

Abstract
Flaviviruses such as West Nile virus (WNV), dengue virus and Zika virus are mosquito-borne pathogens that cause significant human diseases. A novel group of insect-specific flaviviruses (ISFs), which only replicate in mosquitoes, have also been identified. However, little is known about the mechanisms of ISF host restriction. We report the generation of infectious cDNA from two Australian ISFs, Parramatta River virus (PaRV) and Palm Creek virus (PCV). Using circular polymerase extension cloning (CPEC) with a modified OpIE2 insect promoter, infectious cDNA was generated and transfected directly into mosquito cells to produce infectious virus indistinguishable from wild-type virus. When infectious PaRV cDNA under transcriptional control of a mammalian promoter was used to transfect mouse embryo fibroblasts, the virus failed to initiate replication even when cell entry steps were by-passed and the type I interferon response was lacking. We also used CPEC to generate viable chimeric viruses between PCV and WNV. Analysis of these hybrid viruses revealed that ISFs are also restricted from replication in vertebrate cells at the point of entry. The approaches described here to generate infectious ISF DNAs and chimeric viruses provide unique tools to further dissect the mechanisms of their host restriction.
Subject terms: Restriction factors, West nile virus
Introduction
The Flavivirus genus of the Flaviviridae family, encompasses a diverse array of viruses, which are responsible for a number of significant mosquito-transmitted diseases such as West Nile fever and encephalitis, dengue and Zika fever and Japanese encephalitis. These small enveloped viruses contain a ~11 kb positive sense, single-stranded RNA genome with a single open reading frame (ORF) flanked by 5′ and 3′ untranslated regions (UTRs). The viral ORF is translated into a single polyprotein, and post-translationally cleaved into three structural (C, prM and E) and seven non-structural proteins (NS1-NS5)1. Many flaviviruses are transmitted between mosquitoes and vertebrates, relying on replication in both hosts for maintaining their natural transmission cycle. However, a large group of insect-specific flaviviruses (ISFs), which replicate exclusively in mosquitoes have more recently been discovered2–5. These viruses appear to be transmitted vertically between mosquitoes with no requirement for a vertebrate intermediate. The advent of deep sequencing methods, sensitive reverse transcription (RT) PCR assays using flavivirus generic primers and the development of broad-spectrum diagnostic tools, such as monoclonal antibodies (mAbs) to viral dsRNA intermediates, have seen the isolation of many new ISFs from various regions around the world3, 6–11. These interesting viruses thus provide a unique model to investigate the molecular basis of their restriction to an insect host and efficient mode of vertical transmission. This knowledge will provide new insights into the evolution of flaviviruses. There is also the potential for ISFs to benefit public health as natural bio-control agents that suppress the transmission of vertebrate-infecting flaviviruses (VIFs) in mosquito populations3, 4, 9, 12.
The mechanism involved in the insect cell-restricted tropism of ISFs is currently unknown. In-depth investigation into the viral factors contributing to host-restriction of ISFs requires the generation of full-length infectious clones, which can then be readily manipulated to determine the effects of individual genes or RNA sequences on host cell permissiveness. However, this process has been traditionally encumbered by the toxicity of full-length viral cDNAs in bacteria. Various alternative approaches have been employed to overcome this problem including the use of low copy number plasmids13, 14, cosmid vectors15, in vitro ligation16, and insertion of introns17, 18. However, these approaches are time and labour intensive, and are prone to non-specific mutations during plasmid amplification in bacteria or in vitro RNA transcription. Our recently described, novel bacterium-free approach overcomes many of these pitfalls and was used to rapidly assemble flavivirus infectious cDNAs for the Kunjin strain of West Nile virus (WNVKUN)19, 20. Circular Polymerase Extension Cloning (CPEC) operates without the need for restriction enzyme digestion, ligation, or single-stranded homologous recombination21. Our most recent iteration of the CPEC system includes the removal of all bacterial regulatory sequences from the CPEC linker fragment which circularises the flavivirus genome via the two UTR’s, and contains a CMV promoter to drive transcription of the viral RNA19. This system was used to effectively generate WNV chimeric viruses, whereby the systematic and precise exchange of genes between differing strains of WNV was performed to identify the role of non-structural proteins in WNV virulence19. This study highlighted the efficacy of CPEC as a fast and reliable method for manipulating full-length flavivirus infectious DNA.
In its current format, the CPEC system is unsuitable for the preparation of ISF infectious cDNA due to the inability of the CMV promoter to drive the initial transcription of the viral RNA in insect cells. Here we report the generation of ISF infectious cDNAs, which produce full-length viral RNA genomes from a modified Orgyia pseudotsugata multicapsid nucleopolyhedrosis virus immediate-early 2 (OpIE2) promoter. We used Parramatta River virus (PaRV)22 and Palm Creek virus (PCV)3, novel Australian ISF species isolated in our lab from Aedes vigilax and Coquillettidia xanthogaster mosquitoes, respectively. The generation of PaRV and PCV CPEC constructs and the successful recovery of corresponding infectious viruses in insect cells represent major steps forward in ISF research and provides unique tools to identify the stages of vertebrate cell infection where ISF host-restriction takes place. We also report the use of the CPEC approach to generate chimeric viruses between ISFs and VIFs, thus further expanding the tools to investigate mechanisms of ISF restriction as well as providing potential platform for generating recombinant viruses between ISFs and VIFs as candidates for safe vaccines and diagnostic antigens for flaviviral diseases.
Results
CPEC insect promoter optimisation
Due to the restriction of ISF replication to insect cells, a modification of the existing CPEC protocol, which employs a CMV promoter for mammalian systems, was required. The OpIE2 promoter originally described by Blissard and Rohrmann23 was chosen to drive transcription of CPEC-assembled infectious cDNA in insect cells. Previous comparative analyses using this promoter have shown it to be highly active in multiple insect cell lines24, 25. To convert the recently described CPEC system for the generation of infectious flavivirus DNA in vertebrate cells19 to a system for use in mosquito cells, the UTR-linker fragment was modified to replace the existing CMV promoter with the complete OpIE2 promoter. A truncated version of this promoter, lacking the 23 nucleotides comprising the promoter 3′ tail downstream of the transcription start site (OpIE2-CA), was also designed to reduce the number of extra nucleotides added to the 5′ end of the transcribed flavivirus genome (Fig. 1a). The modified OpIE2 UTR-linkers were assembled by CPEC with cDNA fragments from WNVKUN and directly transfected into C6/36 cells. A TCID50 of recovered infectious virus following transfection of WNVKUN CPEC constructs into C6/36 cells indicated that passage 0 (P0) titres were approximately 100-fold higher when using the truncated promoter (OpIE2-CA - 105.42 IU/mL) compared to the full-length version (OpIE2 - 103.35 IU/mL) (Fig. 1b).
Figure 1.
