

Abstract
Histone crotonylation is a new lysine acylation type of post-translational modification (PTM) enriched at active gene promoters and potential enhancers in yeast and mammalian cells. However, lysine crotonylation in nonhistone proteins and plant cells has not yet been studied. In the present study, we performed a global crotonylation proteome analysis of Nicotiana tabacum (tobacco) using high-resolution LC-MS/MS coupled with highly sensitive immune-affinity purification. A total of 2044 lysine modification sites distributed on 637 proteins were identified, representing the most abundant lysine acylation proteome reported in the plant kingdom. Similar to lysine acetylation and succinylation in plants, lysine crotonylation was related to multiple metabolism pathways, such as carbon metabolism, the citrate cycle, glycolysis, and the biosynthesis of amino acids. Importantly, 72 proteins participated in multiple processes of photosynthesis, and most of the enzymes involved in chlorophyll synthesis were modified through crotonylation. Numerous crotonylated proteins were implicated in the biosynthesis, folding, and degradation of proteins through the ubiquitin-proteasome system. Several crotonylated proteins related to chromatin organization are also discussed here. These data represent the first report of a global crotonylation proteome and provide a promising starting point for further functional research of crotonylation in nonhistone proteins.
Introduction
Post-translational modification (PTM) is a covalent modification process resulting from the proteolytic cleavage or addition of a functional group to one amino acid. Thus far, more than 200 PTMs have been characterized (http://www.uniprot.org/help/post-translational_modification). These processes modulate protein functions by altering their localization, activity state and interactions with other proteins. Among all PTMs, lysine acetylation, originally identified in histones1, is one of the most studied PTMs. Early studies on lysine acetylation have focused on nuclear proteins, such as histones and transcriptional factors2, 3. These studies suggested that lysine acetylation was restricted to the nucleus4, 5. The discovery of lysine acetylation on tubulin and mitochondrial proteins suggested an important role for lysine acetylation in cellular biology in addition to chromatin biology6–8. Using high-resolution mass spectrometry, the high abundance of lysine acetylation outside the nucleus has been identified. Lysine acetylation is abundant in most metabolic pathways, such as glycolysis, gluconeogenesis, the tricarboxylic acid (TCA) cycle, and conserved in both eukaryotes and prokaryotes9–13. In addition to lysine acetylation, some new types of PTMs, such as malonylation and lysine succinylation, were identified using mass spectrometry combined with the affinity purification of modified peptides using antibodies directed against these modifications14–20. Similar to lysine acetylation, lysine malonylation and succinylation are important in regulating cellular metabolism, and both processes exist in eukaryotes and prokaryotes21–24.
Histone lysine crotonylation has recently been detected from yeast to humans and is primarily associated with active transcription25. Similar to histone acetylation, crotonylation also occurs on the ε-amino group of lysine but distinguishes itself from acetylation by its four-carbon length and planar orientation. Lysine crotonylation, but not acetylation, preferentially marks “escapee genes” during post-meiotic sex inactivation in mouse testes26, 27. Lysine crotonylation and acetylation sites overlap in histones and are catalysed through p300/CBP, a well-known histone acetyltransferase28. Moreover, Sirtuin family members SIRT1-3, well-studied histone deacetylases, remove crotonylation in a site-specific manner. SIRT3 is present in both mitochondria and nuclei and is expressed in the kidneys and metabolically active tissues29. These studies lead to a question that whether cytoplasmic proteins undergo lysine crotonylation, similar to acetylation, and play an important role in regulating cellular metabolism.
Reflecting their sessile feature, plants rapidly change their endogenous status to adapt to adverse environmental conditions. Compared with the regulation of transcription and translation, PTMs could trigger a much faster response, representing a major concern in plant science. However, studies of lysine acylation of the proteome in plant cells have primarily focused on acetylation and succinylation, confirmed in only a limited number of plant species, including Arabidopsis10, 30, 31, rice11, 32, wheat33, soybean34, pea35, grape36, tomato37, potato38, strawberry39, and Brachypodium distachyon L40. Moreover, relatively few proteins have been modified through acetylation or succinylation. In these plants, both lysine acetylation and succinylation have been implicated in the regulation of diverse metabolic processes, such as carbon metabolism, glycolysis, pyruvate metabolism, the TCA cycle, and photosynthesis33, 37, 40.
Common tobacco (Nicotiana tabacum) is a versatile model organism for fundamental biology research and biotechnology applications41. It is the source of the BY-2 plant cell line, which is a key tool for plant molecular research. Moreover, tobacco is also one of the most widely cultivated non-food crops worldwide. In the present study, we investigated the global lysine crotonylation proteome of tobacco using high-resolution LC-MS/MS coupled with highly sensitive immune-affinity purification. In total, we identified 2044 lysine crotonylation sites in 637 proteins. The identified crotonylated proteins, primarily localized to the chloroplast, cytosol, nucleus, and mitochondria, were primarily involved in carbon metabolism, photosynthesis, protein biosynthesis, folding, degradation, and chromatin organization. To our knowledge, this study is the first to describe lysine crotonylation in the global proteome, thereby expanding the current understanding of the effect of lysine crotonylation on nonhistone proteins.
Results
Detection of lysine-crotonylated proteins in tobacco leaves
To characterize the global crotonylation proteome of tobacco, a proteomic method based on sensitive immune-affinity purification and high-resolution LC-MS/MS was applied to identify crotonylated proteins and their modification sites in tobacco. An overview of the experimental procedures is shown in Fig. 1a. A total of 2044 lysine crotonylation sites distributed in 637 proteins were identified, representing the most abundant lysine acylation proteome reported in the plant kingdom (Table 1). MS/MS information related to these crotonylated peptides were deposited to iProX database with accession number IPX0000889000 (http://www.iprox.org). Detailed information for all identified crotonylated peptides and their corresponding proteins was shown in Supplementary Table S1, the scores for protein and peptide identification were shown in Supplementary Table S2. Among the 637 crotonylated proteins, 357 (56%) proteins contained one or two crotonylation sites, and 80 (13%) proteins had 7 or more crotonylation sites (Fig. 1b). Most peptides ranged from 7 to 28 amino acids in length, consistent with the properties of tryptic peptides (Fig. 1c). To confirm the validation of the MS data, the mass error of all identified peptides was assessed. The distribution of the mass error was near zero, and most of these proteins were less than 0.02 Da, suggesting that the mass accuracy of the MS data met the requirement (Fig. 1d).
Figure 1.
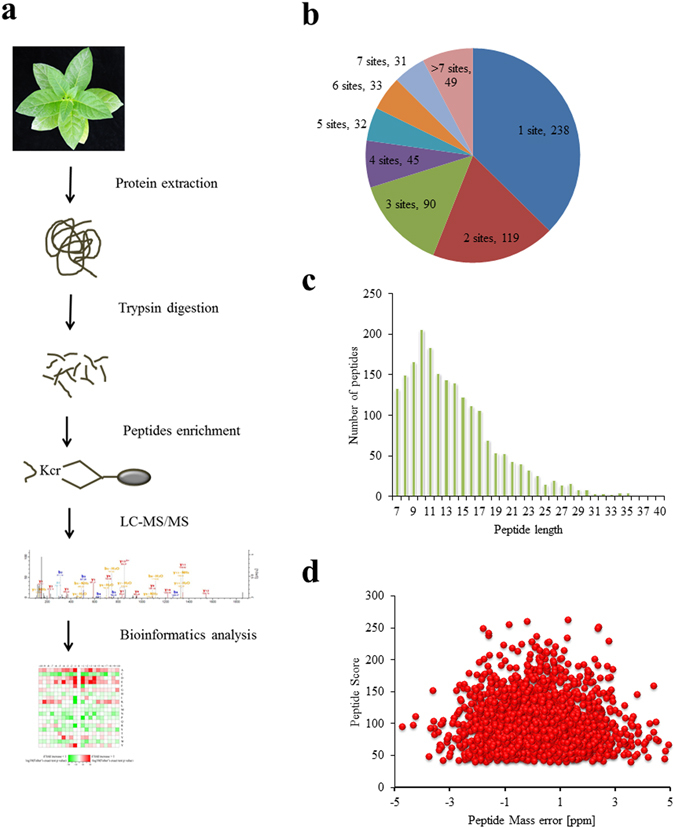
Proteome-wide identification of lysine crotonylation sites in Nicotiana tabacum. (a) Overview of experimental procedures used in the present study. Kcr indicates the crotonylated lysine. (b) Distribution of lysine crotonylation in one protein. (c) Distribution of lysine crotonylation peptides based on their length. (d) Mass error distribution of all crotonylated peptides.
