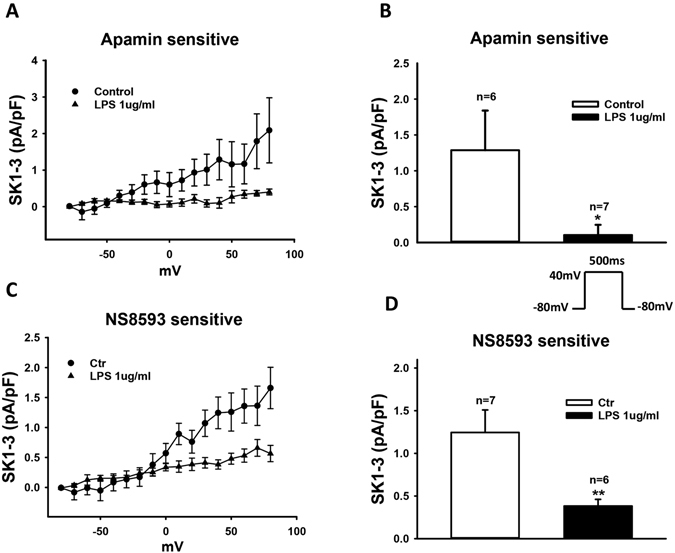
LPS-treatment attenuated SK1-3 channel currents (ISK1-3). Membrane currents were recorded in absence and presence of 100 nM apamin (A and B) or 10 µM NS8593 (C and D), blockers of SK1-3 channels. The blocker-sensitive currents were analyzed as ISK1-3. (A) Current-voltage (I–V) relationship curves of apamin-sensitive currents recorded from −80 to +80 mV with the holding potential of −80 mV. (B) Mean values of apamin-sensitive currents at +40 mV. (C) I–V curves of NS8593-sensitive currents. (D) Mean values of NS8593-sensitive currents at +40 mV. Values given are mean ± SEM. n, number of cells. *p < 0.05, **p < 0.01.
LPS enhanced Na/Ca-exchanger currents (INCX)
We also detected that INCX was significantly enhanced by LPS-treatment (Fig. 8). To separate the INCX, 5 mM NiCl2, a NCX- blocker, was used. LPS of 1 µg/ml enhanced the Ni2+-sensitive currents (INCX) at +60 mV and −100 mV from 1.6 ± 0.3 to 3.5 ± 0.8 pA/pF (p < 0.05) and from −1.7 ± 0.5 to −7.3 ± 2.2 pA/pF (p < 0.05), respectively.
Figure 8.
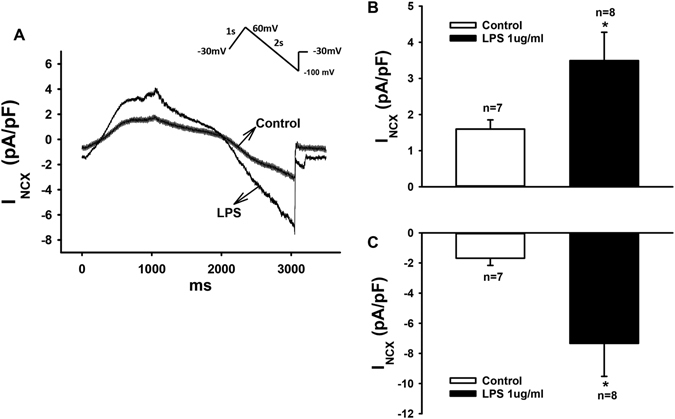
LPS-treatment enhanced Na/Ca exchanger currents (INCX). INCX was evoked by ramp pulses (100 mV/s) at 0.5 Hz from −30 to +60, then to −100 mV with the holding potential of −30 mV. NiCl2 (5 mM) was used to isolate INCX from other currents. (A) Representative traces of Ni2+-sensitive currents in absence and presence of LPS (1 µg/ml for 48 hours). (B,C) Mean values of peak INCX at +60 and −100 mV. Values given are mean ± SEM. n, number of cells. *p < 0.05.
Effects of LPS on other ion channels that may influence APs
In addition to INa, INCX and ISK1-3, many other ion channels are known to be important for APD. Therefore we checked the effects of LPS-treatment on different ion channels that may be involved in the APD-prolongation. LPS enhanced Ito (transient outward current, Figure S5A,B) but failed to change ICa-L (L-type Ca2+ channel currents, Figure S5C,D), IKr (rapidly activating delayed rectifier current, Figure S6A,B), IKs (slowly activating delayed rectifier current, Figure S6C,D), IK-pH (pH-sensitive K+ channel currents, Figure S7A,C) and IKATP (ATP-sensitive K+ channel current, Figure S7D–F).
Effects of LPS on intracellular Ca2+ concentration ([Ca2+]i)
The prolonged APD and enhanced INCX may influence the intracellular Ca2+ level. Therefore we measured [Ca2+]i by Ca2+ imaging with a Ca2+-sensitive fluorescent indicator fluo-3. LPS-treatment (1 µg/ml for 48 h) did not significantly influence systolic and diastolic Ca2+-levels (Figure S8).
Discussion
To our knowledge, this is the first study to demonstrate that hiPSC-CMs possess the functional reaction system involved in endotoxin-induced inflammation and displayed inflammatory responses and ion channel dysfunctions when they were challenged by LPS, suggesting hiPSC-CMs as a successful model of inflammation in cardiomyocytes, which may be useful for future studies on some bacterium-induced inflammatory human cardiac diseases.
