

Abstract
Activating mutations, such as E76K and D61Y, in PTPN11 (SHP2), a protein tyrosine phosphatase implicated in multiple cell signaling processes, are associated with 35% of patients with juvenile myelomonocytic leukemia (JMML), an aggressive childhood myeloproliferative neoplasm (MPN). Effective therapeutic interventions for this malignancy are still lacking. Here we show that the interaction between leukemia-associated mutant Shp2 and Gab2, a scaffolding protein important for cytokine-induced PI3K/Akt signaling, was enhanced, and that the mTOR pathway was elevated in Ptpn11E76K/+ leukemic cells. Importantly, MPN induced by the Ptpn11E76K/+ mutation was markedly attenuated in Ptpn11E76K/+/Gab2−/− double mutant mice — Overproduction of myeloid cells was alleviated, splenomegaly was diminished, and myeloid cell infiltration in non-hematopoietic organs was decreased in these double mutants. Excessive myeloid differentiation of stem cells was also normalized by depletion of Gab2. Acute leukemia progression of MPN was reduced in the double mutant mice, and as such, their survival was much prolonged. Furthermore, treatment of Ptpn11E76K/+ mice with Rapamycin, a specific and potent mTOR inhibitor, mitigated MPN phenotypes. Collectively, this study reveals an important role of the Gab2/PI3K/mTOR pathway in mediating the pathogenic signaling of the PTPN11 gain-of-function mutations, and a therapeutic potential of Rapamycin for PTPN11 mutation-associated JMML.
INTRODUCTION
Juvenile myelomonocytic leukemia (JMML), an aggressive myeloproliferative neoplasm (MPN) of early childhood, is characterized by hypersensitivity of myeloid progenitors to cytokines, such as granulocyte-macrophage colony stimulating factor (GM-CSF) 1, 2, and overproduction of myeloid cells and monocytes. JMML cells retain capabilities to differentiate, but can infiltrate into non-hematopoietic organs, disrupting their functions. This disease can progress to acute myeloid leukemia. While the current standard of care for patients with JMML relies on allogeneic hematopoietic stem cell (HSC) transplant, relapse is the most frequent cause of treatment failure 3–5. New therapeutic interventions are still needed for this fatal disease.
Significant progress in understanding the pathogenesis of JMML has been achieved by deciphering the genetic lesions that occur in patients. The genetic mutations identified in JMML occur in the signaling proteins involved in the RAS/ERK pathway 3–5, providing new opportunities for both diagnosis and therapy. Thirty-five percent of patients with JMML have mutations, such as E76K and D61Y, in the protein tyrosine phosphatase (PTP) PTPN11 (SHP2), a positive regulator of the Ras pathway (see below), while activating mutations in RAS (K-RAS or N-RAS), homozygous inactivation of NF1, a GTPase activating protein that negatively regulates Ras output, and loss-of-function mutations in the E3 ubiquitin ligase c-CBL account for 20%, 15%, and 10–15% of JMML cases, respectively 6–9. PTPN11, RAS, NF1, and c-CBL mutations are usually mutually exclusive in patients. When modeled using murine systems, gain-of-function mutations in Ptpn11, K-Ras, and N-Ras, or loss-of-function in Nf1 and Cbl, are sufficient to induce cytokine hypersensitivity in myeloid progenitors and to produce a JMML-like MPN in mice 10–18.
Ptpn11 (Shp2), a ubiquitously expressed PTP, is implicated in multiple cell signaling processes, such as the Ras/Erk, PI3K/Akt, Jak/STAT, and NF-κB pathways that are activated by diverse growth factors, cytokines, insulin, growth hormones, and cell adhesion molecules 19–21. Shp2 is normally self-inhibited by hydrogen bonding of the backside loop of the N-terminal SH2 (N-SH2) domain with the deep pocket of the PTP domain 22, 23, and gets activated upon binding to signaling partners with phospho-tyrosine (pY) residues. Intriguingly, this phosphatase plays an overall positive role in transducing signals initiated from receptor kinases, particularly in the Ras pathway 19–21. The underlying signaling mechanisms are still not yet well understood. PTPN11 mutations found in JMML are concentrated in the N-SH2 domain and result in amino acid changes at the interphase between N-SH2 and PTP domains, disrupting the inhibitory intramolecular interaction, thus leading to increased Shp2 catalytic activity 7, 24. Although Ras hyperactivation is central in the pathogenesis of JMML, Ras effector or parallel pathways, such as PI3K/Akt signaling, have also been shown to contribute to the disease 25, 26. While activated Ras is able to activate PI3K by a direct interaction with the p110 catalytic subunit 27, cytokines, such as GM-CSF, can also activate PI3K in a Ras-independent manner via a Shp2-containing protein complex including Gab2, Grb2, and PI3K regulatory subunit p85α 17, 28. In the present study, we sought to determine the role of the hematopoietic cell-specific scaffolding protein, Gab2, in gain-of-function Shp2-induced hyperactivation of PI3K/Akt and pathogenesis of MPN in vivo.
MATERIALS AND METHODS
Mice
Conditional knock-in mice (Ptpn11E76K neo/+) bearing the most common PTPN11 mutation found in JMML were generated in a previous study 16. Gab2+/− mice 29 were obtained from Dr. Toshio Hirano (Osaka University, Japan). Mx1-Cre+ transgenic mice were purchased from the Jackson Laboratory. Ptpn11E76K neo/+ mice were used to cross Mx1-Cre+ and Gab2+/− mice to generate Ptpn11E76K neo/+/Mx1-Cre+/Gab+/− mice. These mice were intercrossed to produce various types of mice for subsequent experiments. Mice of each genotype at the same age were numbered and randomly grouped for subsequent analyses. All mice were kept under specific pathogen–free conditions in the Division of Animal Resources at Emory University. All animal procedures complied with the National Institutes of Health Guidelines for the Care and Use of Laboratory Animals and were approved by the Institutional Animal Care and Use Committee.