Table 1.
Comparison of tobacco crotonylation proteome with other published acylation proteome in plants.
| Acylation | No. of acylation sites | No. of acylated proteins | Plant | References |
|---|---|---|---|---|
| Lysine acetylation | 91 | 74 | Arabidopsis thaliana | 10 |
| 699 | 389 | rice | 32 | |
| 416 | 277 | wheat | 33 | |
| 400 | 245 | soybean | 34 | |
| 664 | 358 | pea | 35 | |
| 138 | 97 | grape | 36 | |
| 35 | 31 | potato | 38 | |
| 1392 | 684 | strawberry | 39 | |
| 636 | 353 | Brachypodium distachyon L | 40 | |
| Lysine succinylation | 665 | 261 | rice | 32 |
| 347 | 202 | tomato | 37 | |
| 605 | 262 | Brachypodium distachyon L | 40 | |
| Lysine crotonylation | 2044 | 637 | tobacco | This study |
Motifs and secondary structures of lysine crotonylated peptides
To evaluate the nature of the crotonylated lysines in tobacco, the sequence motifs in all identified crotonylated peptides were investigated using the Motif-X programme. As shown in Supplementary Table S3, a total of nine conserved motifs were retrieved. Particularly, motifs KcrE, EKcr and KcrD (Kcr indicates the crotonylated lysine) were strikingly conserved (Fig. 2a, Supplementary Table S4). Importantly, the significantly conserved amino acids in these motifs, namely E and D, were both negatively charged, which were rarely identified in other PTMs. These motifs are likely to represent a feature of crotonylation in tobacco. Hierarchical cluster analysis was also performed to further analyse these motifs. As shown in the heat map (Fig. 2b), the enrichment of positively charged K residues was observed in the −10 to −5 and +10 to +5 positions, while negatively charged residues D and E were markedly enriched in the −4 to +4 position. Short aliphatic A residues were frequently observed in the −10 to +10 position, while the sulphur-containing C residue was not observed.
Figure 2.
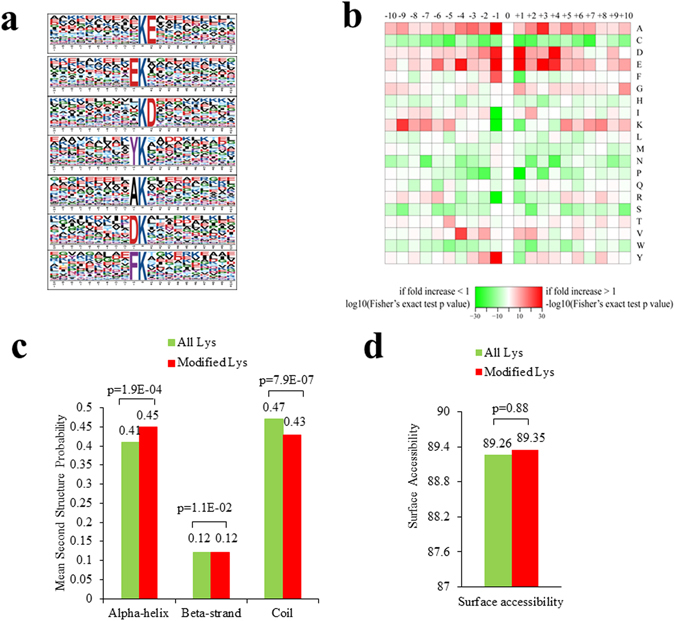
Properties of the lysine crotonylation sites. (a) Sequence probability logos of significantly enriched crotonylation site motifs around the lysine crotonylation sites. (b) Heat map of the amino acid compositions around the lysine crotonylation sites showing the frequency of different types of amino acids around this residue. Red indicates enrichment and green indicates depletion. (c) Probabilities of lysine crotonylation in different protein secondary structures (alpha helix, beta-strand and disordered coil). (d) Predicted surface accessibility of crotonylation sites.
To explore the relationship between lysine crotonylation and protein secondary structures, a structural analysis of all crotonylated proteins was performed using the algorithm NetSurfP. As shown in Fig. 2c, approximately 47% of the crotonylated sites were located in α-helices, and 12% of the sites were located in β-strands. The remaining 42% of the crotonylated sites were located in disordered coils. However, considering the similarity of the distribution pattern between crotonylated lysines and all lysines, there was no tendency towards lysine crotonylation in tobacco. The surface accessibility of the crotonylated lysine sites was also evaluated. The results showed that 91% of the crotonylated lysine sites were exposed to the protein surface, close to that of all lysine residues (Fig. 2d). Therefore, lysine crotonylation likely does not affect the surface properties of modified proteins.
Functional annotation and subcellular localization of crotonylated proteins
To obtain an overview of the crotonylated proteins in tobacco, the Gene Ontology (GO) functional classification of all crotonylated proteins based on their biological processes, molecular functions and subcellular locations was investigated (Supplementary Table S5, Supplementary Table S6). Within the biological processes category, the majority of crotonylated proteins were related to metabolic processes, cellular processes, and single-organism processes, respectively accounting for 36, 27 and 24% of all the crotonylated proteins (Fig. 3a). For the molecular function category, 45 and 40% of the crotonylated proteins were associated with catalytic activity and binding functions, respectively (Fig. 3b). Subcellular localization analysis revealed that most of the crotonylated proteins were localized to the chloroplast (37%), cytosol (30%), nucleus (12%), and mitochondria (5%) (Fig. 3c).
Figure 3.
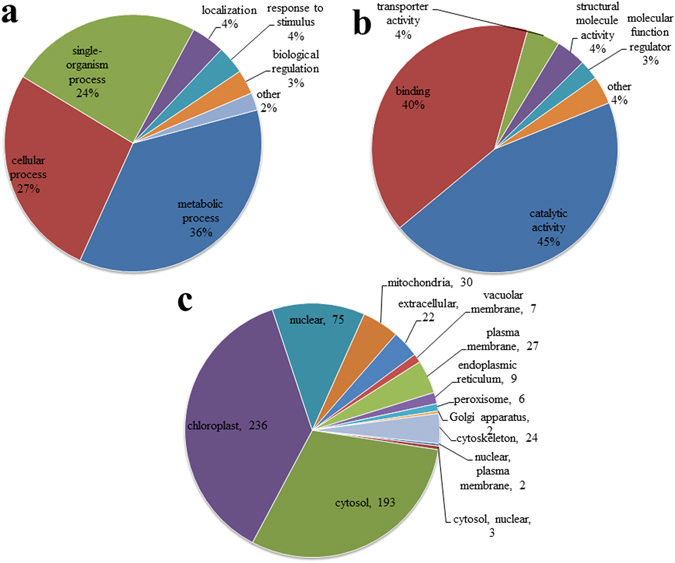
GO classification of the crotonylated proteins based on biology process (a) molecular functional (b) and subcellular localization (c), respectively.
Functional enrichment analysis
To better understand the biological function of these crotonylated proteins, we performed an enrichment analysis of the GO (Supplementary Table S7), Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway (Supplementary Table S8), and Pfam domain databases (Supplementary Table S9). The enrichment analysis of the cellular components revealed that the crotonylated proteins were significantly enriched in the proteasome complex, thylakoid membrane, and photosystem II oxygen evolving complex (Fig. 4a). Based on the enrichment results of the molecular function category, most crotonylated proteins were related to NAD binding, threonine-type peptidase activity, endopeptidase activity, and calcium ion binding (Fig. 4a). In the biological processes category, most of the crotonylated proteins were implicated in oxoacid metabolic processes, protein catabolic processes, cellular amino acid metabolic processes, protein folding, ubiquitin-dependent protein catabolic processes, and photosynthesis (Fig. 4a). The KEGG pathway enrichment analysis showed that a majority of the crotonylated proteins were related to carbon metabolism, carbon fixation in photosynthetic organisms, pyruvate metabolism, proteasome, amino acid biosynthesis, the citrate cycle, glycolysis, porphyrin and chlorophyll metabolism, and photosynthesis (Fig. 4b). Consistent with these observations, Pfam domains, including the NAD(P)-binding domain, ATPase core domain, chlorophyll a/b binding protein domain, aldolase-type TIM barrel, and thioredoxin domain, were significantly enriched in crotonylated proteins (Fig. 4c), implying an important role for lysine crotonylation in these processes.