In our lab, hiPS-CMs have been successfully generated from a healthy donor with the majority of ventricular myocytes. We have demonstrated that the cells expressed cardiac specific proteins, like TNNT2, MYH6, NKX2.5, and ACTN236. They exhibited cardiac striated structure (Figure S2A–C) and contracted either spontaneously or when stimulated. They possess most ion channels reported to exist in human cardiomyocytes and displayed electrophysiological properties similar to that of native cardiomyocytes. Therefore we tried to establish a hiPSC-CM-model mimicking LPS-induced inflammation in cardiac myocytes, which is still lacking.
To know whether hiPSC-CMs can model inflammation induced by LPS, we checked whether the correspondent innate signalings exist in hiPSC-CMs and whether the cells can respond to LPS-challenge. This study demonstrated by qPCR that hiPSC-CMs express the mRNA of the LPS-receptor, TLR4, and its associated signaling proteins, LBP, CD14, MD2, and TIRAP with higher levels at days 35 or 50 after start of differentiation than that at day 0 (Fig. 1). This is consistent with previous findings showing the expression of TLR4 in human cardiomyocytes3, 4. We also detected the mRNA expression of RelA and NFkB1, subunits of the transcription factor NF-kB, which is an important downstream factor of TLR4, being higher at day 50 after differentiation than at day 0.
Activation of NF- kB can induce production of different inflammatory factors. We found out that hiPSC-CMs, like native cardiomyocytes, did express pro-inflammatory and anti-inflammatory as well as chemotactic cytokines when stimulated by LPS in different concentrations for different incubation times (Fig. 2). The well-known pro-inflammatory factor, IL-1ß, and also the chemotactic cytokines, CCL2, CCL5 and IL-8 showed a dose-dependent increase of mRNA expression level after LPS-treatment for 6 hours, suggesting their contribution to the early pro-inflammatory and chemotactic reaction phase of inflammation. On the other hand, the expression of the well-known anti-inflammatory cytokine IL-10 was increased at 48 hour- but not 6 hour-treatment with LPS (Fig. 2B), which is indicative of the initiation of anti-inflammatory processes under prolonged inflammatory circumstances. The mRNA level of TNF-α and IL-6, the important downstream factors of NF- kB, was increased in both early (6 hours) and late (48 hours) LPS-exposure (Fig. 2C,D), which was consistent with their pro- and anti-inflammatory functions. TLR4 expression was also raised by LPS-challenge, consistent with a previous report3. Moreover, immunohistochemistry-analysis showed a nuclear-near signal for NF-kB-targeting antibody after LPS-treatment (Figure S2D), suggesting that the LPS-mediated signaling led to the activation and nuclear translocation of the transcription factor NF-kB.
To further analyze the LPS/NF-kB-signaling, we analyzed IL-6 and its receptors, which have been shown to be involved in some heart diseases35. We first checked the protein-level of IL-6 in hiPSC-CMs treated with LPS. The detectable IL-6 protein levels in hiPSC-CM supernatants were elevated by LPS-treatment for 6 and 48 hours (Figure S1A), which is consistent with the qPCR-data and shows the correlation of transcriptome and protein level in the inflammatory response. It is known that IL-6 initiates inflammatory responses through either the membrane-bound or soluble IL-6 receptors (CD126). The membrane-bound CD126 exists predominantly in hepatocytes, neutrophils, monocytes/macrophages, and some lymphocytes37. The circulating soluble form of CD126, which can be detected in various body fluids, is secreted by monocytes, hepatocytes, and endothelial cells37. Both membrane-bound and soluble CD126 are associated with the signal transducing receptor glycoprotein 130 (gp130, also known as CD130, IL6ST, IL6-beta). The membrane-bound CD126-mediated signaling (classical IL-6 signaling) can be initiated only in cells expressing CD126. In the soluble CD126-mediated signaling (IL-6 trans-signaling), cells do not have to express CD126, as CD130 is widely expressed in most cell types. Therefore, we checked the LPS-treated-hiPSC-CMs for the presence of CD126 and CD130. As we expected, FACS analysis showed no fluorescence signal detectable for CD126 irrespective of LPS-challenge (Figure S1B). However, soluble CD130 (sCD130) was detectable in the hiPSC-CM supernatants and the protein-level was raised by prolonged LPS-exposure (Figure S1C). This may imply that a local IL-6 trans-signaling, like in native cardiomyocytes, can take place in the cells. Taking together, it is quite convincing that in hiPSC-CMs LPS stimulated TLR4 and activated NF-kB signaling, which in turn elevated production of inflammatory factors. The inflammatory factors may trigger inflammatory responses in cells.
The inflammation may result in cell injury. Therefore we measured the cardiac TNNT in the cell culture supernatant from hiPSC-CMs treated by LPS (Fig. 3A). Acute (6 hours) LPS-exposure did not influence the TNNT-level, indicative of no manifest cell injury. When applied for longer time (48 hours), LPS increased the TNNT-level in a dose-dependent manner. This implies that either stronger or longer inflammatory responses may enhance the cell impairment.
The cell injury may result from cell apoptosis and necrosis. Our data demonstrated that short time (6 hours) LPS-treatment increased the apoptosis but not necrosis ratio in a concentration dependent manner, while long time (48 hours) treatment with LPS enhanced necrotic cells ratio with increasing concentrations (Fig. 3B,C). These data, together with the TNNT-analysis, suggest that in the early phase of inflammation the cell injury was slight and resulted mainly from apoptosis induction through LPS associated signal ways. Prolonged LPS exposure led to severe injury because of a higher proportion of necrotic cells.