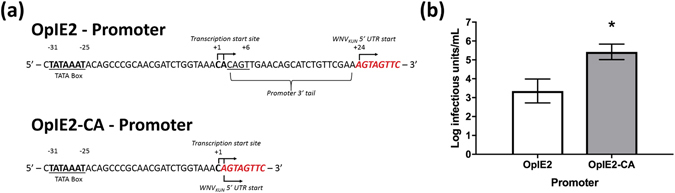
OpIE2 promoter optimisation. (a) Schematic of the 3′ termini of the OpIE2 and OpIE2-CA promoters. (b) TCID50 of P0 supernatants from C6/36 cells transfected with WNVKUN CPEC containing either the OpIE2 or OpIE2-CA promoter (n = 3 biological replicates for each construct). Supernatants harvested 5 days post-transfection indicate that the OpIE2-CA promoter yields approximately 100-fold higher titres than the OpIE2 promoter. Error bars represent standard deviation and asterisks indicate significance (P value < 0.05; two-tailed t-test).
Generation of PaRV infectious DNA construct using circular polymerase extension cloning
The modified OpIE2-CA UTR-linker fragment was selected for generating the PaRV infectious cDNA by CPEC (Fig. 2a). Primers used for constructing the PaRV cDNA library were designed to anneal at the junctions between viral genes predicted from the PaRV genome sequence22 (Fig. 2a). CPEC-derived PaRV (PaRVCPEC) was successfully recovered from two independent transfections of C6/36 cell cultures, and the identity of the progeny virus confirmed by RT-PCR and Sanger sequencing of approximately 1.5 kb of the C-prM-E region of the viral genome. Supernatant from the PaRVCPEC-transfected culture (P0) was also inoculated on to fresh C6/36 cells, and PaRV-specific antigens detected at 3 days post-infection by IFA using anti-PaRV mouse serum22 to demonstrate successful replication of the progeny PaRVCPEC virus (Fig. 2b).
Figure 2.
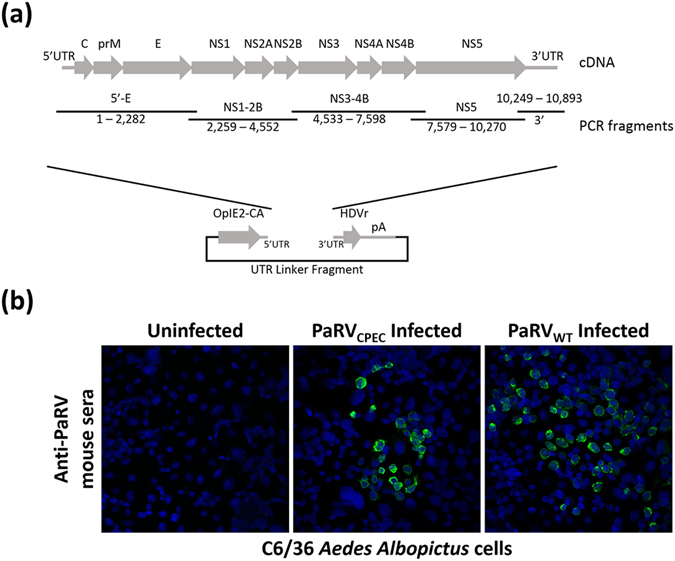
Generation of PaRV using CPEC. (a) A schematic representation for the assembly of infectious DNA for PaRV by CPEC reaction. (b) Visualisation of PaRV replication in mosquito (C6/36) cell monolayers inoculated with either a MOI of 0.1 PaRVWT or undiluted P0 PaRVCPEC. Monolayers were fixed 72 hrs post-infection. IFA analysis was performed by probing with PaRV mouse anti-sera. The nucleus of each cell was stained with Hoechst 33342. Images were taken at ×40 magnification.
CPEC-derived PaRV is phenotypically identical to wild-type PaRV
PaRVCPEC was further assessed by two methods to confirm that it was phenotypically identical to PaRVWT. In a growth kinetics assay, PaRVCPEC displayed a growth profile identical to PaRVWT, with no significant difference in the titres at any time point (two-way ANOVA). Both viruses replicated rapidly in the first 24 hrs (PaRVCPEC 105.78 IU/mL; PaRVWT 105.86 IU/mL), reaching a peak titre at 72 hrs (PaRVCPEC 107.72 IU/mL; PaRVWT 107.91 IU/mL), after which the virus titre plateaued (Fig. 3a) similar to our previously reported results for wild-type PaRV22. The titre of both PaRV viruses at 24 hours was significantly higher when compared to WNVKUN, however, by four days post-infection WNVKUN (108.24 IU/mL) produced a significantly (two-way ANOVA; P < 0.001) higher titre (PaRVWT -107.39 IU/mL; PaRVCPEC - 107.63 IU/mL). This was also consistent with our previous findings22. Further antigenic analysis using a panel of monoclonal antibodies (mAb) that were generated to native PaRV prM and E proteins, confirmed that each epitope was conserved between PaRVWT and PaRVCPEC (Fig. 3b). Comparison of levels of infectious virus derived from P0 cultures transfected with either purified PaRV RNA or a PaRV CPEC reaction also revealed comparable titres (107.80 IU/mL and 106.97 IU/mL, respectively) after a 5 day incubation.
Figure 3.
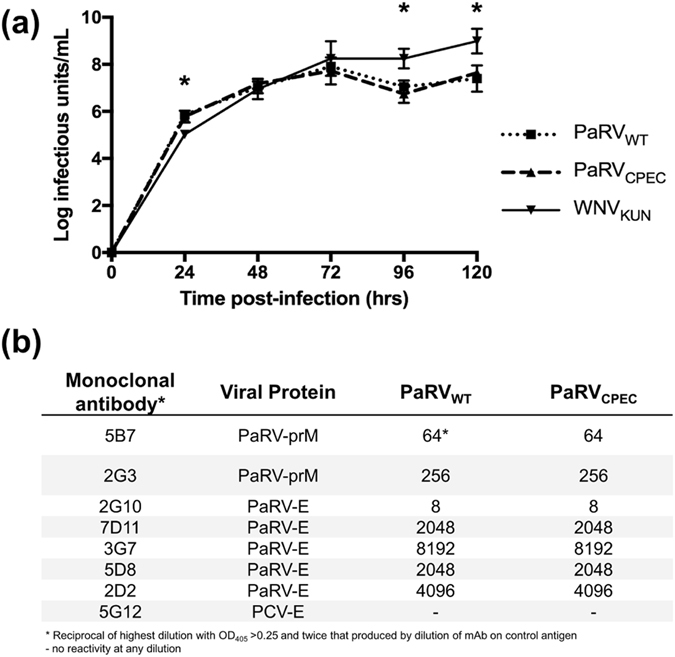
Phenotypic analysis of PaRVWT and PaRVCPEC. (a) Comparative growth kinetics of PaRVWT, PaRVCPEC and WNVKUN in C6/36 cells. C6/36 cells were infected with either PaRVWT, PaRVCPEC or WNVKUN at a MOI of 0.1. Infectious titres at each time point were determined by titration of culture supernatant on to fresh C6/36 cells with infection detected using fixed cell ELISA. Error bars represent standard deviation and asterisks indicate significance (P value < 0.001) as determined by a two-way ANOVA. (b) Comparison of reciprocal titres for the reactivity of a panel of anti-PaRV (5B7, 2G3, 2G10, 7D11, 3G7, 5D8, 2D2 and 1E5) and anti-PCV control (5G12) mAbs to PaRVWT and PaRVCPEC.