Figure 4.
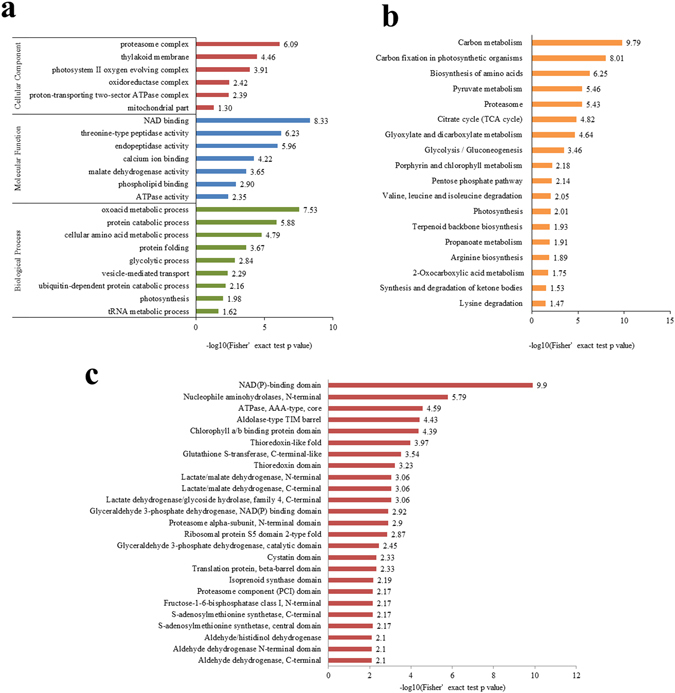
Enrichment analysis of crotonylated proteins. (a) GO-based enrichment analysis of crotonylated proteins in terms of cellular component, molecular function, and biological process. (b) KEGG pathway-based enrichment analysis. (c) Protein domain enrichment analysis. The numbers in X axes represent the value of significant analysis. When the value is greater than 1.3, the p value is less than 0.05, which means the data is statistically significant.
Crotonylated proteins involved in photosynthesis
Notably, 72 crotonylated proteins were implicated in photosynthesis processes, such as light harvesting, the electron transport chain, ATP synthesis and carbon fixation (Table 2). Significantly, 73% (8/11) of the enzymes in the Calvin cycle42, including ribulose-1,5-bisphosphate carboxylase/oxygenase (Rubisco), phosphoglycerate kinase, glyceraldehyde-3-phosphate dehydrogenase, triose phosphate isomerase, fructose-1,6-bisphosphate aldolase, fructose-1,6-bisphosphatase, transketolase, and sedoheptulose-1,7-bisphosphatase, were extensively crotonylated at multiple sites. Among these proteins, Rubisco and phosphoglycerate kinase were crotonylated at 15 and 16 lysine sites, respectively (Table 2, Supplementary Table S1). According to the annotation in UniProt, the 15 crotonylated lysines in Rubisco were distributed around substrate binding sites (Supplementary Fig. S1a). Strikingly, the catalytic sites K201 and key amino acid residues K201 and K334 were precisely crotonylated. The same phenomenon was also observed on phosphoglycerate kinase, whose substrate-binding site and ATP binding site were surrounded with crotonylated lysines (Supplementary Fig. S1b). These results indicated that lysine crotonylation might change enzyme activity, thereby regulating photosynthesis. Moreover, most of the proteins that participated in the synthesis of chlorophyll, including glutamyl-tRNA reductase (HEMA), glutamate-1-semialdehyde 2,1-aminomutase (HEML), 5-aminolevulinate dehydratase (HEMB), uroporphyrinogen III decarboxylase (HEME), coproporphyrinogen III oxidase (HEMF), protoporphyrinogen oxidase (HEMY), magnesium chelatase, magnesium proto IX methyltransferase (CHLM), Mg-protoporphyrin IX monomethylester cyclase (CRD1), 3,8-Divinyl protochlorophyllide a 8-vinyl reductase (DVR), and protochlorophyllide oxidoreductase, were also modified by crotonyl groups (Supplementary Table S1).
Table 2.
Crotonylated proteins involved in photosynthesis pathway.
| Protein | Protein name | Protein | Protein name | |
|---|---|---|---|---|
| Antenna proteins | Q40481 | Chlorophyll a-b binding protein | P27493 | Chlorophyll a-b binding protein 21 |
| Q6RUN3 | Chlorophyll a-b binding protein | P27495 | Chlorophyll a-b binding protein 40 | |
| Q0PWS7 | Chlorophyll a-b binding protein | Q0PWS6 | Chlorophyll a-b binding protein | |
| Q40512 | Chlorophyll a-b binding protein | Q84TM7 | Chlorophyll a-b binding protein | |
| Q0PWS5 | Chlorophyll a-b binding protein | Q5DNZ6 | Chlorophyll a-b binding protein | |
| Photosystems II complex | A0A140G1Q8 | Photosystem II CP43 reaction center protein | P12133 | NAD(P)H-quinone oxidoreductase subunit H |
| Q04126 | photosystem II oxygen-evolving complex | Q40459 | Oxygen-evolving enhancer protein 1 | |
| Q9SMB4 | Photosystem II 22 kDa protein | Q7DM39 | Oxygen-evolving enhancer protein 2-1 | |
| P06411 | Photosystem II CP47 reaction center protein | P18212 | Oxygen-evolving enhancer protein 2-2 | |
| P69686 | Photosystem II D2 protein | Q04127 | Oxygen-evolving enhancer protein 2-3 | |
| Q40519 | Photosystem II 10 kDa polypeptide | Q5EFR4 | oxygen-evolving protein 16 kDa subunit | |
| Q84QE8 | Oxygen evolving complex | Q53UI6 | PsbQ | |
| Cytochrome b6f complex | P06449 | Cytochrome f | P06247 | Cytochrome b6 |
| Q02585 | Cytochrome b6-f complex iron-sulfur subunit 2 | P06249 | Cytochrome b6-f complex subunit 4 | |
| Photosystems I complex | Q84QE7 | Putative photosystem I subunit III | P06405 | Photosystem I P700 chlorophyll a apoprotein A1 |
| Q84QE6 | Photosystem I reaction center subunit X psaK | P06407 | Photosystem I P700 chlorophyll a apoprotein A2 | |
| P62094 | Photosystem I iron-sulfur center | Q9T2H8 | 19.3 kDa photosystem I PSAD protein | |
| D2K7Z2 | Photosystem I reaction center subunit | P35477 | Plastocyanin B’/B” | |
| Ferredoxin–NADP reductase | O04397 | Ferredoxin–NADP reductase | O04977 | Ferredoxin–NADP reductase |
| ATP synthesis complex | A0A140G1S2 | ATP synthase subunit beta | P06286 | ATP synthase subunit c |
| W8SRJ3 | ATP synthase subunit beta | P06290 | ATP synthase subunit b | |
| Q5M9V4 | ATP synthase subunit alpha | P29790 | ATP synthase gamma chain | |
| P00823 | ATP synthase subunit alpha | P32980 | ATP synthase delta chain | |
| P00834 | ATP synthase epsilon chain | |||
| Carbon fixation | P00876 | Ribulose bisphosphate carboxylase large chain | Q006P9 | Malic enzyme |
| A0A075M9F5 | Ribulose bisphosphate carboxylase small chain | A0A077DCL8 | Phosphoenolpyruvate carboxykinase | |
| Q42961 | Phosphoglycerate kinase | A0A076KWG2 | Malate dehydrogenase | |
| P09043 | Glyceraldehyde-3-phosphate dehydrogenase A | Q9XQP4 | NAD-malate dehydrogenase | |
| P09044 | Glyceraldehyde-3-phosphate dehydrogenase B | P27154 | Phosphoenolpyruvate carboxylase | |
| A0A068JFR6 | Triosephosphate isomerase | A0A068JCD2 | Chloroplast fructose-1,6-bisphosphatase | |
| A0A068JD04 | Fructose-bisphosphate aldolase | A0A075F1V0 | Malate dehydrogenase | |
| A0A068JIB0 | Fructose-bisphosphate aldolase | Q006Q0 | Malic enzyme | |
| F2VJ75 | Fructose-bisphosphate aldolase | Q9FSF0 | Malate dehydrogenase | |
| A0A068JD95 | Fructose-1,6-bisphosphatase | A0A0K2GP10 | Glyceraldehyde-3-phosphate dehydrogenase | |
| C3RXI5 | Plastid transketolase | P09094 | Glyceraldehyde-3-phosphate dehydrogenase | |
| A0A076KWG9 | Chloroplast sedoheptulose-1,7-bisphosphatase | Q42962 | Phosphoglycerate kinase | |
| A0A075EZS4 | Glyoxisomal malate dehydrogenase |
Crotonylated proteins involved in protein biosynthesis, folding, ubiquitin-dependent degradation
A total of 47 crotonylated proteins were identified as ribosomal proteins, translation initiation factors, elongation factors, EF-1-alpha-related GTP-binding proteins and aminoacyl-tRNA synthetases (Table 3, Supplementary Table S1), suggesting that lysine crotonylation may be involved in protein biosynthesis. Several lysine residues of HSP70 (HEAT SHOCK 70 PROTEIN), HSP90, ER-resident molecular chaperone BiP 4 (luminal-binding protein 4), and BiP 5, were moddifie by crotonyl groups (Table 3, Supplementary Table S1). These proteins assist in protein folding to avoid abnormal folding and aggregation. Ubiquitin and related proteins, such as ubiquitin extension protein, ubiquitin-conjugating enzyme, and ubiquitin activating enzyme, were also crotonylated (Table 3). Moreover, 14 proteasome subunits, which participated in ubiquitin-dependent protein degradation, were modified through crotonylation (Table 3).