Retroviral vectors and retroviral transduction
Shp2 E76K and Shp2 D61Y cDNAs were subcloned into the murine stem-cell virus (MSCV)-based bicistronic retroviral vector, pMIEG3, in tandem with enhanced green fluorescent protein cDNA (EGFP). Ecotropic retroviral supernatants (pMIEG3, pMIEG3-WT, pMIEG3-E76K, pMIEG3-D61Y) were prepared using Eco-Phoenix packaging cells by the Indiana University Vector Production Facility, as previously described 14.
Generation of BM-derived macrophages
BM cells were cultured in DMEM supplemented with 10% FBS and 20% L cell conditional medium (as a source of mouse colony-stimulating factor-1). Forty-eight hours later, nonadherent cells were collected and seeded into new tissue culture plates. After 5 to 7 days of culture, cells were confirmed to be macrophages as more than 90% of these cells were positive for Mac-1 and F4/80.
Flow cytometric analysis and cell sorting
Multiparameter FACS analyses were performed to determine populations of HSCs (Lineage−Sca-1+c-Kit+Flk2−CD48−CD150+), multipotent progenitor sub-population 1 (MPP1, Lineage−Sca-1+c-Kit+Flk2−CD48−CD150−), MPP2 (Lineage−Sca-1+c-Kit+Flk2−CD48+CD150+), and MPP3 (Lineage−Sca-1+c-Kit+Flk2−CD48+CD150−). Antibodies were purchased from eBiosciences, San Diego, CA, unless otherwise noted. BM cells freshly harvested from femurs and tibias were stained with biotin-labeled antibodies against mouse hematopoietic lineage markers: Mac-1 (M1/70), Gr-1 (RB6-8C5), Ter119 (TER-119), CD3e (145-2C11), B220 (RA3-6B2), and subsequently stained with antibodies conjugated with various fluorochromes: streptavidin eFluor® 450, Sca-1-PE-Cy7 (D7, BD Biosciences, San Jose, CA), c-Kit-APC-eFlour780 (2B8), CD150-Alexa Fluor® 647 (TC15-12FF12.2, BD Biosciences), CD48-FITC (HM48-1), Flk2-PE (A2F10.1, BD Biosciences) for HSCs or MPPs. Specific cell populations were gated based on immune phenotypes for quantification or cell sorting. Fluorescence Minus One (FMO) was used for setting the gating on control samples. Flow cytometric analyses were performed on BD LSR II (BD Biosciences, San Jose). Cell sorting was conducted using BD FACSAria. Data were analyzed using FlowJo software (TreeStar, Ashland).
Apoptosis analysis
Fresh BM cells were stained with biotin-labeled antibodies against lineage markers (Gr-1, Mac-1, Ter119, CD3e, and B220) as described above, followed by staining with: streptavidin eFluor® 450, Sca-1-PE-Cy7 (D7), c-Kit-APC-eFlour780 (2B8). Cells were then incubated with Annexin V-FITC and 7-amino-actinomycin D (7-AAD) using Annexin V-FITC apoptosis detection kit I (BD Biosciences, San Jose, CA). Apoptotic (Annexin V+) cells in the gated Lineage−Sca-1+c-Kit+ (LSK) cell population were quantified by FACS.
Colony-forming unit assay
For the myeloid progenitor assay, freshly harvested mouse BM nucleated cells (2×104 cells/mL) or patient biopsy marrow cells (1×104 cells/mL) were assayed for colony forming units (CFUs) in 0.9% methylcellulose IMDM medium containing 30% FBS, glutamine (10−4 M), β-mercaptoethanol (3.3×10−5 M), and IL-3 or GM-CSF at the indicated concentrations. After 7 days (mouse cells) or 14 days (human cells) of culture at 37 °C in a humidified 5% CO2 incubator, myeloid colonies (CFU-GM and CFU-M) were counted under an inverted microscope.
Rapamycin treatment
Rapamycin (LC Laboratories, Woburn) was dissolved in 100% ethanol at a concentration of 10 mg/mL, diluted in 5% Tween-80 and 5% PEG-400, and administered by intraperitoneal injections at 10 mg/kg body weight, once daily for 12–14 weeks starting on the following day after Ptpn11E76K/+/Mx1-Cre+ mice were administered pI-pC (to induce the Ptpn11E76K/+ mutation).
Statistical analysis
All experiments were repeated two to three times with the indicated numbers of mice. Data are presented as mean±S.D. Statistical significance was determined using unpaired two-tailed Student’s t test. P values < 0.05 were considered to be statistically significant.