Using CPEC to bypass viral entry and IFN response-deficient cells to remove restriction by the innate immune response did not permit PARV replication in vertebrate cells
Barriers to ISF infection and replication in vertebrate cells may occur at any stage of virus entry, replication and release or could be due to restriction by the host innate immune response5, 26. To examine the potential roles for viral entry and innate immune response in restriction of ISFs in vertebrate cells, wild-type (WT) and IFN-α/β receptor knockout (IFNAR−/−) mouse embryonic fibroblasts (MEFs), were transfected with PaRV CPEC DNA driven by a CMV promoter or a similarly prepared WNVKUN CPEC DNA. Infection with PARV and WNVKUN virus was also employed as a control. IFA analysis of both virus-infected and CPEC-transfected cells revealed no observable replication of PaRV in either WT or IFNAR−/− MEFs (Fig. 4). In contrast, both cell lines displayed WNVKUN replication whether infected with virus or transfected with CPEC DNA. To confirm the lack of initiation of PaRV replication in vertebrate cells, purified PaRV virion RNA was also used to transfect WT MEFs, IFNAR−/− MEFs, and BHK cells. IFA analysis of the transfected cells revealed no PaRV replication in any of the vertebrate cell lines while GFP expression was clearly observed in BHK cells transfected with a control WNVKUN replicon RNA encoding GFP (See Supplementary Fig. S1). In contrast, C6/36 cells transfected with PaRV RNA showed clear viral replication. These data demonstrate that PaRV fails to initiate replication even after bypassing viral entry and removing the type I IFN response.
Figure 4.
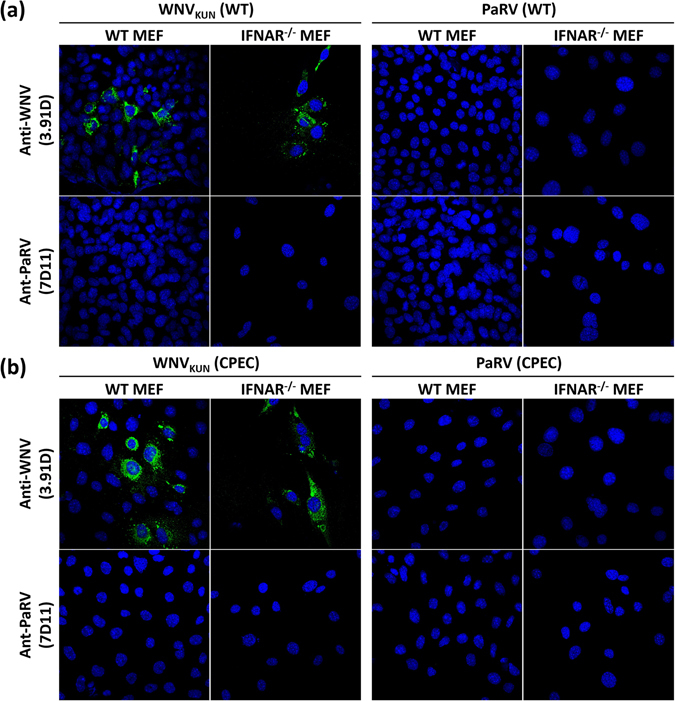
Viral replication analyses in WT and IFNAR−/− MEF cells. (a) Cells infected at an MOI of 1 or transfected CPEC constructs of either WNVKUN or PaRV with a CMV promoter. (b) Cells transfected with genomic RNA of either PaRV, WNVKUN, PCV, PCV/WNVKUN -prME or WNVKUN/PCV-prME. Monolayers were fixed 72 hrs post-infection. IFA analysis was performed by probing with anti-PaRV (7D11) and anti-WNV E (3.91D) mouse monoclonal antibodies. The nucleus of each cell was stained with Hoechst 33342. Images were taken at ×40 magnification.
ISF/VIF chimeric viruses as a tool to study viral host restriction
The generation of chimeric viruses between ISFs and VIFs would provide useful tools to elucidate the molecular basis of ISF host-restriction. However, repeated efforts using CPEC to generate chimeric viral genomes containing either PaRV prME genes on a WNVKUN genomic backbone (WNVKUN/PaRV-prME) or WNVKUN -prME genes on a PaRV genomic backbone (PaRV/WNVKUN-prME) were unsuccessful. Nevertheless, we were able to generate a mutant PaRV virus with an in-frame, 15 nucleotide stretch of the prM gene replaced with the corresponding sequence of WNVKUN (PaRVCPECM1). The progeny virus was confirmed by sequencing of the prM gene (See Supplementary Table S1), and although PaRVCPECM1 contained an additional codon, the virus grew to similar titres to PaRVCPEC (106.3 IU/mL) demonstrating that CPEC could be used to produce infectious, recombinant PaRV.
To assess whether the incompatibility between PaRV and WNV was a trait shared by other ISFs, additional constructs were prepared. Using the same strategy employed for construction of PaRVCPEC, an infectious DNA construct of PCV was also successfully generated by CPEC and characterised (Fig. 5). Similarly, chimeric cDNAs containing the PCV prME genes on a WNVKUN genomic backbone (WNVKUN/PCV-prME) and WNVKUN -prME genes on a PCV genomic backbone (PCV/WNVKUN-prME) were produced by CPEC. In contrast to the non-viable PaRV-WNV chimeric constructs, WNVKUN/PCV-prME and PCV/WNVKUN-prME chimeric cDNAs generated viable viruses in CPEC DNA-transfcted C6/36 cells. IFA analysis of WNVKUN/PCV-prME virus revealed expression of both PCV E and WNVKUN NS1 protein, but not PCV NS1 or WNVKUN E proteins (Fig. 5a). The inverse was observed for the PCV/WNVKUN-prME virus indicating that the correct chimeric viruses were produced. The authenticity of the PCVCPEC, WNVKUN/PCV-prME and PCV/WNVKUN-prME chimeric viruses were further verified as described earlier by Sanger sequencing of a 1 kb C-prME region of the viral RNA isolated from passage P1 C6/36 supernatant.
Figure 5.
Generation and characterisation of chimeric viruses between ISFs and VIFs. (a) Visualisation of C6/36 cells transfected with CPEC constructs of either WNVKUN, PCV, WNVKUN/PCV-prME or PCV/WNVKUN-prME chimeric viruses. IFA analysis was performed by probing with anti-PCV E (5G12), anti-PCV NS1 (3D6/9G4), anti-WNVKUN E (3.91D) and anti-WNVKUN NS1 (3.1112G) mouse monoclonal antibodies. Monolayers were fixed 5 days post-transfection. The nucleus of each cell was stained with Hoechst 33342. Images were taken at ×40 magnification. (b) Comparative growth kinetics of WNVKUN(CPEC), PCVCPEC, WNVKUN/PCV-prME and PCV/WNVKUN-prME in C6/36 cells. C6/36 cells were infected with either virus at a MOI of 0.1. Infectious titres at each time point were determined by titration of culture supernatant on fresh C6/36 cells with infection detected using fixed cell ELISA. Error bars represent standard deviation and asterisks indicate significance (P value < 0.001) as determined by a two-way ANOVA.