It is well-known that some infections, especially some severe infections like sepsis, may cause cardiac dysfunctions including arrhythmias. In this study, hiPSC-CMs treated by LPS displayed APD-prolongation (Fig. 5). APD-prolongation may be arrhythmogenic, which may explain, at least partially, the tachyarrhythmias observed in inflammatory models of animal or in patients with sepsis2, 38–40.
To understand the reason for APD-prolongation, we assessed the changes in ion channel currents that may contribute to APD. We found that peak INa was enhanced but late INa was reduced by LPS (Fig. 6). Reduced late INa should shorten APD, instead of prolonging APD. Next, we observed that LPS increased Ito significantly (Figure S5A,B), which should shorten APD, too. Furthermore, no significant changes in ICa-L (Figure S5C,D), IKr (Figure S6A,B), IKs (Figure S6C,D), pH-sensitive K+ channel currents including acidosis-and alkaline-inhibited K+ channel currents (Figure S7A–C), and KATP (Figure S7D–F) channel currents were detected in LPS-treated cells. All of these data are not consistent with the APD-prolongation. However, we found that LPS reduced ISK1-3 and enhanced INCX (Figs 7 and 8), both of which can result in ADP-prolongation. Increased INCX can also lead to DAD and in turn to tachyarrhythmias. This may be a reason for arrhythmias in patients with sepsis. APD-prolongation and enhanced INCX may elevate intracellular Ca2+-concentration. But we did not observe significant changes of systolic and diastolic Ca2+-levels in cells treated by LPS (1 µg/ml for 48 h). This may suggest extra effects of LPS that counteracted the effects of APD-prolongation and INCX-enhancement on intracellular Ca2+-concentration.
To investigate the reason for changes of ion channel currents, we analyzed the channel mRNA expression levels with the qPCR technique (Fig. 4). The mRNA levels of the studied channels were changed by LPS in a concentration-dependent manner. High concentration of LPS (10 µg/ml) consistently reduced expression of all the channels except NCX1 (Fig. 4), suggesting toxic effects and cell injury. Low concentrations (0.1 or 1 µg/ml) of LPS influenced differentially the ion channel expression. SCN5A was reduced but SCN10A was increased (Fig. 4A and B). Whether the former led to reduction in late INa (Fig. 6C) and the latter to the enhancement of peak INa (Fig. 6B) cannot be decided because the contributions of both channel types to INa were not assessed in this study. The expression of SK2 but not SK3 channel, which have been reported to be the important SK channel isoforms in human cardiomyocytes41, was increased (Fig. 4J). This is not consistent with the reduced ISK1-3 (Fig. 7). Whether the protein levels of SK2 and SK3 changed in LPS-treatment needs to be addressed. No changes of LSC8A1 (NCX1, Fig. 4L) expression were detected, this did not fit the enhanced INCX (Fig. 8) either. The expression of KCND3 (Ito) was increased (Fig. 4E), consistent with the increased currents (Figure S5A,B). The expression of KNCH2 (IKr), CACNA1C (ICa-L) and ABCC8 (KATP , beta-subunit) was also increased (Fig. 4C, I and K) but not consistent with the current measurements (Figure S5–S7). In addition, the expressions of KCNQ1 (IKs), KCNK3 (TASK-1) and KCNJ11 (KATP channel, alpha subunit) were not significantly influenced by LPS (Fig. 4F, G, H), same as the current measurements (Figure S6 and S7). Given the inconsistency in patch-clamp and qPCR measurements, further studies are needed to clarify the mechanisms underlying observed changes in ion channel currents. Nevertheless, the patch-clamp study, a widely accepted and used technique for functional analysis of ion channels, demonstrated in this study clearly that hiPSC-CMs exhibit ion dysfunctions when irritated by LPS.
In summary, by using the established hiPSC-CM platform we assessed intensively the LPS-induced responses including changes in inflammatory cytokines, cell-injury, apoptosis, necrosis, and electrophysiological properties. The results demonstrated that hiPSC-CMs possess the necessary molecular basis of the inflammatory reaction system and displayed inflammatory responses with electrical dysfunctions when challenged by LPS. This indicates that hiPSC-CMs can model some bacterium-induced inflammatory heart diseases, at least in certain aspects and might be a good alternative for mechanistic and therapeutic studies on some inflammatory cardiac diseases.
Study limitations
Some limitations should be considered in extrapolating the data from the current study. hiPSC-CMs from only one healthy donor were used for this study. hiPSC-CMs possess similarities but also distinct differences in their physiological properties when compared to adult human cardiomyocytes. Owing to the limited availability of cells, native human cardiomyocytes were not used for comparison in this study. Therefore we cannot rule out the differences among individuals and between hiPSC-CMs and native human cardiomyocytes regarding the responses to LPS. In addition, the reasons for some discrepancies in ion channel studies were not clarified. Some mRNA expressions are not consistent with the current measurements. Therefore the mechanisms underlying the observed changes of ion channel currents need to be addressed in further studies.
Material and Methods
Ethics statement
The skin biopsy from a healthy donor was obtained with written informed consent. The study was approved by the Ethics Committee of Medical Faculty Mannheim, University of Heidelberg (approval number:2009-350N-MA) and conducted in accordance with the Helsinki Declaration of 1975, as revised in 1983.
Generation of human iPS cells
The human iPS cells (hiPSCs) were generated from primary human fibroblasts derived from skin biopsies of a female healthy donor using lentiviral particles carrying the transactivator tTA and an inducible polycistronic cassette containing the reprogramming factors OCT4, SOX2, KLF4 and c-MYC as previously described42, 43. Generated hiPSCs were cultured under feeder free conditions. To investigate pluripotency, hiPSCs were subjected to a teratoma-formation assay42.