RESULTS
Interaction of Shp2 with Gab2 is enhanced by leukemia-associated Ptpn11 mutations
The hallmark of JMML is Ras hyperactivation and the hypersensitivity of myeloid progenitors to cytokines 1, 2. However, signaling partners that mediate the effects of Ptpn11 mutations in this disease have not been well characterized. We hypothesized that the hematopoietic specific scaffolding protein, Gab2, contributes to Ptpn11 mutation-induced hyperactivation of Ras and PI3K/Akt signaling. To investigate this hypothesis, we transduced BM low density mononuclear cells with wildtype (WT) Shp2 or leukemia-associated mutants Shp2 E76K and Shp2 D61Y. As shown in Figure 1A, in response to GM-CSF stimulation, tyrosine phosphorylation of Gab2 was significantly increased in mutant Shp2-expressing cells as compared to WT Shp2-expressing cells. Moreover, interactions between mutant Shp2 and Gab2 were enhanced by Shp2 mutations. We also generated BM-derived macrophages from Ptpn11E76K conditional knock-in mice (Ptpn11E76K/+/Mx1-Cre+), which developed JMML-like MPN following induction of the Ptpn11E76K mutation 16, and determined the impact of the Ptpn11E76K mutation on GM-CSF-induced signaling. In response to GM-CSF stimulation, activation of Erk, Jak2, and Stat5 was increased in Ptpn11E76K/+ mutant cells (Figure 1B). Previously studies have shown that Gab2 plays an important role in the PI3K/Akt pathway by interacting with the p85 subunit of PI3K 28; we therefore took a closer look at this pathway. As shown in Figure 1C, activation of Akt was increased in Ptpn11E76K/+ cells. Furthermore, activities of the downstream mTOR pathway were also enhanced as evidenced by elevated phosphorylation levels of S6 and 4E-BP1. These observations together indicate that increased interaction of mutated Shp2 with Gab2 might mediate the detrimental effects of Ptpn11 mutations on cytokine signaling.
Figure 1. The Ptpn11E76K mutation enhances cytokine-induced PI3K/Akt/mTOR signaling.

(A) BM progenitors transduced with WT Shp2, Shp2 D61Y, Shp2 E76K, or control vector were sorted and differentiated to macrophages, which were starved in serum and cytokine-free medium for 48 hours and then stimulated with GM-CSF (50 ng/mL) for as long as 60 minutes. Protein extracts were prepared and immunoprecipitated with anti-Gab2 antibody followed by anti-pY immunoblotting. Blots were stripped and reprobed with anti-Shp2 and then anti-Gab2 antibodies. (B) BM-derived macrophages were generated from Ptpn11E76K/+/Mx1-Cre+ and Ptpn11+/+/Mx1-Cre+ mice 6–8 weeks following pI-pC administration. These cells were starved and then stimulated with GM-CSF (50 ng/mL) for the indicated periods of time. Whole cell lysates were prepared. Levels of p-ERK, p-Jak2, and p-Stat5 were determined by immunoblotting analyses. Blots were striped and reprobed with anti-ERK, anti-Jak2, anti-Stat5, and anti-Shp2 antibodies to check for protein loading. (C) The cell lysates were also examined for levels of p-AKT, p-mTOR, p-S6, and p-4E-BP1 by immunoblotting analyses. Blots were stripped and reprobed with anti-AKT and anti-mTOR antibodies to check for protein loading. Experiments were repeated three times with similar results obtained in each.
Important role of Gab2 in mediating the pathogenic effects of the Ptpn11E76K mutation
To further test the role of Gab2 in the pathogenesis and maintenance of MPN induced by Ptpn11 mutations, we generated Ptpn11E76K/+/Gab2−/− double mutant mice from Ptpn11E76K knock-in 16 and Gab2 knockout (Gab2+/−) 29 mice, and monitored disease development and progression in these double mutants. This test is especially interesting given that steady state hematopoiesis in Gab2 deficient mice is not significantly changed although Gab2-depleted stem cells showed decreased repopulation capabilities in competitive repopulation assays 30. Compared to Ptpn11E76K/+ single mutant mice, gross health signs of Ptpn11E76K/+/Gab2−/− double mutants were significantly improved. MPN phenotypes, such as elevated white blood cell (WBC) counts (Supplemental Table 1) and splenomegaly were attenuated in double mutants (Figure 2A). Overproduction of mature myeloid cells (Mac-1+/Gr-1+) and immature myeloid cells (Mac-1+/Gr-1−) was largely corrected (Figure 2B). In addition, myeloid cell infiltration in the liver (Figure 2C), lung, and kidney (data not shown) was diminished in double mutant mice. Histopathological examination confirmed that excessive proliferation of myeloid cells in the BM and spleen was reduced and myeloid cell infiltration in non-hematopoietic organs (liver, lung, and kidney) was significantly decreased in these double mutant mice (Supplemental Figure 1). More importantly, only 11% of Ptpn11E76K/+/Gab2−/− mice progressed into acute leukemias as opposed to 63% in Ptpn11E76K/+ single mutant mice during 12 month follow-up (Figure 2D), and the overall survival of double mutants was prolonged (Figure 2E). These observations suggest that Gab2 plays an important role in mediating the pathogenic effects of the Ptpn11E76K mutation in the development and progression of MPN.
Figure 2. Development and progression of Ptpn11E76K/+ mutation-induced MPN are attenuated by deletion of Gab2.