Growth kinetics assays indicated PCVCPEC replicated rapidly in the first 24 hrs (PCVCPEC 106.52 IU/mL), reaching significantly (two-way ANOVA; P < 0.001) higher tires than WNVKUN(CPEC), WNVKUN/PCV-prME and PCV/WNVKUN-prME. The titre peaked at 48 hrs (PCVCPEC 107.58 IU/mL), after which the virus titre plateaued (Fig. 5b) consistent with our previous results with wild-type PCV12. WNVKUN/PCV-prME displayed a growth profile identical to WNVKUN, with no significant difference in the titres at any time point. Both WNVKUN and WNVKUN/PCV-prME peaked 72 hrs post infection (WNVKUN(CPEC) 106.39 IU/mL, WNVKUN/PCV-prME 106.08 IU/mL). The titres for PCV/WNVKUN-prME were significantly lower than the other viruses tested throughout the study, reaching peak titre 72 hrs post infection (PCV/WNVKUN-prME 103.74 IU/mL). However, in cultures incubated for 7 days the virus reached a titre of 106.3 IU/mL.
To further investigate the role of viral entry in ISF host restriction, BHK, Vero and C6/36 cells were infected with MOI = 1 of P1 WNVKUN(CPEC), PCVCPEC, WNVKUN/PCV-prME or PCV/WNVKUN-prME. IFA analyses of cells revealed that while all four viruses readily replicated in C6/36 cells (See Supplementary Fig. S2), only WNVKUN exhibited productive infection in BHK and Vero cells (Fig. 6). Despite containing the replicative proteins of WNVKUN, WNVKUN/PCV-prME failed to replicate in vertebrate cells. Similarly, PCV/WNVKUN-prME was also unable to replicate in vertebrate cells despite containing WNVKUN structural proteins, which would allow the virus entry into the cells. Similar results were also observed when WT and IFNAR−/− MEF cells were infected with PCVCPEC, WNVKUN/PCV-prME or PCV/WNVKUN-prME at an MOI of 10 (Data not shown). These results suggest that a restriction barrier for PCV likely occurs at both the point of cell entry and at the RNA replication stage.
Figure 6.
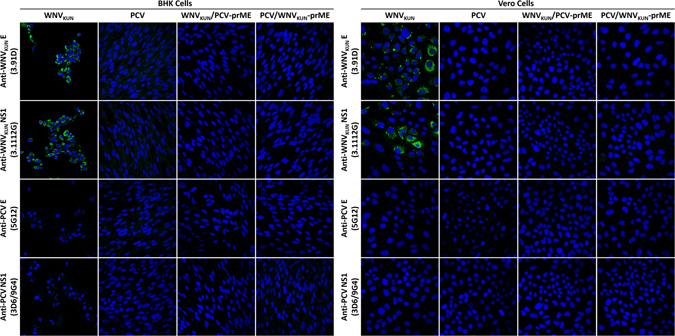
Visualisation of BHK and Vero cells infected with either WNVKUN, PCV, PCV/WNVKUN-prME or WNVKUN/PCV-prME at an MOI of 1. Monolayers were fixed 72 hrs post-infection. IFA analysis was performed by probing with anti-PCV E (5G12), anti-PCV NS1 (3D6/9G4), anti-WNV E (3.91D) and anti-WNV NS1 (3.1112G) mouse monoclonal antibodies. The nucleus of each cell was stained with Hoechst 33342. Images were taken at ×40 magnification.
Discussion
Here we report an infectious ISF cDNA constructed using a modification of our previously described CPEC method19, 20. Infectious PaRV virions were successfully recovered from C6/36 cells transfected with a CPEC-assembled PaRV infectious cDNA containing a modified OpIE2 promoter. The recovered CPEC-derived PaRV was phenotypically identical to the wild-type virus. While attempts to produce chimeric viruses between PaRV and WNVKUN were unsuccessful, demonstration that viable ISF/VIF chimeric viruses could be generated was provided through the successful production of chimeric WNVKUN/PCV-prME and PCV/WNVKUN-prME viruses. Thus, we provide a proof-of-concept that viable ISF/VIFs can be successfully generated. Furthermore, the methods and reagents described here provide a useful set of tools to enable the investigation of viral determinants of host-restriction in ISFs.
Despite the power of reverse genetics as a tool for understanding the aspects of viral replication and pathogenesis, as well as vaccine development, few infectious clones of insect-specific viruses have been reported. The first publication describing an insect-specific virus infectious clone was for a full-length cDNA clone of Culex flavivirus (CxFV)27. Additionally, infectious clones of Eilat virus (EILV), a unique insect-specific alphavirus, were transfected into vertebrate cells to elucidate that its host-restriction was present even as early as RNA replication28, 29. The EILV infectious clones were also chimerised with Sindbis virus (SINV) and chikungunya virus (CHIKV) structural genes to determine that EILV was restricted both at entry and genomic RNA replication levels in vertebrate cells and for developing novel and safe means of generating diagnostic antigen and vaccines for CHIKV30–32. More recently, an infectious clone of Niénokoué virus (NIEV), a flavivirus isolated from mosquitoes sampled in Côte d’Ivoire, and a chimeric virus containing the NIEV structural genes on a yellow fever virus (YFV) backbone were used to identify the stages of ISF restriction in vertebrate cells33. Infectious clones of two mosquito-specific negeviruses have also been reported34, 35. In each of these examples, infectious clones and chimeric constructs (for EILV) were generated using standard cDNA subcloning techniques into suitable plasmid(s) along with the incorporation of an SP6 or T7 RNA polymerase promoter, followed by in vitro transcription of the RNA transcript and subsequent transfection of the transcript into cells36, 37.