Generation of hiPSC-CMs
Frozen aliquots of hiPSCs were thawed and cultured without feeder cells and differentiated into hiPSC-CMs as described with some modifications44. Culture dishes and wells were coated with Matrigel (Corning). Culture medium of hiPSCs was TeSR-E8 (Stemcell Technologies) and for hiPSC-CMs we used RPMI 1640 Glutamax (Life Technologies) containing sodium pyruvate, Penicillin/Streptomycin, B27 (Life Technologies) and ascorbic acid (Sigma Aldrich). Adding of CHIR99021 (Stemgent), BMP-4 (R&D Systems), Activin A (R&D Systems), FGF-2 (MiltenyiBiotec) and IWP-4 (Stemgent) at different time points induced the cells to differentiate into hiPSC-CMs during 3 weeks. During the third week a lactate (Sigma, Germany) containing RPMI-medium without glucose and glutamine (WKS, Germany) was added to select for cardiomyocyte cells. At 30 to 60 days of culture with basic culture medium, cardiomyocytes were dissociated from 24 well plates and plated as single cells on matrigel-coated 3.5 cm petri dishes for patch-clamp measurements. The cells were incubated with 300 µl (150 U) collagenase CLS I (Worthington, Germany) for 40 min at 37 °C, washed with PBS and incubated with 0.05% Trypsin-EDTA (Life Technologies) for 4 min at 37 °C. After adding of RPMI medium containing 10% FCS, cells were centrifuged at 250 × g for 2 min at room temperature, the supernatant discarded and the cells resuspended with basic culture medium. The cells were plated on the 3.5 cm petri dishes at a density of 2–4 × 104 cells/dish for subsequent patch-clamp experiments.
Polymerase-Chain-Reaction Assays
For quantitative evaluation of the steady-state mRNA expression in hiPSC-CM cultures, total RNA was prepared using the RNeasy mini kit (Qiagen, Hilden, Germany), including DNAse treatment. The RNA was reverse transcribed to cDNA with oligo (dT)15 primers using AMV reverse transcriptase according to standard protocols (Roche Applied Science, Germany, AMV Cat. No.11495062001). The cDNA was amplified by qPCR on a Stratagene MX 3005 P real time cycler (Stratagene, USA) using a PCR-mix with hot start Taq DNA polymerase and SYBR-Green (SibirRox Hot Mastermix, Bioron, Germany, Cat No. 119405) in the presence of sense and antisense primers (400 nM each). The sense- and antisense-primers for all human genes were supplied as RT² qPCR Primer Assays from Qiagen, Germany (see supplementary material Table 1, list of genes, RefSeq numbers and primers). The PCR condition consisted of 95 °C for 5 min (denaturation of DNA and activation of Taq polymerase), followed by 40 cycles of 95 °C for 15 sec and 60 °C for 1 min (annealing and extension), followed by melting-curve analysis to verify the correctness of the amplicon. Relative quantification of mRNA expression was calculated as follows: The expression of the mRNA of the gene of interest relative to the housekeeping gene GAPDH in samples from treated or untreated (Control) cells was calculated by the ΔΔCT method, based on the threshold cycle (CT), as fold change = 2−Δ(ΔCT), where ΔCT = CTgene of interest − CTGAPDH and Δ(ΔCT) = ΔCTtreated −ΔCT Control 45. Four equally treated wells were pooled to one biological replica to enrich the RNA-concentration. Every experiment was made with at least 2 biological replica. qPCR analysis of each biological replica was measured in duplicate (2 technical replica). The GenBank NCBI reference sequence number (RefSeq No.) and catalog number of all the investigated genes are listed in Table S1.
Enzyme-Linked Immunosorbent Assays (ELISA)
Concentrations of human interleukin 6, sCD130 and troponin t were measured in the cell culture supernatants by ELISA (RayBiotech:ELH-IL6, ELH-sgp130 and ELH-troponin t) according to the manufacturer’s instructions. The dilution of the supernatants was 1:2.
Flow cytometric analysis (FACS)
Cardiomyocytes were dissociated from cell culture plates as described in “Generation of hiPSC-CMs”. Afterwards cells were washed with PBS, fixed with 1% formaldehyde solution (Merck) and permeabilized with perm/wash buffer for 10 minutes (1:10 dilution, BD Bioscienes cat# 554723). Then cells were incubated with different antibodies for 30 minutes at 4 °C in the dark. Finally antibody solution was washed out with PBS. Samples were measured immediately by FACS analysis, which was performed with CANTO II (BD). Compensation-measurement was considered if necessary by using OneCompeBeads (eBioscience). All antibodies were checked for correct concentration and non-specific fluorescence signal by previously testing with titration (1:10000 to 1:10 dilution) and comparison to negative control respectively by using isotype controls. The antibodies used for FACS analysis were AlexaFluor 647-conjugated cardiac Troponin T (1:10000 dilution, BD Biosciences cat# 565744), and PerCP/Cy5.5-conjugated CD 126 antibody (1:20 dilution, BioLegend cat# 352812). Apoptosis- and necrosis- ratio of LPS-treated hiPSC-CMs were measured with the Annexin-V-APC/7-AAD Kit (BioLegend cat# 640930, 420201).