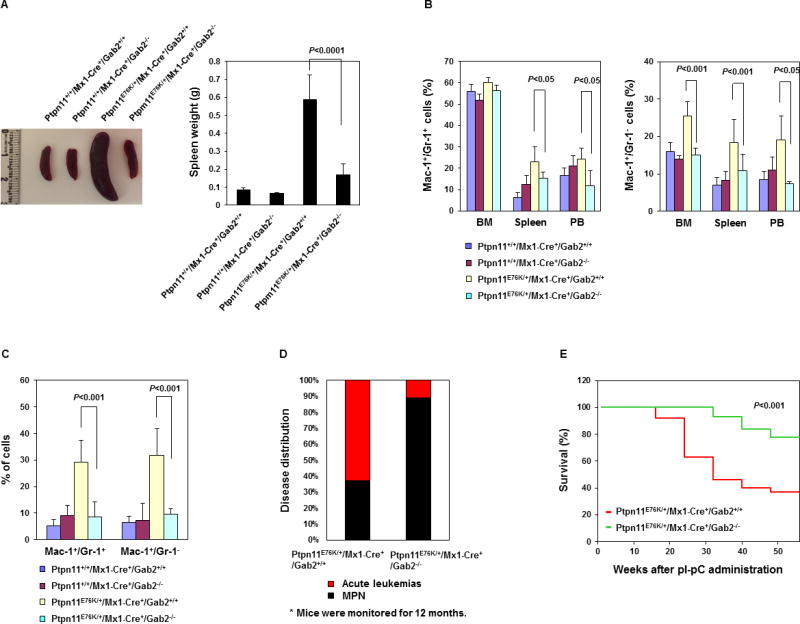
Ptpn11+/+/Mx1-Cre+/Gab2+/+, Ptpn11+/+/Mx1-Cre+/Gab2−/−, Ptpn11E76K/+/Mx1-Cre+/Gab2+/+, and Ptpn11E76K/+/Mx1-Cre+/Gab2−/− mice were analyzed 10~14 weeks after pI-pC administration. Spleen weights (A) (n=5–10/group), percentages of Mac-1+/Gr-1+ and Mac-1+/Gr-1− cells in the BM, spleen, peripheral blood (B), and liver (C) (n=4–10/group) were determined. Data are shown as mean±S.D. (D) A cohort of pI-pC-administered Ptpn11E76K/+/Mx1-Cre+/Gab2+/+ (n=27) and Ptpn11E76K/+/Mx1-Cre+/Gab2−/− (n=18) mice were monitored for acute leukemia progression for 12 months as we previously described 16. The percentages of the mice that progressed to acute leukemias were documented. (E) Kaplan–Meier survival curves of Ptpn11E76K/+/Mx1-Cre+/Gab2+/+ and Ptpn11E76K/+/Mx1-Cre+/Gab2−/− mice.
MPN is known to be a clonal stem cell disorder; to further determine whether the rescue effects of Gab2 deletion occurred at the stem cell level, we analyzed HSCs in double mutants and compared to those in Ptpn11E76K/+ single mutants. The frequency and absolute number of HSCs in the BM of Ptpn11E76K/+ mice were reduced due to the hyperactivation and exhaustion of the reserve capacity, consistent with previous findings 16. The frequency and absolute number (Supplemental Figure 2A–2C) of HSCs at steady state in Ptpn11E76K/+/Gab2−/− mice were not significantly changed as compared to Ptpn11E76K/+ mice. However, apoptosis of hematopoietic stem/progenitor cells (Lineage−Sca-1+c-Kit+, LSK cells) in Ptpn11E76K/+/Gab2−/− double mutant mice was nearly restored to the physiological level (Supplemental Figure 2D and 2E) as opposed to significantly reduced apoptosis in Ptpn11E76K/+ single mutant mice.
The myeloid malignancy induced by Ptpn11 mutations is characterized by cytokine hypersensitivity of myeloid progenitors 1, 2; we therefore assessed the sensitivity of myeloid progenitors to GM-CSF and IL-3 in Ptpn11E76K/+/Gab2−/− double and Ptpn11E76K/+ single mutant mice. As demonstrated in Figure 3A and Supplemental Figure 3, Gab2 depletion in Ptpn11E76K/+ progenitors significantly decreased the hypersensitivity to GM-CSF and IL-3. Moreover, cytokine-independent baseline colony formation of myeloid progenitors was abolished in Ptpn11E76K/+/Gab2−/− double mutants. To further verify these results, we sorted stem cell-enriched LSK cells and assessed their responses to cytokine-induced differentiation. Compared to Ptpn11E76K/+ cells, myeloid differentiation/expansion (Figure 3B and Supplemental Figure 4 and Figure 3C) of Ptpn11E76K/+/Gab2−/− cells in the presence of IL-3 was significantly decreased. These data together suggest that Gab2 depletion attenuates Ptpn11E76K/+ mutation-induced myeloid malignancy mainly by decreasing the cytokine sensitivity of myeloid progenitors.
Figure 3. Depletion of Gab2 corrects the hypersensitive growth pattern of Ptpn11E76K/+ myeloid progenitors.

(A) BM cells (2×104 cells/mL) freshly harvested from the mice with the indicated genotypes (n=3/group) were assessed by CFU assays 8 weeks after pI-pC administration with a range of GM-CSF concentrations. (B) BM-derived macrophages were generated from the mice with the indicated genotypes (n=3/group) and cultured in DMEM supplemented with 10% FBS and 20% L cell conditional medium for 3 days. Cell growth rates were determined using a One Solution Cell Proliferation Assay kit. (C) LSK (Lineage−Sca-1+c-Kit+) cells were sorted from the mice with the indicated genotypes (n=6/group) and cultured in the presence of IL-3 (2 ng/mL). After 7 days of in vitro culture, percentages of myeloid (Mac-1+/Gr-1+) cells differentiated from LSK cells were assayed by FACS. Three independent experiments were performed with similar results obtained in each. Data shown are mean±S.D. from one experiment.