We believe that the generation of an infectious ISF cDNA construct using CPEC, as described herein, represents a significant improvement in the methodologies for generating recombinant ISFs. Our inclusion of the OpIE2 insect promoter to drive the transcription of the PaRV genome negated the requirement for in vitro transcription. However, initial transfections of PaRVCPEC containing the full-length promoter sequence yielded significantly (P value < 0.05) lower titres compared to PaRVWT, suggesting that the OpIE2 promoter required optimisation in the context of driving transcription of the PaRV genome. Indeed, the 5′ and 3′ UTRs, which flank the coding regions of the flaviviral genome, are a prerequisite for initiating RNA replication38. Both UTRs contain highly-defined stem-loop structures which have been shown to interact with viral replicative proteins such as NS5 to initiate RNA transcription39–41. Importantly, the functionality of the 5′ UTR is likely to be dependent on the specificity of the upstream promoter to initiate transcription from the first nucleotide of the viral genome. Thus, it is likely the extra nucleotides added to the 5′ end of viral RNA following transcription via the full length OpIE2 promoter was rendering the virus less replication competent. Previous deletion analyses of the OpIE2 promoter had shown that only nucleotides up to −275 from the transcription start site were necessary for promoter function42. The truncation of 23 nucleotides from the 3′ end in the modified promoter (OpIE2-CA), between the transcription start site and 5′ terminal nucleotide of the viral genome sequence, ensured an authentic 5′ UTR sequence of the PaRV RNA with no additional bases. OpIE2 promoter transcription has been shown to initiate equally from either the +1 or +2 transcription start nucleotides resulting in some transcripts containing a single additional nucleotide at the 5′ terminus. However, previous studies have shown that a single additional nucleotide at the distal end of the flavivirus 5′ UTR is lost early during viral replication and had no significant side effects43. Thus, the increase in viral titre following the optimisation of the OpIE2 promoter was most likely due to the deletion of the promoter 3′ tail region immediately downstream of the transcription start site resulting an authentic 5′ UTR sequence in the transcribed PaRV genome. The availability of CPEC to easily generate infectious DNAs of ISFs under transcription control of both insect and mammalian promoters also provides a powerful tool to investigate the mechanisms of ISF host restriction. Thus, transfection of IFNAR−/− MEFs with the PaRV CPEC construct incorporating a CMV promoter allowed us to assess viral replication efficiency without the requirement of the virus to enter cells and in the absence of downstream JAK-STAT signalling pathways and induction of IFN-stimulated genes (ISGs)44.
To create tools for the identification of the viral factors associated with the mechanisms underlying host-restriction in ISFs, chimeric CPEC constructs of PaRV and WNVKUN with swapped prM-E genes were designed. The capsid gene was not included in the exchanged structural genes, as it has been shown to contain key elements such as cyclisation sequences, which are critical for the appropriate folding and configuration of flaviviral UTRs during replication45, 46. Our failure to generate infectious PaRV/WNVKUN chimeras, despite repeated attempts and serial passaging of the transfected cultures, may be due to a lack of recognition of key cleavage motifs at the junctions between the PaRV and WNVKUN genome components (i.e. C-prM and E-NS1) by host signalases. However, in silico analyses of the proposed cleavage sites in the deduced polyprotein sequence of the non-viable chimeric viral genomes revealed that the predicted cleavage efficiency by signalase was similar to that of the corresponding sites in the wild-type parental viruses (Data not shown). Interestingly the recovery of viable virus from PaRVCPECM1, which contains a shorter substitution from WNVKUN suggested that chimerisation between the two viruses is also possible on a much smaller scale.
Our success in generating an infectious chimeric virus containing genes from WNVKUN on a PCV genetic backbone is the first report of a viable ISF chimera expressing VIF structural genes. Previous attempts to generate chimeric viruses using ISFs and VIFs have proved difficult47, with only recent developments leading to the generation of a chimeric VIF with ISF structural genes33. Our demonstration that the WNVKUN/PCV-prME chimera could not infect vertebrate cells despite containing WNVKUN replicative genes and UTRs, indicated that the ISF structural proteins were associated with host restriction at the stage of cell entry. Indeed, the structure of the receptor-binding domain III of the E protein of most ISFs is radically altered compared to that of VIFs, suggesting it may be associated with binding to mosquito-specific cell receptors22, 26. In contrast, the failure of the PCV/WNVKUN-prME chimera to initiate replication in vertebrate cells, despite possessing the structural proteins of WNVKUN, indicated that additional barriers to ISF replication exist in vertebrate cells, such as IFN-independent antiviral responses and/or incompatible virus-host cell interactions that are required for virus replication48. These findings are consistent with a recent report showing an absence of viral replication in IFNAR−/− MEFs inoculated with the ISF Kamiti River virus (KRV). However, that study did show trace levels of virus replication in IRF 3, 5, 7−/− MEFs, further suggesting a role for IFN-independent innate responses in restricting KRV replication48, 49. A more recent study using a NIEV reporter replicon and a YFV/NIEV chimeric virus also showed a lack of viral replication in vertebrate cells inoculated with the YFV/NIEV chimeric virus or transfected with NIEV replicon RNA33. However, limited replication with a lack of infectious particle production was observed in cells transfected with YFV/NIEV RNA. The authors also concluded that ISFs were unable to enter vertebrate cells and that an additional intracellular barrier to replication was likely associated with a lack of interaction between ISFs and vertebrate host cell factors.
In this study the development of a CPEC system to generate infectious ISF DNAs and chimeric viruses, not only allowed the identification of multiple stages of ISF growth restriction in vertebrate cells, but also provides an approach to rapidly prepare infectious cDNA constructs for a variety of ISFs and other insect-specific viruses. This will allow the fast and efficient production of mutant and chimeric viruses to further dissect the molecular mechanisms of host restriction in ISFs. Our generation of a panel of mAbs reactive to ISF viral proteins further provides a complimentary set of reagents to facilitate these studies.
Materials and Methods
Cell Culture
C6/36 (Aedes albopictus) cells were cultured at 28 °C in RPMI 1640 medium supplemented with 5% foetal bovine serum (FBS). Wild-type (WT) and interferon-α/β receptor deficient (IFNAR−/−) mouse embryonic fibroblasts (MEF), baby hamster kidney (BHK) and African green monkey (Vero) cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 5% FBS and grown at 37 °C with 5% CO2. All media contained 50 U penicillin/mL, 50 mg streptomycin/mL and 2 mM L-glutamine.
Virus Culture
Parramatta River virus (PaRV - NC_027817.1), Palm Creek virus (PCV - KC505248.1) and West Nile virus strain Kunjin (WNVKUN - AY274504) viral stocks were propagated in C6/36 cells incubated at 28 °C for 5–7 days. Viral titres were assessed by infection of C6/36 cells with 10-fold serial dilutions of supernatant in 96-well plates and incubation for 5 days. The cell supernatant was aspirated and the monolayers fixed with acetone (20% acetone, 0.02% bovine serum albumin (BSA) in phosphate buffered saline (PBS). PaRV was detected by enzyme-linked immunosorbent assay (ELISA) using the PaRV-specific monoclonal antibody (mAb) 7D11, PCV using PCV-specific mAb 5G12 and WNVKUN using mAb 4G250. Virus titres were calculated as 50% tissue culture infective dose (TCID50) using the methods described by Reed and Muench51.
Preparation of monoclonal antibodies to PaRV and PCV
All animal experiments were conducted according to the guidelines set out in the Australian Code for the Care and Use of Animals for Scientific Purposes 8th edition (2013) and were approved by The University of Queensland Animal Ethics Committee - approval #SCMB/329/15/ARC. All animal procedures where necessary were performed under ketamine:xylazil anaesthesia. Six-week old BALB/c mice (Animal Resources Centre, Murdoch, Western Australia, Australia) were immunised twice via the subcutaneous route with purified PaRV or PCV virions, along with the inulin-based adjuvant Advax (Vaxine Ltd, Adelaide, Australia). Mice were kept on clean bedding and given food and water ad libitum. The mice were boosted with PaRV or PCV virions by intravenous injection four days prior to harvesting of the spleen. Fusion of the spleen cells with NS0 myeloma cells (European Collection of Cell Cultures) was performed as previously described52. Hybridomas secreting antibodies reactive to PaRV- or PCV-infected C6/36 cells were identified by ELISA using previously described methods53. The target protein of each mAb was determined using PaRV- or PCV-infected cell lysates in Western blot using previously published methods53. Reaction of selected monoclonal antibodies with antigens of wild-type and CPEC derived-PaRV were analysed by fixed cell ELISA53.