Immunofluorescence staining
Cardiomyocytes were dissociated from cell culture plates as described in “Generation of hiPSC-CMs”. The separated cells were resuspended with serum-free medium and transferred to 4 chamber glass slides (Corning). After resting for 48 h the cells were washed with PBS. Fixation was performed using 4% paraformaldehyde (Roti-Histofix Roth cat# P087.4) for 10 minutes. Afterwards the cells were permeabilized for 10 minutes with 0,5% Triton X-100 (Sigma cat# T-9284) and blocked with PBS containing 1% bovine serum albumin (Sigma cat# A-6003). After each step they were washed with PBS. Finally cells were incubated with several antibodies at 4 °C in the dark. After one hour antibodies were washed out with PBS. Nuclear staining was performed with Vectashield mounting medium with DAPI (Linaris cat# H-1200). The antibodies used in this study were FITC-conjugated cardiac troponin T antibody (1:66 dilution, biorbyt cat# orb187249), cy5-conjugated titin antibody (1:66 dilution, BiossInc cat# bs-9861R-Cy5), Alexa Fluor 488-conjugated NfκB-p65 subunit antibody (1:40 dilution, BD biosciences cat# 558421). Pictures were taken with Leica DMRE fluorescence microscope, DFC3000G/DFC 450c camera equipped with Leica Application suite 4.4 software.
Measurement of intracellular calcium concentration
To measure the intracellular Ca2+ concentration ([Ca2+]i), cells were loaded with the fluorescent Ca2+-indicator Fluo-3 AM. First, 1.5 ml PSS (see below) was added into a petri dish with hiPSC-CMs cultured for 2 to 4 days. The following steps were executed under consideration of the light sensitivity of the fluorescent Ca2+ Indicator Fluo-3. Then, 50 µg of the membrane permeable acetoxymethyl ester derivative of Fluo-3 was dissolved in 44 µl of the Pluronic F-127 stock solution (20% w/v in DMSO) to get a 1 mM Fluo-3 AM stock solution, which can be stored at −20 °C for a maximum of 1 week. Next, 15 µl of the Fluo-3 AM stock solution were added into 1.5 ml PSS resulting in a final concentration of 10 µM Fluo-3 and the dish was agitated carefully. The cells were incubated at room temperature for 10 minutes in an optically opaque box to protect from light. Thereafter, the PSS was carefully sucked out and discarded and the cells were washed with PSS for 4–5 times. Finally, the cells in PSS were kept at room temperature for about 30 minutes for de-esterification before measurements. After de-esterification the fluorescence of the cells was measured by using Cairn Optoscan (Cairn Research, UK) calcium imaging system. Fluorescence is excited by 488 nm and emitted at 520 nm. Changes in [Ca2+]I were described by
, where k d = dissociation constant of Fluo-3 (864-nmol/L), F = Fluo-3 fluorescence; F max = Ca2+-saturated fluorescence obtained at the end of each experiment46.
Patch-clamp
Standard patch-clamp recording techniques were used to measure the action potential (AP) and ion channel currents in the whole-cell configuration. Patch electrodes were pulled from borosilicate glass capillaries (MTW 150F; world Precision Instruments, Inc., Sarasota, FL) using a DMZ-Universal Puller (Zeitz-Instrumente Vertriebs GmbH, Martinsried, Germany) and filled with pre-filtered pipette solution (see below). Pipette resistance ranged from 1–2 MΩ. Electrode offset potentials were zero-adjusted before a Giga-seal was formed. After a Giga-seal was obtained, fast capacitance was first compensated and then the membrane under the pipette tip was disrupted by negative pressure to establish the whole-cell configuration. To determine the cell capacitance, a voltage pulse from −80 to −85 was given to record the cell capacitance transient current. Then we integrated the area under transient current curve and divide the area value by 5 mV to get the whole cell capacitance in pF. Thereafter the membrane capacitance (Cm) and series resistance (Rs) were compensated (60–80%). Liquid junction potentials were not corrected. Signals were acquired at 10 kHz and filtered at 2 kHz with the Axon 200B amplifier and Digidata 1440A digitizer hardware as well as pClamp 10.2 software (Molecular Devices, Sunnyvale, CA). Myocytes were held at different potentials and different voltage clamp protocols were used measuring different currents. Measured currents were normalized to the membrane capacitance. Current-voltage (I-V) relationships were generated by plotting the current density against voltages. For assessing the activation and inactivation of peak INa, the membrane conductance or relative current (Im/Imax) were plotted against voltages and then fitted with Boltzmann distribution to obtain the half maximal voltage (V0.5) of activation or inactivation. The membrane conductance (Gm) was calculated as Gm = I/(Em − Erev), where I is macroscopic current, Em is the test membrane potential, and Erev is the reversal potential. To measure the recovery from inactivation of sodium channels, the protocol of double-pulse with increasing intervals was used. The peak INa elicited by the second pulse was normalized to that elicited by the first pulse and plotted against the intervals between the two pulses and then fitted with mono-exponential growth to get the time constant of recovery. The TTX sensitive late INa was measured as the area under the current curve integrated from 50 to 350 ms after the beginning of the depolarization pulse. To isolate different ion channel currents, specific channel blockers were used and the blocker-sensitive currents were analyzed. To minimize the effects of run-down of recorded currents on the results of experiments, we carefully monitored the time-dependent change of currents. Recordings were started after the current reached a steady state, normally within 3 to 5 minutes.
The bath solution for AP and K+ channel current measurements contained (mmol/l): 130 NaCl, 5.9 KCl, 2.4 CaCl2, 1.2 MgCl2, 11 glucose, 10 HEPES, pH 7.4 (NaOH). For the Ito measurements, 10 µM nifedipine, 10 µM TTX and 1 µM E-4031 were added in the bath solution to block ICa-L, INa and IKr. For IKr and IKs measurements, 10 µM nifedipine, 10 µM TTX and 5 mM 4-AP were added. The pipette solution contains 10 mM HEPES, 126 mM KCl, 6 mM NaCl, 1.2 mM MgCl2, 5 mM EGTA, 11 mM glucose and 1 mM MgATP, pH 7.4 (KOH). For measuring SK channel currents, appropriate CaCl2 was added to get the free-Ca2+ concentration of 0.5 µM according to the calculation by the software MAXCHELATOR (http://web.stanford.edu/~cpatton/downloads.htm). For measuring IKATP (ATP-sensitive K+ channel current), the ATP-free pipette solution was used.