Gab2 depletion attenuates the detrimental effects of the Ptpn11E76K mutation on cytokine signaling
The signaling mechanisms underlying the effects of Gab2 deletion on the pathogenesis of Ptpn11E76K mutation-induced MPN were determined. Gab2 is an important scaffolding protein in cytokine signaling pathways 28. We generated BM-derived macrophages from Ptpn11E76K/+ single and Ptpn11E76K/+/Gab2−/− double mutant mice and analyzed GM-CSF signaling in these cells. As shown in Figure 4A, in response to GM-CSF stimulation, activation of Erk and Stat5 was not significantly changed in the double mutant cells. However, Akt activity determined by phosphorylation of Serine473 and Threonine308 was decreased in the double mutant cells as compared to that in Ptpn11E76K/+ single mutant cells, suggesting that the rescue effects of Gab2 deficiency on Ptpn11E76K mutation-associated aberrant hematopoietic cell development might be attributable to the reduced Akt pathway. To verify this notion, we analyzed the downstream mTOR signaling in primary MPN cells isolated from diseased Ptpn11E76K/+ mice and Ptpn11E76K/+/Gab2−/− double mutants. As shown in Figure 4B, phosphorylation of S6 and 4E-BP1 in the MPN cells of Ptpn11/Gab2 double mutants was reduced relative to that in Ptpn11E76K/+ single mutant mice.
Figure 4. Gab2 deletion reverses the enhanced PI3K/Akt/mTOR signaling caused by the Ptpn11E76K/+ mutation.

(A) BM-derived macrophages generated from pI-pC administered Ptpn11E76K/+/Mx1-Cre+/Gab2+/+ and Ptpn11E76K/+/Mx1-Cre+/Gab2−/− mice were starved in serum and cytokine-free medium for 48 hours and then stimulated with GM-CSF (50 ng/mL) for the indicated periods of time. Whole cell lysates were prepared. Levels of p-Erk, p-Akt, and p-Stat5 were determined by immunoblotting analyses. Blots were striped and reprobed with anti-Erk, anti-Akt, anti-Shp2, and anti-Gab2 antibodies to check for protein loading and Gab2 deletion efficiency. Three independent experiments with three pairs of mice were performed. Similar results were obtained in each. (B) MPN cell lysates prepared from Ptpn11E76K/+/Mx1-Cre+/Gab2+/+ and Ptpn11E76K/+/Mx1-Cre+/Gab2−/− mice 10 weeks after pI-pC administration when MPN was fully developed were immunoblotted with p-S6 and p-4E-BP1. Shp2 levels in the tissue lysates were examined by anti-Shp2 immunoblotting. Each lane represents an individual mouse.
Therapeutic effects of Rapamycin for the MPN induced by the Ptpn11E76K mutation
Given that MPN was ameliorated in Ptpn11E76K/+/Gab2−/− double mutant mice and that the mTOR signaling activities were decreased in double mutant cells, we reasoned that targeting the mTOR pathway might be effective in controlling Ptpn11 activating mutation-induced myeloid malignancy. To test this hypothesis, we treated Ptpn11E76K knock-in mice with Rapamycin, a potent and selective inhibitor of mTOR, and monitored disease phenotypes in these mice. As shown in Figure 5A, treatment of Rapamycin at an optimal dosage significantly decreased WBC counts in diseased Ptpn11E76K/+ mice. Importantly, control mice did not show overt hematopoietic suppression, highlighting a potential therapeutic index. Splenomegaly was also reduced in treated Ptpn11E76K/+ mice (Figure 5B). Furthermore, cytokine hypersensitivity of myeloid progenitors was abolished in Rapamycin-treated Ptpn11E76K/+ mice (Figure 5C and Supplemental Figure 5), and the colonies derived in colony assays were much smaller (Figure 5D). Additionally, we analyzed the mTOR signaling pathway in the BM cells isolated from Rapamycin-treated Ptpn11E76K/+ and control mice, and found that phosphorylation of S6 and 4E-BP1 was barely detectable (Figure 5E). These results collectively demonstrate therapeutic effects of Rapamycin in the mouse model of Ptpn11 mutation-associated myeloid malignancy.
Figure 5. Rapamycin treatment mitigates MPN in Ptpn11E76K/+ mice.

(A) WBC counts in the peripheral blood of Ptpn11E76K/+/Mx1-Cre+ mice were determined before and after 12–14 weeks of treatment with Rapamycin (10 mg/kg body weight, once daily) or the vehicle (n=5/group). (B) Spleen weights were determined after 12–14 weeks of treatment with Rapamycin or vehicle. (C) BM cells were harvested from Ptpn11+/+/Mx1-Cre+ and Ptpn11E76K/+/Mx1-Cre+ mice after 12–14 weeks of treatment with Rapamycin or vehicle. These cells were assessed by CFU assays at the indicated GM-CSF concentrations. Experiments were performed three times with similar results obtained in each. Data shown are mean±S.D. from one experiment in triplicates. (D) Representative colonies growing in the medium containing GM-CSF (0.1 ng/mL) are shown. The average number of the cells per colony was determined by dividing the total number of the cells harvested from each plate with the number of the colonies in that plate (n=3/group). Experiments were performed in three pairs of mice with similar results obtained in each. Data shown are mean±S.D. from one experiment in triplicates. (E) Spleen lysates were prepared from Ptpn11+/+/Mx1-Cre+ and Ptpn11E76K/+/Mx1-Cre+ mice after 12–14 weeks of treatment with Rapamycin or vehicle. Levels of p-S6, p-4E-BP1, and Shp2 were determined by immunoblotting analyses. Each lane represents an individual mouse.