Table 3.
Crotonylated proteins involved in protein biosynthesis, folding, Ubiquitin-dependent degradation.
| Protein | Protein name | Protein | Protein name | |
|---|---|---|---|---|
| Ribosome subunits | P06379 | 50S ribosomal protein L2, chloroplastic | P06374 | 30S ribosomal protein S16, chloroplastic |
| O80361 | 50S ribosomal protein L4, chloroplastic | P69660 | 30S ribosomal protein S18, chloroplastic | |
| O80362 | 50S ribosomal protein L10, chloroplastic | P69660 | 30S ribosomal protein S18, chloroplastic | |
| P06382 | 50S ribosomal protein L14, chloroplastic | P25998 | 60S ribosomal protein L8 | |
| P06386 | 50S ribosomal protein L20, chloroplastic | A0A0D3QSL6 | 60S ribosomal protein L17 | |
| P06391 | 50S ribosomal protein L23, chloroplastic | Q07761 | 60S ribosomal protein L23a | |
| P30956 | 50S ribosomal protein L28, chloroplastic | Q285L8 | 40S ribosomal protein S3a | |
| P30956 | 50S ribosomal protein L28, chloroplastic | P29345 | 40S ribosomal protein S6 (Fragment) | |
| P02376 | 30S ribosomal protein S19, chloroplastic | A0A077D9P0 | 40S ribosomal protein S17-like protein | |
| P06355 | 30S ribosomal protein S2, chloroplastic | Q6TKQ9 | Ribosomal protein L3B | |
| P06357 | 30S ribosomal protein S3, chloroplastic | Q6TKR0 | Ribosomal protein L3A | |
| P06359 | 30S ribosomal protein S4, chloroplastic | Q9FSF6 | Ribosomal protein L11-like (Fragment) | |
| P62732 | 30S ribosomal protein S7, chloroplastic | A0A076L4N7 | Cytoplasmic ribosomal protein S13 | |
| P62129 | 30S ribosomal protein S12, chloroplastic | A0A076L2E2 | Ribosomal protein S25 | |
| P06373 | 30S ribosomal protein S15, chloroplastic | |||
| Translation initiation factors | Q40554 | Eukaryotic translation initiation factor 3 subunit A | A0A075EYQ6 | Eukaryotic translation initiation factor 5A |
| P56821 | Eukaryotic translation initiation factor 3 subunit B | A0A077D849 | Eukaryotic translation initiation factor 5A | |
| Q40471 | Eukaryotic initiation factor 4A-9 | A0A075QVP3 | Eukaryotic translation initiation factor NCBP-like protein | |
| A0A075QPA9 | Eukaryotic initiation factor iso4E | A0A075EYP9 | Translation initiation factor IF1 | |
| Elongation factors | P93769 | Elongation factor 1-alpha | Q9FEL2 | Elongation factor 2 |
| Q40581 | EF-1-alpha-related GTP-binding protein | Q9FEL3 | Elongation factor 2 | |
| A0A077DCL2 | Elongation factor 1-delta-like isoform 2 | P68158 | Elongation factor Tu, chloroplastic | |
| P93352 | Elongation factor 2 | |||
| Aminoacyl-tRNA synthetases | A0A077D7Q3 | Cytoplasmic asparagine-tRNA ligase 1 | Q43794 | Glutamate–tRNA ligase, chloroplastic/mitochondrial |
| Q9FEL1 | Lysyl-tRNA synthetase | |||
| Molecular chaperones | Q03684 | Luminal-binding protein 4 | I7GVS5 | Heat shock protein 70 |
| Q03685 | Luminal-binding protein 5 | Q67BD0 | Heat shock protein 70-3 | |
| G9MD86 | Heat shock protein 90 | P36182 | Heat shock protein 82 | |
| G9MD87 | Heat shock protein 90 | Q9ZT13 | 101 kDa heat shock protein | |
| Q14TB1 | Heat shock protein 90 | |||
| Ubiquitin | A0A075F2H4 | Ubiquitin-conjugating enzyme E2 36-like protein | Q40578 | Ubiquinol oxidase 2, mitochondrial |
| B6A8D0 | Ubiquitin | Q45FL8 | Ubiquitin extension protein | |
| B6V765 | Ubiquitin specific protease 12 | Q5M9U1 | NADH-ubiquinone oxidoreductase chain 6 | |
| O49905 | Polyubiquitin | Q75VJ8 | Ubiquitin activating enzyme 2 | |
| Proteasome subunits | L7UU40 | 26S proteasome ATPase regulatory subunit 6 | Q93X34 | Proteasome subunit alpha type |
| Proteasome subunits | P93395 | Proteasome subunit beta type-6 | Q93X35 | Proteasome subunit alpha type |
| P93768 | Probable 26S proteasome non-ATPase regulatory subunit 3 | Q93X37 | Putative alpha5 proteasome subunit | |
| Q93X30 | Proteasome subunit beta type | Q93X38 | Putative alpha4 proteasome subunit | |
| Q93X31 | Putative beta5 proteasome subunit | Q93X39 | Putative alpha3 proteasome subunit | |
| Q93X32 | Putative beta4 proteasome subunit | Q9XG77 | Proteasome subunit alpha type-6 | |
| Q93X33 | Putative beta 3 proteasome subunit | Q9XGH8 | Putative preprocysteine proteinase |
Protein interaction network of the crotonylated proteins in tobacco
To further identify the cellular processes regulated through crotonylation in tobacco, the crotonylated protein interaction network was established using an algorithm in Cytoscape software. A total of 264 acetylated proteins were mapped to the protein interaction database (Supplementary Table S10), presenting a global view of the diverse cellular functions of crotonylated proteins in tobacco. As shown in Fig. 5, crotonylated protein involved in ribosome, proteasome, carbon metabolism, oxidative phosphorylation, and terpenoid backbone biosynthesis were retrieved, comprising a dense protein interaction network. The physiological interactions among these crotonylated protein complexes likely contribute to their cooperation and coordination in tobacco.
Figure 5.
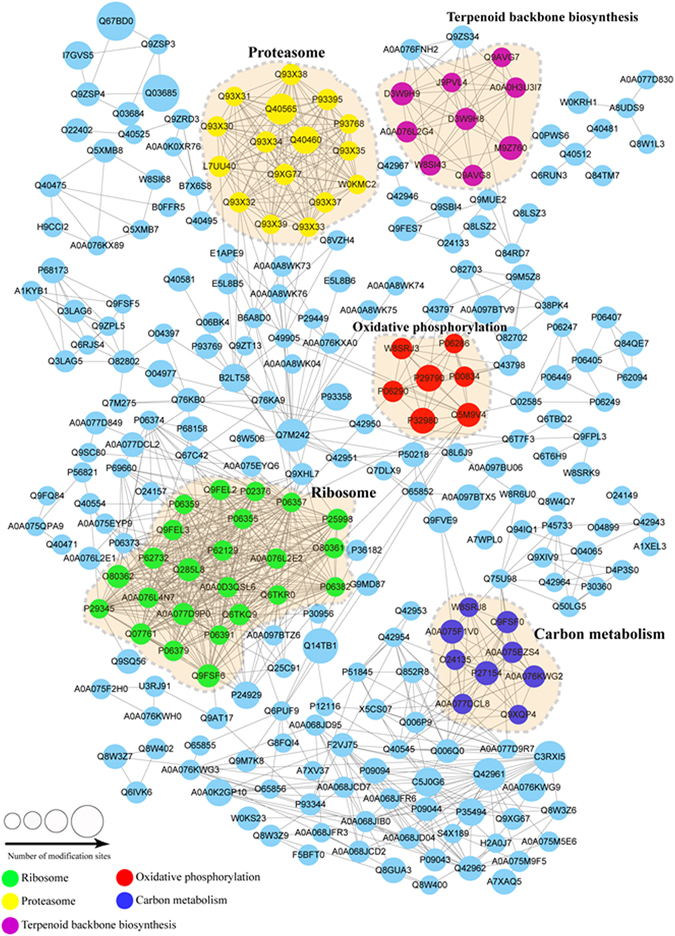
Interaction networks of the crotonylated proteins in tobacco.