The bath solution for peak sodium current measurements contained (mmol/l): 20 NaCl, 110 CsCl, 1.8 CaCl2, 1 MgCl2, 10 Hepes, 10 glucose, 0.001 nifedipine, pH 7.4 (CsOH). Microelectrodes were filled with (mmol/l): 10 NaCl, 135 CsCl, 2 CaCl2, 3 MgATP, 2 TEA-Cl, 5 EGTA, 10 HEPES, pH7.2 (CsOH).
The bath solution for late sodium current measurements contained (mmol/l): 135 NaCl, 20 CsCl, 1.8 CaCl2, 1 MgCl2, 10 Hepes, 10 glucose, 0.001 nifedipine, pH 7.4 (CsOH). Microelectrodes were filled with (mmol/l): 10 NaCl, 135 CsCl, 2 CaCl2, 3 MgATP, 2 TEA-Cl, 5 EGTA, 10 HEPES, pH7.2 (CsOH).
The bath solution for ICa-L recordings contained (mmol/l): 140 TEA-Cl, 5 CaCl2, 1 MgCl2, 10 3R4S-chromanol 293B, 10 Hepes, 0.01 TTX, 2 4-AP, pH 7.4 (CsOH). Microelectrodes were filled with (mmol/l): 10 NaCl, 135 CsCl, 2 CaCl2, 3 MgATP, 2 TEA-Cl, 5 EGTA, 10 HEPES, pH7.2 (CsOH).
The bath solution for INCX measurements contained (mmol/l): 135 NaCl, 10 CsCl, 2 CaCl2, 1 MgCl2, 10 Hepes, 10 glucose, 0.01 nifedipine, 0.1 niflumic acid, 0.05 lidocaine, 0.02 dihydroouabain, pH 7.4 (CsOH). Microelectrodes were filled with (mmol/l): 10 NaOH, 150CsOH, 2 CaCl2, 1 MgCl2, 75 aspartic acid, 5 EGTA, pH7.2 (CsOH).
Drugs
E-4031, chromanol 293B, nifedipine, NiCl2, NS8593, niflunic acid, LPS (Lipopolysaccharides from E. coli, source strain ATCC 12740, serotype 0127:B8, gel filtrated, gamma irradiated, cell culture tested, Sigma L 4516), lidocaine, glybenclamide and dihydroouabain are from Sigma Aldrich, 4-AP from RBI, apamin from Alomone Labs, TTX from Carl Roth. E-4031, NiCl2, TTX, 4-AP, apamin, niflumic acid, glybenclamide and dihydrooubain were dissolved in H2O. Nifedipine, NS8593 and chromanol 293B were dissolved in DMSO, lidocaine in ethanol and LPS in RBMI medium. Stock solutions were kept at −20 °C.
Statistics
If not otherwise indicated data are shown as mean ± SEM and were analyzed using InStat© (GraphPad, San Diego, USA) and SigmaPlot 11.0 (Systat GmbH, Germany). By analyzing the data with the Kolmogorov Smirnov test it was decided whether parametric or non-parametric tests were used for analysis. For parametric data one-way ANOVA with Dunnett’s post-test for multiple comparisons (all treated groups versus control) or test of trend were performed. For non-parametric data the Kruskal-Wallis test with Dunn’s multiple comparisons post-test was used. For repeated measurements, e.g. analysis of current-voltage relationships, parametric one-way repeated measures ANOVA with Dunnett’s multiple comparisons post-test was applied. Unpaired Student’s t-test was used for comparisons of two independent groups with normal distribution. p < 0.05 (two-tailed) was considered significant, p < 0.10 (two-tailed) was considered significant by tendency.
Electronic supplementary material
Acknowledgements
This work was supported by the DZHK (German Center for Cardiovascular Research), the BMBF (German Ministry of Education and Research) (grants to MB, JU, TW and WZ). We thank Claudia Liebetrau for her assistance. We are grateful to Stefanie Uhlig for her support in FACS-analysis.
Author Contributions
G.Y., I.E.-B., S.L., X.-B.Z. designed the study; G.Y., Z.Z., I.E.-B., H.L., S.L., X.L., F.B., L.C., J.U. performed experiments and analysis; U.R., W.-H.Z., T.W., M.B., I.A. participated in study conception and design, interpretation of the data and revising the manuscript critically for important intellectual content; G.Y., I.E.-B., S.L., X.-B.Z. drafted the manuscript.
Competing Interests
The authors declare that they have no competing interests.
Footnotes
Gökhan Yücel and Zhihan Zhao contributed equally to this work.