Generation and characterisation of a modified OpIE2 insect promoter
OpIE2 promoter sequences characterised by Blissard and Rohrmann23 were synthesised as gBlocks Gene Fragments (IDT). These fragments were cloned into a previously generated plasmid containing a sequence which, when expressed by itself, linked the UTR regions of the viral genome together19. A Gibson Assembly Master Mix (NEB) was used. Constructs were transformed into DH5α competent E. coli, and colony PCR using Taq DNA Polymerase (NEB) conducted to screen for viable colonies. Plasmids were extracted from overnight cultures of positive colonies using a NucleoSpin Plasmid Miniprep kit (Macherey-Nagel). Extracted plasmids were sequenced by the Australian Genome Research Centre.
Generation of viruses by CPEC
CPEC constructs were generated based on previously described methods19. Briefly, viral RNA was extracted using a NucleoSpin RNA Virus kit (Macherey-Nagel) and converted to cDNA using a qScript cDNA SuperMix (Quantabio). For each CPEC assembly, 0.1 pmol of each viral cDNA fragment was added to a Q5 PCR reaction (NEB) as per the manufacturer’s instructions. Primers used are available upon request. Thermal cycling was carried out at 98 °C for 2 mins (one cycle), 98 °C for 30 secs, 55 °C for 30 secs, 72 °C for 6 mins (2 cycles), 98 °C for 30 secs, 55 °C for 30 secs, 72 °C for 8 mins (ten cycles). The entire CPEC reaction was transfected into cells and the passage 0 (P0) cell culture supernatants harvested and stored at −80 °C, five days post-transfection. Additionally, cDNA of any progeny virus was generated as before and amplified with Q5 High-Fidelity DNA Polymerase (NEB) prior to sequencing by the Australian Genome Research Centre.
Growth Kinetics
C6/36 cells seeded at a density of 1 × 105 were inoculated in triplicate with a P1 CPEC-derived and P7 wild-type virus stock at a multiplicity of infection (MOI) of 0.1. After incubation at 28 °C for 1 hr the inoculum was removed and the monolayer washed three times with sterile PBS before re-incubating at 28 °C with fresh RPMI 1640 with 2% FBS. Supernatant was harvested at 2, 24, 48, 72, 96 and 120 hrs post infection. Viral titres from each time point were determined using a TCID50 assay as previously described. A two way-ANOVA was performed on the results using Graphpad Prism.
IFA
Cells seeded at a density of 1 × 105 on glass coverslips in a 24-well plate were transfected or infected as required. Following a 72 hr incubation, the coverslips were fixed with ice cold 100% acetone and air dried before storing at −20 °C. Prior to staining, coverslips were blocked with blocking buffer (0.05 M Tris/HCl (pH 8.0), 1 mM EDTA, 0.15 M NaCl, 0.05% (v/v) Tween-20, 0.2% w/v casein) for 1 hr at room temperature. Coverslips were then incubated for 1 hr with primary antibody in blocking buffer. The mAbs used for this work included anti-WNV E (3.91D54 or 4G255), anti-WNV NS1 (3.1112 G54 or 4G4), anti-PaRV E (7D11), anti-PCV E (5G12) and anti-PCV NS1 (3D6/9G4)3. Following 3 washes with PBS containing 0.05% Tween-20 (PBST), antibody binding was detected by incubation for 1 hour with Alexafluor 488-conjugated goat anti-mouse IgG (H + L) (Invitrogen) diluted 1:1000 in blocking buffer. A Hoechst 33342 nuclear stain (Invitrogen) was applied at 1:1000 for 5 mins at room temperature. Following a final 3 washes with PBST, the coverslips were mounted onto glass microscope slides using ProLong Gold Anti-fade (Invitrogen). All coverslips were viewed under the ZEISS LSM 510 META confocal microscope.
Cell Transfection
C6/36 cells were transfected with DNA using Effectene (Qiagen) or RNA using TransMessenger (Qiagen), as per the manufacturer’s instructions. Wild-type and IFNAR−/− MEF cells were transfected using Lipofectamine 2000 (Invitrogen), as per the manufacturer’s instructions. RNA from a GFP-nanoLuc fusion protein-expressing WNV replicon generated using methods previously described by Khromykh and Westaway56, was used as a transfection control for vertebrate cells.
Electronic supplementary material
Acknowledgements
We would like to thank Agathe Colmant, Jessica Harrison and Caitlin O’Brien (all University of Queensland) for expert technical advice and assistance. We thank Renee Traves (University of Queensland) and Parthiban Periasamy (University of Queensland) for expert technical advice. We also thank Prof. Nikolai Petrovsky (Flinders University) for supplying the Advax adjuvant. This study was funded by the Australian Research Council (ARC DP120103994) and an Australian Government Research Training Program Scholarship to TBHP.
Author Contributions
Conceived and designed the experiments: T.B.H.P., Y.X.S., J.H.P., H.B.O., A.A.K., R.A.H.; Conducted experimental procedures: T.B.H.P., J.H.P., H.B.O., B.J.M., L.J.V., N.D.N.; Wrote the first draft of the paper: T.B.H.P.; All authors contributed to the structure and arguments for the paper. All authors reviewed and approved the final manuscript.
Competing Interests
The authors declare that they have no competing interests.
Footnotes
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Thisun B. H. Piyasena, Yin X. Setoh and Jody Hobson-Peters contributed equally to this work.
Change history
6/5/2019
A correction to this article has been published and is linked from the HTML and PDF versions of this paper. The error has not been fixed in the paper.