Discussion
Histone crotonylation is a new lysine acylation type of PTM enriched at active gene promoters and potential enhancers in mammalian cells25. Crotonylation is catalysed through histone acetyltransferase p300/CBP28, ‘read’ by YEATS2 and AF9, ‘erased’ by Sirtuin family members SIRT1-3 in yeast and mammals29, 43–46. However, the lysine crotonylation of nonhistone proteins and in plant cells has not yet been studied. To determine whether lysine crotonylation also exists in plants and to study its function in cellular processes, a global crotonylation tobacco proteome was realized using high-resolution LC-MS/MS coupled with highly sensitive immune-affinity purification. A total of 2044 lysine crotonylation sites distributed in 637 proteins were identified, representing the most abundant lysine acylation proteome reported in the plant kingdom. These crotonylated proteins were associated with diverse biological processes, including multiple metabolic pathways, chromatin organization, protein biosynthesis, folding, and degradation. The protein interaction network analysis also suggested that a wide range of interactions involved in these biological processes was likely modulated through protein crotonylation.
Carbon is one of the most important macroelements, providing the backbone for biological macromolecules. Lysine acetylation and succinylation in plants have been implicated in carbon metabolism, glycolysis, pyruvate metabolism, TCA cycle, pentose phosphate pathway, glyoxylate and dicarboxylate metabolism32, 33, 37, 40. The results of the present study showed that numerous enzymes in these metabolism pathways were also modified through crotonylation. In plants, one of the most important metabolic processes is photosynthesis. In the present study, there are 236 crotonylated proteins were localized to the chloroplast. Among these proteins, a total of 72 proteins were involved in photosynthesis processes. For example, 10, 14, 4, 8, 2, 9, and 25 proteins, identified as members of antenna proteins, photosystems II complex, cytochrome b6f complex, photosystems I complex, ferredoxin-NADP reductase, ATP synthesis complex, and the carbon fixation pathway, respectively. Significantly, 73% (8/11) enzymes in the Calvin cycle42 were extensively crotonylated at multiple sites, with an average of 10. For example, ribulose-1,5-bisphosphate carboxylase/oxygenase (Rubisco), the key carbon fixation enzyme, was crotonylated at 15 amino acid sites. The key amino acid residues of Rubisco, K201 and K334 which were identified as acetylated resulting in the downregulation of Rubisco activity47, also modified through crotonylation. This result suggested that crotonylation might change Rubisco activity in coordination with acetylation. Moreover, the two Rubisco activase isoforms48, involved in the light activation of Rubisco, were also crotonylated at 24 sites. Moreover, 67% (10/15) of the enzymes involved in chlorophyll synthesis49 were also modified through crotonylation. To our knowledge, until recently, there have been no reports of lysine acylation in chlorophyll metabolism. These results suggested that lysine crotonylation might play a role in regulating carbon metabolism and photosynthesis.
Proteins are macromolecules that, in addition to carbohydrates, perform a vast array of functions within organisms. Proteins comprise amino acids and are synthesized through translation. In plants, proteins can be degraded in two ways - proteolysis in the vacuole or via the ubiquitin-proteasome system. The data in the present study revealed that lysine crotonylation was related to the synthesis and degradation of multiple amino acids, such as lysine, valine, leucine and isoleucine. The ribosome serves as the factory of protein synthesis. In the present study, we identified 47 crotonylated proteins associated with translation, including 29 ribosome subunits, 8 translation initiation factors, 7 elongation factors, and 3 aminoacyl-tRNA synthetases. After synthesis in the ribosome, the polypeptide chain rapidly folds into its characteristic and functional three-dimensional structure from a random coil. This process is accomplished through the assistance of chaperones, such as the ER-resident molecular chaperone BiP, the HSP70 family, and the HSP90 family50–55. The data in the present study showed that lysine residues in members of HSP70 and HSP90 were extensively crotonylated in tobacco. Moreover, Bip 4 and Bip 5 were also extensively modified through crotonylation, suggesting an important role for lysine crotonylation in protein folding. If several rounds of chaperone-assisted folding are futile, unfolded or misfolded proteins are recognized and targeted by ubiquitin and subsequently degraded by proteasomes56, 57. In the present study, we found ubiquitin, ubiquitin extension protein, ubiquitin-conjugating enzyme, and ubiquitin-activating enzyme, are all modified through crotonylation. Furthermore, 14 proteasome subunits were also crotonylated. These results indicated the likely involvement of lysine crotonylation in regulating protein synthesis, folding, and ubiquitin-dependent degradation.
The organization of the eukaryotic genome into nucleosomes dramatically impacts the regulation of gene expression. The structure of the nucleosome core is relatively invariant in eukaryotic organisms, and includes a 147-bp segment of DNA and two copies of each of the four core histone proteins58. Histone chaperone nucleosome assembly protein 1 (Nap1) has been implicated in nucleosome assembly by eliminating competing, nonnucleosomal histone-DNA interactions59. The data presented here showed that tobacco histones H1, H2A, H2B, H3, and H4, and nucleosome assembly proteins Nap1;2, Nap1;3, and Nap1;4, were modified through crotonylation, indicating a potential role for lysine crotonylation in nucleosome assembly or disassembly. As complementary evidence, topoisomerase I, required for efficient nucleosome disassembly at gene promoter regions60, was also crotonylated in the present study. Nucleosomes are folded through a series of higher-order structures to eventually form a chromosome. An important factor in higher-order organization is the nuclear matrix, which serves as a scaffold for loops of chromatin61. Nuclear matrix has been proposed to play a role in regulating transcription, DNA replication, and RNA processing62. Chromosomal DNA was anchored to nuclear matrix by its matrix-associated regions (MARs), bound by matrix attachment region-binding protein63. Histone acetyltransferase (HAT) p300 and deacetylase SIRT1 interacts with matrix attachment region-binding protein SAF-A and SATB1, respectively, and thereby regulates gene expression64, 65. Surprisingly, in the present study, a matrix attachment region binding filament-like protein (MFP1) was identified as crotonylated at 20 amino acid sites, and even its homologue was also crotonylated at 8 amino acid sites. MFP1 is a conserved nuclear and chloroplast DNA-binding protein in plants; however, its physiological function is not understood66–68. Considering that p300 and SIRT1 possess crotonylation and decrotonylation activities, respectively, in animals25, 28, 29, it is an interesting assumption that the crotonylated or decrotonylated form of MFP1 was also associated with the regulation of gene expression. In addition to these crotonylated protein that might be associated with the assembly of nucleosome and chromatin, we identified a G-strand-specific single-stranded telomere-binding protein (GTBP), associated with maintaining telomere stability, also modified through crotonyl groups69, 70. These results indicated the likely involvement of lysine crotonylation in chromatin organization and gene regulation at least in tobacco.
In summary, the present study provided the first global lysine crotonylation proteome in tobacco. These data revealed lots of crotonylated proteins associated with diverse aspects of cellular process, particularly carbon metabolism, photosynthesis, protein biosynthesis, folding, degradation, and chromatin organization. These finding raised some questions that if the crotonylation of these proteins are related to biological functions and that if crotonylation changes in different situations. All these questions should be addressed in the future work. Nevertheless, the results presented here may provide a promising starting point for further functional research of crotonylation in nonhistone proteins.
Materials and Methods
Plant materials and growth conditions
Tobacco were grown in a greenhouse at 25 °C and a photoperiod of 16/8 h (light/dark). The leaves were excised from 4-week-old seedlings with three biological replicates and immediately used for protein extraction.