Rapamycin corrects the hypersensitive growth pattern of JMML patient cells with the PTPN11E76K mutation
BM cells from patients with JMML display a characteristic hypersensitive growth pattern, resulting in greater production of granulocyte-macrophage colonies in response to GM-CSF 1, 2. We next determined whether Rapamycin could inhibit colony formation and proliferation of JMML patient cells with PTPN11 activating mutations at an optimal dosage that does not inhibit normal marrow progenitors. As shown in Figure 6A, GM-CSF-induced colony formation of myeloid blasts from JMML patients with PTPN11 activating mutations was more sensitive to Rapamycin than healthy donor progenitors. Consistent with this data, PTPN11 mutation-positive JMML cells cultured in GM-CSF-containing liquid medium was also more sensitive than healthy donor BM cells to the inhibition by Rapamycin at 2.5 nM (Figure 6B). The finding that Rapamycin is able to overcome the dominant effects of oncogenic mutant SHP2 in primary human leukemia cells support for a potential therapeutic benefit of this drug in PTPN11 mutation-associated JMML.
Figure 6. Rapamycin abrogates cytokine-induced overgrowth of JMML patient cells with the PTPN11E76K mutation.

(A) BM cells (1×104 cells/mL) from five JMML patients with the indicated PTPN11 mutations and three normal donors were plated in methylcellulose medium containing GM-CSF (1.0 ng/mL) and Rapamycin at the indicated concentrations. Colonies were enumerated 14 days later and normalized against the number of the colonies derived without Rapamycin treatment. (B) BM cells from five JMML patients with the indicated PTPN11 mutations and normal donors were cultured in IMDM containing GM-CSF (1.0 ng/mL) and Rapamycin (2.5 nM) or vehicle. Cell numbers were determined using the One Solution Cell Proliferation Assay kit. Data shown are mean±S.D. of all samples tested.
DISCUSSION
Hyperactivation of the Ras/Erk signaling pathway is known to underlie the pathogenesis of JMML 3–5. Interestingly, the PI3K/Akt signaling pathway is also dysregulated in this disease, and this can be dependent and independent of the Ras pathway 25. PTPN11 gain-of-function mutations are associated with 35% of JMML. However, it has not been well characterized how PTPN11 mutations lead to Ras-dependent and/or independent hyperactivation of the PI3K/Akt signaling. In this study, we demonstrate that the interaction between JMML-associated mutant Shp2 E76K and Gab2, a scaffolding protein important for cytokine-induced PI3K/Akt signaling 28, was enhanced, and that the mTOR pathway was highly activated in Ptpn11E76K/+ leukemic cells. Importantly, MPN induced by the Ptpn11E76K/+ mutation was markedly attenuated in Ptpn11E76K/+/Gab2−/− double mutant mice. These observations indicate that Gab2 may play a key role downstream of mutated Shp2 in mediating the leukemogenic effects of Ptpn11 mutations. Ptpn11 gain-of-function mutations appear to disturb cytokine signaling through multiple mechanisms. These mutations result in increased catalytic activity of Shp2 7, 24 and also enhanced binding to signaling partners 13, 31 due to the disruption of the inhibitory intramolecular interaction and the conformational change of the Shp2 protein. The gain-of-function mutations of Shp2 enhance Ras activity likely through elevated catalytic activity of Shp2 since Shp2 catalytic activity plays a positive role in the Ras pathway although the detailed mechanism is still elusive 19–21. Hyperactivation of Ras leads to increased PI3K/Akt signaling due to the well-established crosstalk between the two pathways 27. In addition, mutated Shp2 may directly induce increased PI3K/Akt signaling through enhanced binding with the scaffolding protein Gab2 that couples the p85 subunit of PI3K to the cytokine receptor 28 and the plasma membrane. In this signaling complex, mutant Shp2 may serve as an adaptor protein independent of its catalytic activity. Indeed, this phosphatase can function in cytokine signaling in both catalytically-dependent and –independent manners 32.
Another important finding is this study is the pre-clinical therapeutic efficacy of Rapamycin in the mouse model of Ptpn11 mutation-induced MPN. We showed that administration of Rapamycin at a tolerable dose significantly alleviated the MPN burden in Ptpn11E76K/+ mice, and also that this drug at a low dose selectively suppressed PTPN11 mutation-positive JMML patient cells over healthy donor cells in vitro. Given that Rapamycin is a clinically-used immunosuppressant and anti-cancer drug for adult patients, this drug may be further developed, used alone or in combination with MEK inhibitors that target the hyperactivated Ras/Erk pathway, as a novel therapeutic strategy for PTPN11 mutation-associated JMML. This notion is also supported by the previous observation that PTEN expression, a negative regulator of PI3K/Akt/mTOR signaling, frequently was reduced in primary JMML patient samples 33 although the gene mutation status of the JMML specimens was not determined. The elevated Ras-independent activation of PI3/Akt/mTOR signaling may be specific for Ptpn11 mutation-induced MPN since this is caused by enhanced interaction of mutated Shp2 with Gab2. It remains to be determined whether the PI3/Akt/mTOR pathway may also be an effective target for the treatment of other subtypes of JMML associated with the RAS, NF1, or CBL mutations.
Supplementary Material
Acknowledgments
We are grateful to Dr. Toshio Hirano for Gab2+/− mice. This work was supported by National Institutes of Health grants HL130995 and DK092722 and a Hyundai Hope on Wheels scholar grant (to C.K.Q.).