Contributor Information
Alexander A. Khromykh, Email: alexander.khromykh@uq.edu.au
Roy A. Hall, Email: roy.hall@uq.edu.au
Electronic supplementary material
Supplementary information accompanies this paper at doi:10.1038/s41598-017-03120-1
References
- 1.Chambers TJ, Hahn CS, Galler R, Rice CM. Flavivirus genome organization, expression, and replication. Annu Rev Microbiol. 1990;44:649–688. doi: 10.1146/annurev.mi.44.100190.003245. [DOI] [PubMed] [Google Scholar]
- 2.Stollar V, Thomas VL. An agent in the Aedes aegypti cell line (Peleg) which causes fusion of Aedes albopictus cells. Virology. 1975;64:367–377. doi: 10.1016/0042-6822(75)90113-0. [DOI] [PubMed] [Google Scholar]
- 3.Hobson-Peters J, et al. A new insect-specific flavivirus from northern Australia suppresses replication of West Nile virus and Murray Valley encephalitis virus in co-infected mosquito cells. PLoS One. 2013;8:e56534. doi: 10.1371/journal.pone.0056534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bolling BG, Olea-Popelka FJ, Eisen L, Moore CG, Blair CD. Transmission dynamics of an insect-specific flavivirus in a naturally infected Culex pipiens laboratory colony and effects of co-infection on vector competence for West Nile virus. Virology. 2012;427:90–97. doi: 10.1016/j.virol.2012.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Blitvich BJ, Firth AE. Insect-specific flaviviruses: a systematic review of their discovery, host range, mode of transmission, superinfection exclusion potential and genomic organization. Viruses. 2015;7:1927–1959. doi: 10.3390/v7041927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.O’Brien CA, et al. Viral RNA intermediates as targets for detection and discovery of novel and emerging mosquito-borne viruses. PLoS Negl Trop Dis. 2015;9:e0003629. doi: 10.1371/journal.pntd.0003629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Farfan-Ale JA, et al. Detection of RNA from a novel West Nile-like virus and high prevalence of an insect-specific flavivirus in mosquitoes in the Yucatan Peninsula of Mexico. Am J Trop Med Hyg. 2009;80:85–95. doi: 10.4269/ajtmh.2009.80.85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Huhtamo E, et al. Novel insect-specific flavivirus isolated from northern Europe. Virology. 2012;433:471–478. doi: 10.1016/j.virol.2012.08.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kenney JL, Solberg OD, Langevin SA, Brault AC. Characterization of a novel insect-specific flavivirus from Brazil: potential for inhibition of infection of arthropod cells with medically important flaviviruses. J Gen Virol. 2014;95:2796–2808. doi: 10.1099/vir.0.068031-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Crabtree MB, Sang RC, Stollar V, Dunster LM, Miller BR. Genetic and phenotypic characterization of the newly described insect flavivirus, Kamiti River virus. Arch Virol. 2003;148:1095–1118. doi: 10.1007/s00705-003-0019-7. [DOI] [PubMed] [Google Scholar]
- 11.Cook S, et al. Novel virus discovery and genome reconstruction from field RNA samples reveals highly divergent viruses in dipteran hosts. PLoS One. 2013;8:e80720. doi: 10.1371/journal.pone.0080720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hall-Mendelin S, et al. The insect-specific Palm Creek virus modulates West Nile virus infection in and transmission by Australian mosquitoes. Parasit Vectors. 2016;9:414. doi: 10.1186/s13071-016-1683-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bredenbeek PJ, et al. A stable full-length yellow fever virus cDNA clone and the role of conserved RNA elements in flavivirus replication. J Gen Virol. 2003;84:1261–1268. doi: 10.1099/vir.0.18860-0. [DOI] [PubMed] [Google Scholar]
- 14.Shan C, et al. An Infectious cDNA Clone of Zika Virus to Study Viral Virulence, Mosquito Transmission, and Antiviral Inhibitors. Cell Host Microbe. 2016;19:891–900. doi: 10.1016/j.chom.2016.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yun SI, Kim SY, Rice CM, Lee YM. Development and application of a reverse genetics system for Japanese encephalitis virus. J Virol. 2003;77:6450–6465. doi: 10.1128/JVI.77.11.6450-6465.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Audsley M, et al. Virulence determinants between New York 99 and Kunjin strains of West Nile virus. Virology. 2011;414:63–73. doi: 10.1016/j.virol.2011.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tsetsarkin, K. A. et al. A Full-Length Infectious cDNA Clone of Zika Virus from the 2015 Epidemic in Brazil as a Genetic Platform for Studies of Virus-Host Interactions and Vaccine Development. mBio7, doi:10.1128/mBio.01114-16 (2016). [DOI] [PMC free article] [PubMed]
- 18.Schwarz, M. C. et al. Rescue of the 1947 Zika Virus Prototype Strain with a Cytomegalovirus Promoter-Driven cDNA Clone. mSphere1, doi:10.1128/mSphere.00246-16 (2016). [DOI] [PMC free article] [PubMed]
- 19.Setoh YX, et al. Systematic analysis of viral genes responsible for differential virulence between American and Australian West Nile virus strains. J Gen Virol. 2015;96:1297–1308. doi: 10.1099/vir.0.000069. [DOI] [PubMed] [Google Scholar]
- 20.Edmonds J, et al. A novel bacterium-free method for generation of flavivirus infectious DNA by circular polymerase extension reaction allows accurate recapitulation of viral heterogeneity. J Virol. 2013;87:2367–2372. doi: 10.1128/JVI.03162-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Quan J, Tian J. Circular polymerase extension cloning of complex gene libraries and pathways. PLoS One. 2009;4:e6441. doi: 10.1371/journal.pone.0006441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.McLean BJ, et al. A novel insect-specific flavivirus replicates only in Aedes-derived cells and persists at high prevalence in wild Aedes vigilax populations in Sydney, Australia. Virology. 2015;486:272–283. doi: 10.1016/j.virol.2015.07.021. [DOI] [PubMed] [Google Scholar]
- 23.Blissard GW, Rohrmann GF. Location, sequence, transcriptional mapping, and temporal expression of the gp64 envelope glycoprotein gene of the Orgyia pseudotsugata multicapsid nuclear polyhedrosis virus. Virology. 1989;170:537–555. doi: 10.1016/0042-6822(89)90445-5. [DOI] [PubMed] [Google Scholar]
- 24.Pfeifer TA, Hegedus DD, Grigliatti TA, Theilmann DA. Baculovirus immediate-early promoter-mediated expression of the Zeocin resistance gene for use as a dominant selectable marker in dipteran and lepidopteran insect cell lines. Gene. 1997;188:183–190. doi: 10.1016/S0378-1119(96)00756-1. [DOI] [PubMed] [Google Scholar]
- 25.Kempf J, et al. Expression of the human mu opioid receptor in a stable Sf9 cell line. J Biotechnol. 2002;95:181–187. doi: 10.1016/S0168-1656(02)00008-1. [DOI] [PubMed] [Google Scholar]
- 26.Hall RA, et al. Commensal Viruses of Mosquitoes: Host Restriction, Transmission, and Interaction with Arboviral Pathogens. Evol Bioinform Online. 2016;12:35–44. doi: 10.4137/EBO.S40740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Isawa H, et al. Construction of an infectious cDNA clone of Culex flavivirus, an insect-specific flavivirus from Culex mosquitoes. Arch Virol. 2012;157:975–979. doi: 10.1007/s00705-012-1240-z. [DOI] [PubMed] [Google Scholar]