Protein Extraction
The samples were grinded to powder in liquid nitrogen, and subsequently mixed with extraction buffer (8 M urea, 2 mM EDTA, 3 μM TSA, 50 mM NAM, 10 mM DTT and 1% Protease Inhibitor Cocktail, Millipore). The remaining debris was removed through centrifugation at 20,000 g for 10 min at 4 °C. Finally, the proteins were precipitated using cold 15% TCA for 2 h at −20 °C. After centrifugation at 4 °C for 10 min, the supernatant was discarded. The remaining precipitate was washed three times with cold acetone. The protein was redissolved in buffer (8 M urea, 100 mM NH4CO3, pH 8.0) and the protein concentration was determined using the 2-D Quant kit (GE Healthcare) according to the manufacturer’s instructions.
Trypsin Digestion
For digestion, the protein solution was reduced with 10 mM DTT for 1 h at 37 °C and alkylated with 20 mM IAA for 45 min at room temperature in darkness. For trypsin digestion, the protein sample was diluted after adding 100 mM NH4CO3 to a urea concentration of less than 2 M. Finally, trypsin was added at 1:50 trypsin-to-protein mass ratio for the first digestion overnight and a 1:100 trypsin-to-protein mass ratio for a second 4-h digestion.
HPLC Fractionation
The sample was subsequently fractionated through high pH reverse-phase HPLC using an Agilent 300 Extend C18 column (5 μm particles, 4.6 mm ID, 250 mm length). Briefly, the peptides were separated into 80 fractions using a gradient of 2% to 60% acetonitrile in 10 mM ammonium bicarbonate, pH 10, over 80 min. Subsequently, the peptides were combined into 6 fractions and dried using vacuum centrifugation.
Affinity Enrichment
To enrich Kcro peptides, tryptic peptides dissolved in NETN buffer (100 mM NaCl, 1 mM EDTA, 50 mM Tris-HCl, and 0.5% NP-40, pH 8.0) were incubated with pre-washed antibody beads (PTM Biolabs) at 4 °C overnight with gentle shaking. The beads were washed four times with NETN buffer and twice with ddH2O. The bound peptides were eluted from the beads using 0.1% TFA. The eluted fractions were combined and vacuum-dried. The resulting peptides were cleaned with C18 ZipTips (Millipore) according to the manufacturer’s instructions, followed by LC-MS/MS analysis.
Quantitative Proteomic Analysis by LC-MS/MS
The peptides were dissolved in 0.1% FA and directly loaded onto a reversed-phase pre-column (Acclaim PepMap 100, Thermo Scientific). Peptide separation was performed using a reversed-phase analytical column (Acclaim PepMap RSLC, Thermo Scientific). The gradient comprised an increase from 6% to 22% solvent B (0.1% FA in 98% ACN) for 24 min, 22% to 40% for 8 min and climbing to 80% in 5 min, subsequently holding at 80% for the last 3 min, all at a constant flow rate of 300 nl/min on an EASY-nLC 1000 UPLC system, the resulting peptides were analysed using the Q ExactiveTM Plus hybrid quadrupole-Orbitrap mass spectrometer (ThermoFisher Scientific). The peptides were subjected to NSI source followed by tandem mass spectrometry (MS/MS) in Q ExactiveTM plus (Thermo) coupled online to the UPLC. Intact peptides were detected in the Orbitrap at a resolution of 70,000. The peptides were selected for MS/MS using NCE setting as 30; ion fragments were detected using Orbitrap at a resolution of 17,500. A data-dependent procedure that alternated between one MS scan followed by 20 MS/MS scans was applied for the top 20 precursor ions above a threshold ion count of 5E3 in the MS survey scan with 15.0 s dynamic exclusion. The electrospray voltage applied was 2.0 kV. Automatic gain control (AGC) was used to prevent overfilling of the Orbitrap; 5E4 ions were accumulated for generation of MS/MS spectra. For MS scans, the m/z scan range was 350 to 1800. Fixed first mass was set as 100 m/z.
Database Search
The resulting MS/MS data was processed using MaxQuant with integrated Andromeda search engine (v.1.5.1.8). Tandem mass spectra were searched against UniProt tobacco database concatenated with reverse decoy database. Trypsin/P was specified as cleavage enzyme allowing up to 4 missing cleavages, 5 modifications per peptide and 5 charges. Mass error was set to 10 ppm for precursor ions and 0.02 Da for fragment ions. Carbamidomethylation on Cys was specified as fixed modification and oxidation on Met, crotonylation on Lys and crotonylation on protein N-terminal were specified as variable modifications. False discovery rate (FDR) thresholds for protein, peptide and modification sites were specified at 1%. Minimum peptide length was set at 7. All the other parameters in MaxQuant were set to default values. The site localization probability was set as >0.75.
Bioinformatics Methods
Motif-X software (http://motif-x.med.harvard.edu/) was used to analyse the model of sequences constituted with amino acids in specific positions of acetyl-21-mers (10 amino acids upstream and downstream of the site) in all protein sequences71. For further hierarchical clustering based on categories, all the acetylation substance categories obtained after enrichment were first collated along with their p-values, and subsequently filtered for those categories at least enriched in one of the clusters with a p-value < 0.05. This filtered p-value matrix was transformed by the function x = −log (p-value), and the x values for each category were z-transformed. These z scores were subsequently clustered using one-way hierarchical clustering (Euclidean distance, average linkage clustering) in the Genesis programme. The cluster membership was visualized using a heat map through the “heatmap.2” function in the “gplot2” R-package. Secondary structures were predicted using NetSurfP. Gene Ontology (GO) annotation proteome was derived from the UniProt-GOA database (http://www.ebi.ac.uk/GOA/). The proteins were classified using Gene Ontology annotation based on three categories: biological process, cellular component and molecular function. The protein subcellular localization was analysed using Wolfpsort (http://www.genscript.com/wolf-psort.html). The KEGG was used to annotate protein pathways. GO term, protein domain, and KEGG pathway enrichment were performed using the DAVID bioinformatics resources 6.7. Fisher’s exact test was used to examine the enrichment or depletion (two-tailed test) of specific annotation terms among members of resulting protein clusters. Correction for multiple hypothesis testing was performed using standard false discovery rate control methods. Any terms with adjusted p-values below 0.05 in any of the clusters were treated as significant. The Search Tool for Retrieval of Interacting Genes/Proteins (STRING) database (http://string-db.org/) was used for PPI analysis. Cytoscape (version 3.0) software was used to display the network72.
Electronic supplementary material
Acknowledgements
This work was supported by Shandong Provincial Natural Science Foundation (ZR2015YL065, ZR2014CQ025), State Tobacco Monopoly Bureau (110201601024(LS-04)), Hongyunhonghe Tobacco (Group) Co., Ltd. (HYHH2016YL02), and Yunnan Tobacco Company of China National Tobacco Corporation (2016YL02).
Author Contributions
J.Y. and F.W. designed research; H.S., X.L., F.L., W.L., J.Z. and Z.X. performed search; J.Y., H.S., L.S. and Y.L. analyzed data; H.S. wrote the paper.
Competing Interests
The authors declare that they have no competing interests.
Footnotes
Electronic supplementary material
Supplementary information accompanies this paper at doi:10.1038/s41598-017-03369-6
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Fenglong Wang, Email: wangfenglong@caas.cn.
Jinguang Yang, Email: yangjinguang@caas.cn.