Footnotes
Authorship contributions
W.L., W.M.Y., and J.Z., conducted the research and summarized the data. R.J.C., M.L.L., Z.Z., and K.D.B. provided critical reagents, discussed the work, and edited the manuscript. C.K.Q. designed the experiments and provided technical training to the first three authors. W.L. and C.K.Q. wrote the manuscript.
Conflict-of-interest disclosure
The authors declare no competing financial interests.
References
- 1.Birnbaum RA, O’Marcaigh A, Wardak Z, Zhang YY, Dranoff G, Jacks T, et al. Nf1 and Gmcsf interact in myeloid leukemogenesis. Molecular cell. 2000 Jan;5(1):189–195. doi: 10.1016/s1097-2765(00)80415-3. [DOI] [PubMed] [Google Scholar]
- 2.Emanuel PD, Bates LJ, Castleberry RP, Gualtieri RJ, Zuckerman KS. Selective hypersensitivity to granulocyte-macrophage colony-stimulating factor by juvenile chronic myeloid leukemia hematopoietic progenitors. Blood. 1991 Mar 1;77(5):925–929. [PubMed] [Google Scholar]
- 3.Chang TY, Dvorak CC, Loh ML. Bedside to bench in juvenile myelomonocytic leukemia: insights into leukemogenesis from a rare pediatric leukemia. Blood. 2014 Oct 16;124(16):2487–2497. doi: 10.1182/blood-2014-03-300319. [DOI] [PubMed] [Google Scholar]
- 4.Liu X, Sabnis H, Bunting KD, Qu CK. Molecular targets for the treatment of juvenile myelomonocytic leukemia. Adv Hematol. 2012;2012:308252. doi: 10.1155/2012/308252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Emanuel PD. Juvenile myelomonocytic leukemia and chronic myelomonocytic leukemia. Leukemia. 2008 Jul;22(7):1335–1342. doi: 10.1038/leu.2008.162. [DOI] [PubMed] [Google Scholar]
- 6.Loh ML, Vattikuti S, Schubbert S, Reynolds MG, Carlson E, Lieuw KH, et al. Mutations in PTPN11 implicate the SHP-2 phosphatase in leukemogenesis. Blood. 2004 Mar 15;103(6):2325–2331. doi: 10.1182/blood-2003-09-3287. [DOI] [PubMed] [Google Scholar]
- 7.Tartaglia M, Niemeyer CM, Fragale A, Song X, Buechner J, Jung A, et al. Somatic mutations in PTPN11 in juvenile myelomonocytic leukemia, myelodysplastic syndromes and acute myeloid leukemia. Nat Genet. 2003 Jun;34(2):148–150. doi: 10.1038/ng1156. [DOI] [PubMed] [Google Scholar]
- 8.Loh ML, Sakai DS, Flotho C, Kang M, Fliegauf M, Archambeault S, et al. Mutations in CBL occur frequently in juvenile myelomonocytic leukemia. Blood. 2009 Aug 27;114(9):1859–1863. doi: 10.1182/blood-2009-01-198416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Muramatsu H, Makishima H, Jankowska AM, Cazzolli H, O’Keefe C, Yoshida N, et al. Mutations of an E3 ubiquitin ligase c-Cbl but not TET2 mutations are pathogenic in juvenile myelomonocytic leukemia. Blood. 2010 Mar 11;115(10):1969–1975. doi: 10.1182/blood-2009-06-226340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhang Y, Taylor BR, Shannon K, Clapp DW. Quantitative effects of Nf1 inactivation on in vivo hematopoiesis. The Journal of clinical investigation. 2001 Sep;108(5):709–715. doi: 10.1172/JCI12758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chan IT, Kutok JL, Williams IR, Cohen S, Kelly L, Shigematsu H, et al. Conditional expression of oncogenic K-ras from its endogenous promoter induces a myeloproliferative disease. The Journal of clinical investigation. 2004 Feb;113(4):528–538. doi: 10.1172/JCI20476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Braun BS, Tuveson DA, Kong N, Le DT, Kogan SC, Rozmus J, et al. Somatic activation of oncogenic Kras in hematopoietic cells initiates a rapidly fatal myeloproliferative disorder. Proc Natl Acad Sci U S A. 2004 Jan 13;101(2):597–602. doi: 10.1073/pnas.0307203101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Araki T, Mohi MG, Ismat FA, Bronson RT, Williams IR, Kutok JL, et al. Mouse model of Noonan syndrome reveals cell type- and gene dosage-dependent effects of Ptpn11 mutation. Nat Med. 2004 Aug;10(8):849–857. doi: 10.1038/nm1084. [DOI] [PubMed] [Google Scholar]