Electronic supplementary material
Supplementary information accompanies this paper at doi:10.1038/s41598-017-03147-4
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Kakihana Y, Ito T, Nakahara M, Yamaguchi K, Yasuda T. Sepsis-induced myocardial dysfunction: pathophysiology and management. Journal of intensive care. 2016;4:22. doi: 10.1186/s40560-016-0148-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Patel N, Shenoy A, Dous G, Kamran H, El-Sherif N. Sepsis-Induced Takotsubo Cardiomyopathy Leading to Torsades de Pointes. Case reports in cardiology. 2016;2016:2384752. doi: 10.1155/2016/2384752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Frantz S, et al. Toll4 (TLR4) expression in cardiac myocytes in normal and failing myocardium. The Journal of clinical investigation. 1999;104:271–280. doi: 10.1172/JCI6709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tavener SA, et al. Immune cell Toll-like receptor 4 is required for cardiac myocyte impairment during endotoxemia. Circulation research. 2004;95:700–707. doi: 10.1161/01.RES.0000144175.70140.8c. [DOI] [PubMed] [Google Scholar]
- 5.Natanson C, et al. Role of endotoxemia in cardiovascular dysfunction and mortality. Escherichia coli and Staphylococcus aureus challenges in a canine model of human septic shock. The Journal of clinical investigation. 1989;83:243–251. doi: 10.1172/JCI113866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ognibene FP, Parker MM, Natanson C, Shelhamer JH, Parrillo JE. Depressed left ventricular performance. Response to volume infusion in patients with sepsis and septic shock. Chest. 1988;93:903–910. doi: 10.1378/chest.93.5.903. [DOI] [PubMed] [Google Scholar]
- 7.Parker MM, et al. Profound but reversible myocardial depression in patients with septic shock. Annals of internal medicine. 1984;100:483–490. doi: 10.7326/0003-4819-100-4-483. [DOI] [PubMed] [Google Scholar]
- 8.Bruni FD, Komwatana P, Soulsby ME, Hess ML. Endotoxin and myocardial failure: role of the myofibril and venous return. The American journal of physiology. 1978;235:H150–156. doi: 10.1152/ajpheart.1978.235.2.H150. [DOI] [PubMed] [Google Scholar]
- 9.Hinshaw LB, et al. Effects of coronary hypotension and endotoxin on myocardial performance. The American journal of physiology. 1974;227:1051–1057. doi: 10.1152/ajplegacy.1974.227.5.1051. [DOI] [PubMed] [Google Scholar]
- 10.Dhainaut JF, et al. Coronary hemodynamics and myocardial metabolism of lactate, free fatty acids, glucose, and ketones in patients with septic shock. Circulation. 1987;75:533–541. doi: 10.1161/01.CIR.75.3.533. [DOI] [PubMed] [Google Scholar]
- 11.Cunnion RE, Schaer GL, Parker MM, Natanson C, Parrillo JE. The coronary circulation in human septic shock. Circulation. 1986;73:637–644. doi: 10.1161/01.CIR.73.4.637. [DOI] [PubMed] [Google Scholar]
- 12.Suffredini AF, et al. The cardiovascular response of normal humans to the administration of endotoxin. The New England journal of medicine. 1989;321:280–287. doi: 10.1056/NEJM198908033210503. [DOI] [PubMed] [Google Scholar]
- 13.Flesch M, et al. Effects of endotoxin on human myocardial contractility involvement of nitric oxide and peroxynitrite. Journal of the American College of Cardiology. 1999;33:1062–1070. doi: 10.1016/S0735-1097(98)00660-3. [DOI] [PubMed] [Google Scholar]
- 14.Parrillo JE, et al. A circulating myocardial depressant substance in humans with septic shock. Septic shock patients with a reduced ejection fraction have a circulating factor that depresses in vitro myocardial cell performance. The Journal of clinical investigation. 1985;76:1539–1553. doi: 10.1172/JCI112135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Natanson C, et al. Endotoxin and tumor necrosis factor challenges in dogs simulate the cardiovascular profile of human septic shock. The Journal of experimental medicine. 1989;169:823–832. doi: 10.1084/jem.169.3.823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Vincent JL, et al. Administration of anti-TNF antibody improves left ventricular function in septic shock patients. Results of a pilot study. Chest. 1992;101:810–815. doi: 10.1378/chest.101.3.810. [DOI] [PubMed] [Google Scholar]
- 17.Kumar A, et al. Tumor necrosis factor alpha and interleukin 1beta are responsible for in vitro myocardial cell depression induced by human septic shock serum. The Journal of experimental medicine. 1996;183:949–958. doi: 10.1084/jem.183.3.949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kumar A, et al. Role of nitric oxide and cGMP in human septic serum-induced depression of cardiac myocyte contractility. The American journal of physiology. 1999;276:R265–276. doi: 10.1152/ajpregu.1999.276.1.R265. [DOI] [PubMed] [Google Scholar]
- 19.Potz, B. A., Sellke, F. W. & Abid, M. R. Endothelial ROS and Impaired Myocardial Oxygen Consumption in Sepsis-induced Cardiac Dysfunction. Journal of intensive and critical care2 (2016). [DOI] [PMC free article] [PubMed]
- 20.Plociennikowska, A. et al. Contribution of CD14 and TLR4 to changes of the PI(4,5)P2 level in LPS-stimulated cells. Journal of leukocyte biology, doi:10.1189/jlb.2VMA1215-577R (2016). [DOI] [PubMed]
- 21.Kopp F, Kupsch S, Schromm AB. Lipopolysaccharide-binding protein is bound and internalized by host cells and colocalizes with LPS in the cytoplasm: Implications for a role of LBP in intracellular LPS-signaling. Biochimica et biophysica acta. 2016;1863:660–672. doi: 10.1016/j.bbamcr.2016.01.015. [DOI] [PubMed] [Google Scholar]
- 22.Lu Y, et al. TLR4 plays a crucial role in MSC-induced inhibition of NK cell function. Biochemical and biophysical research communications. 2015;464:541–547. doi: 10.1016/j.bbrc.2015.07.002. [DOI] [PubMed] [Google Scholar]