References
- 1.Allfrey VG, Faulkner R, Mirsky AE. Acetylation and methylation of histones and their possible role in the regulation of rna synthesis. Proceedings of the National Academy of Sciences of the United States of America. 1964;51:786–794. doi: 10.1073/pnas.51.5.786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gu W, Roeder RG. Activation of p53 sequence-specific DNA binding by acetylation of the p53 C-terminal domain. Cell. 1997;90:595–606. doi: 10.1016/S0092-8674(00)80521-8. [DOI] [PubMed] [Google Scholar]
- 3.Roth SY, Denu JM, Allis CD. Histone acetyltransferases. Annual review of biochemistry. 2001;70:81–120. doi: 10.1146/annurev.biochem.70.1.81. [DOI] [PubMed] [Google Scholar]
- 4.Lee KK, Workman JL. Histone acetyltransferase complexes: one size doesn’t fit all. Nature reviews. Molecular cell biology. 2007;8:284–295. doi: 10.1038/nrm2145. [DOI] [PubMed] [Google Scholar]
- 5.Shahbazian MD, Grunstein M. Functions of site-specific histone acetylation and deacetylation. Annual review of biochemistry. 2007;76:75–100. doi: 10.1146/annurev.biochem.76.052705.162114. [DOI] [PubMed] [Google Scholar]
- 6.Hubbert C, et al. HDAC6 is a microtubule-associated deacetylase. Nature. 2002;417:455–458. doi: 10.1038/417455a. [DOI] [PubMed] [Google Scholar]
- 7.Onyango P, Celic I, McCaffery JM, Boeke JD, Feinberg AP. SIRT3, a human SIR2 homologue, is an NAD-dependent deacetylase localized to mitochondria. Proceedings of the National Academy of Sciences of the United States of America. 2002;99:13653–13658. doi: 10.1073/pnas.222538099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schwer B, North BJ, Frye RA, Ott M, Verdin E. The human silent information regulator (Sir)2 homologue hSIRT3 is a mitochondrial nicotinamide adenine dinucleotide-dependent deacetylase. The Journal of cell biology. 2002;158:647–657. doi: 10.1083/jcb.200205057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Choudhary C, et al. Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science (New York, N.Y.) 2009;325:834–840. doi: 10.1126/science.1175371. [DOI] [PubMed] [Google Scholar]
- 10.Finkemeier I, Laxa M, Miguet L, Howden AJ, Sweetlove LJ. Proteins of diverse function and subcellular location are lysine acetylated in Arabidopsis. Plant Physiol. 2011;155:1779–1790. doi: 10.1104/pp.110.171595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nallamilli BR, et al. Global analysis of lysine acetylation suggests the involvement of protein acetylation in diverse biological processes in rice (Oryza sativa) PLoS One. 2014;9:e89283. doi: 10.1371/journal.pone.0089283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kim SC, et al. Substrate and functional diversity of lysine acetylation revealed by a proteomics survey. Mol Cell. 2006;23:607–618. doi: 10.1016/j.molcel.2006.06.026. [DOI] [PubMed] [Google Scholar]
- 13.Zhao S, et al. Regulation of cellular metabolism by protein lysine acetylation. Science (New York, N.Y.) 2010;327:1000–1004. doi: 10.1126/science.1179689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Peng C, et al. The first identification of lysine malonylation substrates and its regulatory enzyme. Molecular & cellular proteomics: MCP. 2011;10:M111.012658. doi: 10.1074/mcp.M111.012658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhang Z, et al. Identification of lysine succinylation as a new post-translational modification. Nature chemical biology. 2011;7:58–63. doi: 10.1038/nchembio.495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Xie Z, et al. Lysine succinylation and lysine malonylation in histones. Molecular & cellular proteomics: MCP. 2012;11:100–107. doi: 10.1074/mcp.M111.015875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Park J, et al. SIRT5-mediated lysine desuccinylation impacts diverse metabolic pathways. Mol Cell. 2013;50:919–930. doi: 10.1016/j.molcel.2013.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Weinert BT, et al. Lysine succinylation is a frequently occurring modification in prokaryotes and eukaryotes and extensively overlaps with acetylation. Cell reports. 2013;4:842–851. doi: 10.1016/j.celrep.2013.07.024. [DOI] [PubMed] [Google Scholar]
- 19.Xie L, et al. First succinyl-proteome profiling of extensively drug-resistant Mycobacterium tuberculosis revealed involvement of succinylation in cellular physiology. Journal of proteome research. 2015;14:107–119. doi: 10.1021/pr500859a. [DOI] [PubMed] [Google Scholar]
- 20.Pan J, Chen R, Li C, Li W, Ye Z. Global Analysis of Protein Lysine Succinylation Profiles and Their Overlap with Lysine Acetylation in the Marine Bacterium Vibrio parahemolyticus. Journal of proteome research. 2015;14:4309–4318. doi: 10.1021/acs.jproteome.5b00485. [DOI] [PubMed] [Google Scholar]
- 21.Hirschey MD, Zhao Y. Metabolic Regulation by Lysine Malonylation, Succinylation, and Glutarylation. Molecular & cellular proteomics: MCP. 2015;14:2308–2315. doi: 10.1074/mcp.R114.046664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Colak G, et al. Proteomic and Biochemical Studies of Lysine Malonylation Suggest Its Malonic Aciduria-associated Regulatory Role in Mitochondrial Function and Fatty Acid Oxidation. Molecular & cellular proteomics: MCP. 2015;14:3056–3071. doi: 10.1074/mcp.M115.048850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Qian L, et al. Global Profiling of Protein Lysine Malonylation in Escherichia coli Reveals Its Role in Energy Metabolism. Journal of proteome research. 2016;15:2060–2071. doi: 10.1021/acs.jproteome.6b00264. [DOI] [PubMed] [Google Scholar]
- 24.Heiling S, et al. Using the knowns to discover the unknowns: MS-based dereplication uncovers structural diversity in 17-hydroxygeranyllinalool diterpene glycoside production in the Solanaceae. The Plant journal: for cell and molecular biology. 2016;85:561–577. doi: 10.1111/tpj.13119. [DOI] [PubMed] [Google Scholar]
- 25.Tan M, et al. Identification of 67 histone marks and histone lysine crotonylation as a new type of histone modification. Cell. 2011;146:1016–1028. doi: 10.1016/j.cell.2011.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Montellier E, Rousseaux S, Zhao Y, Khochbin S. Histone crotonylation specifically marks the haploid male germ cell gene expression program: post-meiotic male-specific gene expression. BioEssays: news and reviews in molecular, cellular and developmental biology. 2012;34:187–193. doi: 10.1002/bies.201100141. [DOI] [PubMed] [Google Scholar]
- 27.Baumann K. Post-translational modifications: Crotonylation versus acetylation. Nature reviews. Molecular cell biology. 2015;16:265. doi: 10.1038/nrm3992. [DOI] [PubMed] [Google Scholar]
- 28.Sabari BR, et al. Intracellular crotonyl-CoA stimulates transcription through p300-catalyzed histone crotonylation. Mol Cell. 2015;58:203–215. doi: 10.1016/j.molcel.2015.02.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bao, X. et al. Identification of ‘erasers’ for lysine crotonylated histone marks using a chemical proteomics approach. eLife3, doi:10.7554/eLife.02999 (2014). [DOI] [PMC free article] [PubMed]
- 30.Wu X, et al. Lysine acetylation is a widespread protein modification for diverse proteins in Arabidopsis. Plant Physiol. 2011;155:1769–1778. doi: 10.1104/pp.110.165852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Konig AC, Hartl M, Boersema PJ, Mann M, Finkemeier I. The mitochondrial lysine acetylome of Arabidopsis. Mitochondrion. 2014;19(Pt B):252–260. doi: 10.1016/j.mito.2014.03.004. [DOI] [PubMed] [Google Scholar]
- 32.He D, et al. Global Proteome Analyses of Lysine Acetylation and Succinylation Reveal the Widespread Involvement of both Modification in Metabolism in the Embryo of Germinating Rice Seed. Journal of proteome research. 2016;15:879–890. doi: 10.1021/acs.jproteome.5b00805. [DOI] [PubMed] [Google Scholar]
- 33.Zhang Y, et al. Comprehensive profiling of lysine acetylproteome analysis reveals diverse functions of lysine acetylation in common wheat. Scientific reports. 2016;6:21069. doi: 10.1038/srep21069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Smith-Hammond CL, Swatek KN, Johnston ML, Thelen JJ, Miernyk JA. Initial description of the developing soybean seed protein Lys-N(epsilon)-acetylome. Journal of proteomics. 2014;96:56–66. doi: 10.1016/j.jprot.2013.10.038. [DOI] [PubMed] [Google Scholar]
- 35.Smith-Hammond CL, Hoyos E, Miernyk JA. The pea seedling mitochondrial Nepsilon-lysine acetylome. Mitochondrion. 2014;19(Pt B):154–165. doi: 10.1016/j.mito.2014.04.012. [DOI] [PubMed] [Google Scholar]