- 14.Chan RJ, Leedy MB, Munugalavadla V, Voorhorst CS, Li Y, Yu M, et al. Human somatic PTPN11 mutations induce hematopoietic-cell hypersensitivity to granulocyte-macrophage colony-stimulating factor. Blood. 2005 May 1;105(9):3737–3742. doi: 10.1182/blood-2004-10-4002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chan G, Kalaitzidis D, Usenko T, Kutok JL, Yang W, Mohi MG, et al. Leukemogenic Ptpn11 causes fatal myeloproliferative disorder via cell-autonomous effects on multiple stages of hematopoiesis. Blood. 2009 Apr 30;113(18):4414–4424. doi: 10.1182/blood-2008-10-182626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Xu D, Liu X, Yu WM, Meyerson HJ, Guo C, Gerson SL, et al. Non-lineage/stage-restricted effects of a gain-of-function mutation in tyrosine phosphatase Ptpn11 (Shp2) on malignant transformation of hematopoietic cells. J Exp Med. 2011 Sep 26;208(10):1977–1988. doi: 10.1084/jem.20110450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mohi MG, Williams IR, Dearolf CR, Chan G, Kutok JL, Cohen S, et al. Prognostic, therapeutic, and mechanistic implications of a mouse model of leukemia evoked by Shp2 (PTPN11) mutations. Cancer Cell. 2005 Feb;7(2):179–191. doi: 10.1016/j.ccr.2005.01.010. [DOI] [PubMed] [Google Scholar]
- 18.Sanada M, Suzuki T, Shih LY, Otsu M, Kato M, Yamazaki S, et al. Gain-of-function of mutated C-CBL tumour suppressor in myeloid neoplasms. Nature. 2009 Aug 13;460(7257):904–908. doi: 10.1038/nature08240. [DOI] [PubMed] [Google Scholar]
- 19.Chan G, Kalaitzidis D, Neel BG. The tyrosine phosphatase Shp2 (PTPN11) in cancer. Cancer Metastasis Rev. 2008 Jun;27(2):179–192. doi: 10.1007/s10555-008-9126-y. [DOI] [PubMed] [Google Scholar]
- 20.Nabinger SC, Chan RJ. Shp2 function in hematopoietic stem cell biology and leukemogenesis. Current opinion in hematology. 2012 Jul;19(4):273–279. doi: 10.1097/MOH.0b013e328353c6bf. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Xu D, Qu CK. Protein tyrosine phosphatases in the JAK/STAT pathway. Front Biosci. 2008;13:4925–4932. doi: 10.2741/3051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Eck MJ, Pluskey S, Trub T, Harrison SC, Shoelson SE. Spatial constraints on the recognition of phosphoproteins by the tandem SH2 domains of the phosphatase SH-PTP2. Nature. 1996;379(6562):277–280. doi: 10.1038/379277a0. [DOI] [PubMed] [Google Scholar]
- 23.Hof P, Pluskey S, Dhe-Paganon S, Eck MJ, Shoelson SE. Crystal structure of the tyrosine phosphatase SHP-2. Cell. 1998;92(4):441–450. doi: 10.1016/s0092-8674(00)80938-1. [DOI] [PubMed] [Google Scholar]
- 24.Keilhack H, David FS, McGregor M, Cantley LC, Neel BG. Diverse biochemical properties of Shp2 mutants. Implications for disease phenotypes. J Biol Chem. 2005 Sep 2;280(35):30984–30993. doi: 10.1074/jbc.M504699200. [DOI] [PubMed] [Google Scholar]
- 25.Goodwin CB, Li XJ, Mali RS, Chan G, Kang M, Liu Z, et al. PI3K p110delta uniquely promotes gain-of-function Shp2-induced GM-CSF hypersensitivity in a model of JMML. Blood. 2014 May 1;123(18):2838–2842. doi: 10.1182/blood-2013-10-535104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gritsman K, Yuzugullu H, Von T, Yan H, Clayton L, Fritsch C, et al. Hematopoiesis and RAS-driven myeloid leukemia differentially require PI3K isoform p110alpha. The Journal of clinical investigation. 2014 Apr;124(4):1794–1809. doi: 10.1172/JCI69927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rodriguez-Viciana P, Warne PH, Dhand R, Vanhaesebroeck B, Gout I, Fry MJ, et al. Phosphatidylinositol-3-OH kinase as a direct target of Ras. Nature. 1994 Aug 18;370(6490):527–532. doi: 10.1038/370527a0. [DOI] [PubMed] [Google Scholar]
- 28.Gu H, Neel BG. The “Gab” in signal transduction. Trends Cell Biol. 2003 Mar;13(3):122–130. doi: 10.1016/s0962-8924(03)00002-3. [DOI] [PubMed] [Google Scholar]
- 29.Nishida K, Wang L, Morii E, Park SJ, Narimatsu M, Itoh S, et al. Requirement of Gab2 for mast cell development and KitL/c-Kit signaling. Blood. 2002 Mar 1;99(5):1866–1869. doi: 10.1182/blood.v99.5.1866. [DOI] [PubMed] [Google Scholar]
- 30.Zhang Y, Diaz-Flores E, Li G, Wang Z, Kang Z, Haviernikova E, et al. Abnormal hematopoiesis in Gab2 mutant mice. Blood. 2007 Jul 1;110(1):116–124. doi: 10.1182/blood-2006-11-060707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yu WM, Daino H, Chen J, Bunting KD, Qu CK. Effects of a Leukemia-associated Gain-of-Function Mutation of SHP-2 Phosphatase on Interleukin-3 Signaling. J Biol Chem. 2006 Mar 3;281(9):5426–5434. doi: 10.1074/jbc.M507622200. [DOI] [PubMed] [Google Scholar]
- 32.Yu WM, Hawley TS, Hawley RG, Qu CK. Catalytic-dependent and -independent roles of SHP-2 tyrosine phosphatase in interleukin-3 signaling. Oncogene. 2003 Sep 4;22(38):5995–6004. doi: 10.1038/sj.onc.1206846. [DOI] [PubMed] [Google Scholar]
- 33.Liu YL, Castleberry RP, Emanuel PD. PTEN deficiency is a common defect in juvenile myelomonocytic leukemia. Leuk Res. 2009 May;33(5):671–677. doi: 10.1016/j.leukres.2008.09.036. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.