- 23.Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006;126:663–676. doi: 10.1016/j.cell.2006.07.024. [DOI] [PubMed] [Google Scholar]
- 24.Takahashi K, et al. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell. 2007;131:861–872. doi: 10.1016/j.cell.2007.11.019. [DOI] [PubMed] [Google Scholar]
- 25.Yu J, et al. Induced pluripotent stem cell lines derived from human somatic cells. Science. 2007;318:1917–1920. doi: 10.1126/science.1151526. [DOI] [PubMed] [Google Scholar]
- 26.Zhang J, et al. Functional cardiomyocytes derived from human induced pluripotent stem cells. Circulation research. 2009;104:e30–41. doi: 10.1161/CIRCRESAHA.108.192237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Germanguz I, et al. Molecular characterization and functional properties of cardiomyocytes derived from human inducible pluripotent stem cells. Journal of cellular and molecular medicine. 2011;15:38–51. doi: 10.1111/j.1582-4934.2009.00996.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ma J, et al. High purity human-induced pluripotent stem cell-derived cardiomyocytes: electrophysiological properties of action potentials and ionic currents. American journal of physiology. Heart and circulatory physiology. 2011;301:H2006–2017. doi: 10.1152/ajpheart.00694.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Moretti A, et al. Patient-specific induced pluripotent stem-cell models for long-QT syndrome. The New England journal of medicine. 2010;363:1397–1409. doi: 10.1056/NEJMoa0908679. [DOI] [PubMed] [Google Scholar]
- 30.Itzhaki I, et al. Modelling the long QT syndrome with induced pluripotent stem cells. Nature. 2011;471:225–229. doi: 10.1038/nature09747. [DOI] [PubMed] [Google Scholar]
- 31.Hoekstra M, Mummery CL, Wilde AA, Bezzina CR, Verkerk AO. Induced pluripotent stem cell derived cardiomyocytes as models for cardiac arrhythmias. Frontiers in physiology. 2012;3:346. doi: 10.3389/fphys.2012.00346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sharma A, et al. Human induced pluripotent stem cell-derived cardiomyocytes as an in vitro model for coxsackievirus B3-induced myocarditis and antiviral drug screening platform. Circulation research. 2014;115:556–566. doi: 10.1161/CIRCRESAHA.115.303810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kishimoto T, Akira S, Taga T. Interleukin-6 and its receptor: a paradigm for cytokines. Science. 1992;258:593–597. doi: 10.1126/science.1411569. [DOI] [PubMed] [Google Scholar]
- 34.Taga T, Kishimoto T. Gp130 and the interleukin-6 family of cytokines. Annual review of immunology. 1997;15:797–819. doi: 10.1146/annurev.immunol.15.1.797. [DOI] [PubMed] [Google Scholar]
- 35.Fernandes CJ, Jr., Akamine N, Knobel E. Cardiac troponin: a new serum marker of myocardial injury in sepsis. Intensive care medicine. 1999;25:1165–1168. doi: 10.1007/s001340051030. [DOI] [PubMed] [Google Scholar]
- 36.El-Battrawy I, et al. Hyperthermia Influences the Effects of Sodium Channel Blocking Drugs in Human-Induced Pluripotent Stem Cell-Derived Cardiomyocytes. PloS one. 2016;11:e0166143. doi: 10.1371/journal.pone.0166143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wang Q, et al. Soluble interleukin-6 receptor-mediated innate immune response to DNA and RNA viruses. Journal of virology. 2013;87:11244–11254. doi: 10.1128/JVI.01248-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Aoki Y, et al. Role of ion channels in sepsis-induced atrial tachyarrhythmias in guinea pigs. British journal of pharmacology. 2012;166:390–400. doi: 10.1111/j.1476-5381.2011.01769.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zila I, et al. Heart rate variability and inflammatory response in rats with lipopolysaccharide-induced endotoxemia. Physiological research. 2015;64(Suppl 5):S669–676. doi: 10.33549/physiolres.933226. [DOI] [PubMed] [Google Scholar]
- 40.Kuipers S, Klein Klouwenberg PM, Cremer OL. Incidence, risk factors and outcomes of new-onset atrial fibrillation in patients with sepsis: a systematic review. Critical care. 2014;18:688. doi: 10.1186/s13054-014-0688-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Skibsbye L, et al. Small-conductance calcium-activated potassium (SK) channels contribute to action potential repolarization in human atria. Cardiovascular research. 2014;103:156–167. doi: 10.1093/cvr/cvu121. [DOI] [PubMed] [Google Scholar]
- 42.Larribere L, et al. NF1 loss induces senescence during human melanocyte differentiation in an iPSC-based model. Pigment cell & melanoma research. 2015;28:407–416. doi: 10.1111/pcmr.12369. [DOI] [PubMed] [Google Scholar]
- 43.Maherali N, et al. A high-efficiency system for the generation and study of human induced pluripotent stem cells. Cell stem cell. 2008;3:340–345. doi: 10.1016/j.stem.2008.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tiburcy M, Zimmermann WH. Modeling myocardial growth and hypertrophy in engineered heart muscle. Trends in cardiovascular medicine. 2014;24:7–13. doi: 10.1016/j.tcm.2013.05.003. [DOI] [PubMed] [Google Scholar]
- 45.Schmittgen TD, Livak KJ. Analyzing real-time PCR data by the comparative C(T) method. Nature protocols. 2008;3:1101–1108. doi: 10.1038/nprot.2008.73. [DOI] [PubMed] [Google Scholar]
- 46.Trafford AW, Diaz ME, Eisner DA. A novel, rapid and reversible method to measure Ca buffering and time-course of total sarcoplasmic reticulum Ca content in cardiac ventricular myocytes. Pflugers Archiv: European journal of physiology. 1999;437:501–503. doi: 10.1007/s004240050808. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.