- 36.Melo-Braga MN, et al. Modulation of protein phosphorylation, N-glycosylation and Lys-acetylation in grape (Vitis vinifera) mesocarp and exocarp owing to Lobesia botrana infection. Molecular & cellular proteomics: MCP. 2012;11:945–956. doi: 10.1074/mcp.M112.020214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jin W, Wu F. Proteome-Wide Identification of Lysine Succinylation in the Proteins of Tomato (Solanum lycopersicum) PLoS One. 2016;11:e0147586. doi: 10.1371/journal.pone.0147586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Salvato F, et al. The potato tuber mitochondrial proteome. Plant Physiol. 2014;164:637–653. doi: 10.1104/pp.113.229054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fang X, et al. Global analysis of lysine acetylation in strawberry leaves. Front Plant Sci. 2015;6:739. doi: 10.3389/fpls.2015.00739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhen S, et al. First Comprehensive Proteome Analyses of Lysine Acetylation and Succinylation in Seedling Leaves of Brachypodium distachyon L. Scientific reports. 2016;6:31576. doi: 10.1038/srep31576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sierro N, et al. The tobacco genome sequence and its comparison with those of tomato and potato. Nature communications. 2014;5:3833. doi: 10.1038/ncomms4833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Michelet L, et al. Redox regulation of the Calvin-Benson cycle: something old, something new. Front Plant Sci. 2013;4:470. doi: 10.3389/fpls.2013.00470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhang, Q. et al. Structural Insights into Histone Crotonyl-Lysine Recognition by the AF9 YEATS Domain. Structure (London, England: 1993)24, 1606–1612, doi:10.1016/j.str.2016.05.023 (2016). [DOI] [PMC free article] [PubMed]
- 44.Li Y, et al. Molecular Coupling of Histone Crotonylation and Active Transcription by AF9 YEATS Domain. Mol Cell. 2016;62:181–193. doi: 10.1016/j.molcel.2016.03.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhao D, et al. YEATS2 is a selective histone crotonylation reader. Cell research. 2016;26:629–632. doi: 10.1038/cr.2016.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Andrews FH, et al. The Taf14 YEATS domain is a reader of histone crotonylation. Nature chemical biology. 2016;12:396–398. doi: 10.1038/nchembio.2065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gao X, et al. Downregulation of Rubisco Activity by Non-enzymatic Acetylation of RbcL. Molecular plant. 2016;9:1018–1027. doi: 10.1016/j.molp.2016.03.012. [DOI] [PubMed] [Google Scholar]
- 48.Henderson JN, Hazra S, Dunkle AM, Salvucci ME, Wachter RM. Biophysical characterization of higher plant Rubisco activase. Biochimica et biophysica acta. 2013;1834:87–97. doi: 10.1016/j.bbapap.2012.09.006. [DOI] [PubMed] [Google Scholar]
- 49.Nagata N, Tanaka R, Satoh S, Tanaka A. Identification of a vinyl reductase gene for chlorophyll synthesis in Arabidopsis thaliana and implications for the evolution of Prochlorococcus species. Plant Cell. 2005;17:233–240. doi: 10.1105/tpc.104.027276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Margaritopoulou T, et al. HSP90 canonical content organizes a molecular scaffold mechanism to progress flowering. The Plant journal: for cell and molecular biology. 2016;87:174–187. doi: 10.1111/tpj.13191. [DOI] [PubMed] [Google Scholar]
- 51.Tillmann B, et al. Hsp90 Is Involved in the Regulation of Cytosolic Precursor Protein Abundance in Tomato. Molecular plant. 2015;8:1128. doi: 10.1016/j.molp.2015.05.011. [DOI] [PubMed] [Google Scholar]
- 52.Xu ZS, et al. Heat shock protein 90 in plants: molecular mechanisms and roles in stress responses. International journal of molecular sciences. 2012;13:15706–15723. doi: 10.3390/ijms131215706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yu A, et al. Roles of Hsp70s in Stress Responses of Microorganisms, Plants, and Animals. Biomed Res Int. 2015;2015:510319. doi: 10.1155/2015/510319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Valente MA, et al. The ER luminal binding protein (BiP) mediates an increase in drought tolerance in soybean and delays drought-induced leaf senescence in soybean and tobacco. Journal of experimental botany. 2009;60:533–546. doi: 10.1093/jxb/ern296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Liu JX, Howell SH. Managing the protein folding demands in the endoplasmic reticulum of plants. The New phytologist. 2016;211:418–428. doi: 10.1111/nph.13915. [DOI] [PubMed] [Google Scholar]
- 56.Liu Y, Li J. Endoplasmic reticulum-mediated protein quality control in Arabidopsis. Front Plant Sci. 2014;5:162. doi: 10.3389/fpls.2014.00162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sadanandom A, Bailey M, Ewan R, Lee J, Nelis S. The ubiquitin-proteasome system: central modifier of plant signalling. The New phytologist. 2012;196:13–28. doi: 10.1111/j.1469-8137.2012.04266.x. [DOI] [PubMed] [Google Scholar]
- 58.Cutter AR, Hayes JJ. A brief review of nucleosome structure. FEBS letters. 2015;589:2914–2922. doi: 10.1016/j.febslet.2015.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Andrews AJ, Chen X, Zevin A, Stargell LA, Luger K. The histone chaperone Nap1 promotes nucleosome assembly by eliminating nonnucleosomal histone DNA interactions. Mol Cell. 2010;37:834–842. doi: 10.1016/j.molcel.2010.01.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Durand-Dubief M, Persson J, Norman U, Hartsuiker E, Ekwall K. Topoisomerase I regulates open chromatin and controls gene expression in vivo. The EMBO journal. 2010;29:2126–2134. doi: 10.1038/emboj.2010.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bode J, Benham C, Knopp A, Mielke C. Transcriptional augmentation: modulation of gene expression by scaffold/matrix-attached regions (S/MAR elements) Critical reviews in eukaryotic gene expression. 2000;10:73–90. doi: 10.1615/CritRevEukarGeneExpr.v10.i1.90. [DOI] [PubMed] [Google Scholar]
- 62.Nickerson JA, Blencowe BJ, Penman S. The architectural organization of nuclear metabolism. International review of cytology. 1995;162a:67–123. doi: 10.1016/s0074-7696(08)61229-2. [DOI] [PubMed] [Google Scholar]
- 63.Cockerill PN, Garrard WT. Chromosomal loop anchorage of the kappa immunoglobulin gene occurs next to the enhancer in a region containing topoisomerase II sites. Cell. 1986;44:273–282. doi: 10.1016/0092-8674(86)90761-0. [DOI] [PubMed] [Google Scholar]
- 64.Martens JH, Verlaan M, Kalkhoven E, Dorsman JC, Zantema A. Scaffold/matrix attachment region elements interact with a p300-scaffold attachment factor A complex and are bound by acetylated nucleosomes. Mol Cell Biol. 2002;22:2598–2606. doi: 10.1128/MCB.22.8.2598-2606.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Xue Z, et al. SIRT1 deacetylates SATB1 to facilitate MAR HS2-MAR epsilon interaction and promote epsilon-globin expression. Nucleic acids research. 2012;40:4804–4815. doi: 10.1093/nar/gks064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Meier I, Phelan T, Gruissem W, Spiker S, Schneider D. MFP1, a novel plant filament-like protein with affinity for matrix attachment region DNA. Plant Cell. 1996;8:2105–2115. doi: 10.1105/tpc.8.11.2105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Samaniego R, Jeong SY, Meier I, de la Espina SM. Dual location of MAR-binding, filament-like protein 1 in Arabidopsis, tobacco, and tomato. Planta. 2006;223:1201–1206. doi: 10.1007/s00425-005-0168-x. [DOI] [PubMed] [Google Scholar]
- 68.Harder PA, Silverstein RA, Meier I. Conservation of matrix attachment region-binding filament-like protein 1 among higher plants. Plant Physiol. 2000;122:225–234. doi: 10.1104/pp.122.1.225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Lee YW, Kim WT. Tobacco GTBP1, a homolog of human heterogeneous nuclear ribonucleoprotein, protects telomeres from aberrant homologous recombination. Plant Cell. 2010;22:2781–2795. doi: 10.1105/tpc.110.076778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Lee YW, Kim WT. Roles of NtGTBP1 in telomere stability. Plant Signal Behav. 2011;6:523–525. doi: 10.4161/psb.6.4.14749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Schwartz D, Gygi SP. An iterative statistical approach to the identification of protein phosphorylation motifs from large-scale data sets. Nat Biotechnol. 2005;23:1391–1398. doi: 10.1038/nbt1146. [DOI] [PubMed] [Google Scholar]
- 72.Cline MS, et al. Integration of biological networks and gene expression data using Cytoscape. Nature protocols. 2007;2:2366–2382. doi: 10.1038/nprot.2007.324. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.