- 28.Nasar F, et al. Eilat virus, a unique alphavirus with host range restricted to insects by RNA replication. Proc Natl Acad Sci USA. 2012;109:14622–14627. doi: 10.1073/pnas.1204787109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nasar F, Haddow AD, Tesh RB, Weaver SC. Eilat virus displays a narrow mosquito vector range. Parasit Vectors. 2014;7:595. doi: 10.1186/s13071-014-0595-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Erasmus JH, et al. Utilization of an Eilat Virus-Based Chimera for Serological Detection of Chikungunya Infection. PLoS Negl Trop Dis. 2015;9:e0004119. doi: 10.1371/journal.pntd.0004119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nasar F, Gorchakov RV, Tesh RB, Weaver SC. Eilat virus host range restriction is present at multiple levels of the virus life cycle. J Virol. 2015;89:1404–1418. doi: 10.1128/JVI.01856-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Erasmus JH, et al. A chikungunya fever vaccine utilizing an insect-specific virus platform. Nat Med. 2017;23:192–199. doi: 10.1038/nm.4253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Junglen S, et al. Host Range Restriction of Insect-Specific Flaviviruses Occurs at Several Levels of the Viral Life Cycle. mSphere. 2017;2:e00375–00316. doi: 10.1128/mSphere.00375-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gorchakov RV, Tesh RB, Weaver SC, Nasar F. Generation of an infectious Negev virus cDNA clone. J Gen Virol. 2014;95:2071–2074. doi: 10.1099/vir.0.066019-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kurnia YW, et al. Generation of an infectious cDNA clone of Okushiri virus and its derivative capable of expressing an exogenous gene. J Insect Biotechnol Sericology. 2016;85:2039–2047. doi: 10.11416/jibs.85.2_039. [DOI] [Google Scholar]
- 36.Rice CM, Levis R, Strauss JH, Huang HV. Production of infectious RNA transcripts from Sindbis virus cDNA clones: mapping of lethal mutations, rescue of a temperature-sensitive marker, and in vitro mutagenesis to generate defined mutants. J Virol. 1987;61:3809–3819. doi: 10.1128/jvi.61.12.3809-3819.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Volkova E, Gorchakov R, Frolov I. The efficient packaging of Venezuelan equine encephalitis virus-specific RNAs into viral particles is determined by nsP1-3 synthesis. Virology. 2006;344:315–327. doi: 10.1016/j.virol.2005.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Alvarez DE, Lodeiro MF, Luduena SJ, Pietrasanta LI, Gamarnik AV. Long-range RNA-RNA interactions circularize the dengue virus genome. J Virol. 2005;79:6631–6643. doi: 10.1128/JVI.79.11.6631-6643.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Choi KH, et al. The structure of the RNA-dependent RNA polymerase from bovine viral diarrhea virus establishes the role of GTP in de novo initiation. Proc Natl Acad Sci USA. 2004;101:4425–4430. doi: 10.1073/pnas.0400660101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Liu L, et al. Flavivirus RNA cap methyltransferase: structure, function, and inhibition. Front Biol (Beijing) 2010;5:286–303. doi: 10.1007/s11515-010-0660-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lodeiro MF, Filomatori CV, Gamarnik AV. Structural and functional studies of the promoter element for dengue virus RNA replication. J Virol. 2009;83:993–1008. doi: 10.1128/JVI.01647-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Theilmann DA, Stewart S. Molecular analysis of the trans-activating IE-2 gene of Orgyia pseudotsugata multicapsid nuclear polyhedrosis virus. Virology. 1992;187:84–96. doi: 10.1016/0042-6822(92)90297-3. [DOI] [PubMed] [Google Scholar]
- 43.Khromykh AA, Westaway EG. Completion of Kunjin virus RNA sequence and recovery of an infectious RNA transcribed from stably cloned full-length cDNA. J Virol. 1994;68:4580–4588. doi: 10.1128/jvi.68.7.4580-4588.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Platanias LC. Mechanisms of type-I- and type-II-interferon-mediated signalling. Nat Rev Immunol. 2005;5:375–386. doi: 10.1038/nri1604. [DOI] [PubMed] [Google Scholar]
- 45.Khromykh AA, Meka H, Guyatt KJ, Westaway EG. Essential role of cyclization sequences in flavivirus RNA replication. J Virol. 2001;75:6719–6728. doi: 10.1128/JVI.75.14.6719-6728.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Liu ZY, et al. Novel cis-acting element within the capsid-coding region enhances flavivirus viral-RNA replication by regulating genome cyclization. J Virol. 2013;87:6804–6818. doi: 10.1128/JVI.00243-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Saiyasombat R, Carrillo-Tripp J, Miller WA, Bredenbeek PJ, Blitvich BJ. Substitution of the premembrane and envelope protein genes of Modoc virus with the homologous sequences of West Nile virus generates a chimeric virus that replicates in vertebrate but not mosquito cells. Virol J. 2014;11:150. doi: 10.1186/1743-422X-11-150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Nandakumar R, et al. Hepatitis C virus replication in mouse cells is restricted by IFN-dependent and -independent mechanisms. Gastroenterology. 2013;145:1414–1423 e1411. doi: 10.1053/j.gastro.2013.08.037. [DOI] [PubMed] [Google Scholar]
- 49.Tree MO, et al. Insect-specific flavivirus infection is restricted by innate immunity in the vertebrate host. Virology. 2016;497:81–91. doi: 10.1016/j.virol.2016.07.005. [DOI] [PubMed] [Google Scholar]
- 50.Gentry MK, Henchal EA, McCown JM, Brandt WE, Dalrymple JM. Identification of distinct antigenic determinants on dengue-2 virus using monoclonal antibodies. Am J Trop Med Hyg. 1982;31:548–555. doi: 10.4269/ajtmh.1982.31.548. [DOI] [PubMed] [Google Scholar]
- 51.Reed LJ, Muench H. A simple method of estimating fifty per cent endpoints. Am J Epidemiol. 1938;27:493–497. doi: 10.1093/oxfordjournals.aje.a118408. [DOI] [Google Scholar]
- 52.Hall RA, Burgess GW, Kay BH. Type-specific monoclonal antibodies produced to proteins of Murray Valley encephalitis virus. Immunol Cell Biol. 1988;66(Pt 1):51–56. doi: 10.1038/icb.1988.6. [DOI] [PubMed] [Google Scholar]
- 53.Clark DC, et al. In situ reactions of monoclonal antibodies with a viable mutant of Murray Valley encephalitis virus reveal an absence of dimeric NS1 protein. J Gen Virol. 2007;88:1175–1183. doi: 10.1099/vir.0.82609-0. [DOI] [PubMed] [Google Scholar]
- 54.Adams SC, et al. Glycosylation and antigenic variation among Kunjin virus isolates. Virology. 1995;206:49–56. doi: 10.1016/S0042-6822(95)80018-2. [DOI] [PubMed] [Google Scholar]
- 55.Henchal EA, Gentry MK, McCown JM, Brandt WE. Dengue virus-specific and flavivirus group determinants identified with monoclonal antibodies by indirect immunofluorescence. Am J Trop Med Hyg. 1982;31:830–836. doi: 10.4269/ajtmh.1982.31.830. [DOI] [PubMed] [Google Scholar]
- 56.Khromykh AA, Westaway EG. Subgenomic replicons of the flavivirus Kunjin: construction and applications. J Virol. 1997;71:1497–1505. doi: 10.1128/jvi.71.2.1497-1505.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.