

Abstract
Chronic Lymphocytic Leukemia (CLL) is a progressive malignancy of mature B-cells that involves the peripheral blood (PB), lymph nodes (LNs) and bone marrow (BM). While the majority of CLL cells are in a resting state, small populations of proliferating cells exist; however, the anatomical site of active cell proliferation remains to be definitively determined. Based on findings that CLL cells in LNs have increased expression of B-cell activation genes, we tested the hypothesis that the fraction of “newly born” cells would be highest in the LNs. Using a deuterium oxide (2H) in vivo labeling method in which patients consumed deuterated (heavy) water (2H2O), we determined CLL cell kinetics in concurrently obtained samples from LN, PB, and BM. The LN was identified as the anatomical site harboring the largest fraction of newly born cells, compared to PB and BM. In fact, the calculated birth rate in the LN reached as high a 3.3% of the clone per day. Subdivision of the bulk CLL population by flow cytometry identified the subpopulation with the CXCR4dimCD5bright phenotype as containing the highest proportion of newly born cells within each compartment, including the LN, identifying this subclonal population as an important target for novel treatment approaches.
Introduction
Chronic Lymphocytic Leukemia (CLL) and Small Lymphocytic Lymphoma (SLL) are B-cell malignancies that mainly affect the elderly.1 CLL and SLL are considered different presentations of the same disease.2, 3 CLL is defined as ≥5 000 monoclonal B-cells per μL in the peripheral blood (PB) with or without involvement of the lymphoid organs including the lymph nodes (LNs). In SLL, the affected cells are primarily in the LNs with ≤5 000 monoclonal B-cells per μL in the PB. Here we will refer to CLL as comprising both CLL and SLL.
Patients with CLL have a variable disease course with a third of patient's never needing treatment. In contrast, other patients need treatment soon after diagnosis and a subset of these only reach short remissions and undergo rapid decline and death thereafter.4, 5 Progressive CLL is often characterized by the usage of unmutated IGHV genes, high expression of CD49d, and genomic alterations that lead to a more rapid clonal expansion and inferior response to chemoimmunotherapy.4, 6-9
CLL is characterized by a large population of resting cells which may be resistant to apoptosis and a smaller, but actively proliferating cell population.10 The identification of the site of proliferation is of interest for understanding the process by which CLL progresses to more aggressive disease. Previous work using in vivo deuterium (2H) incorporation estimated that between 0.1 and 1% of the CLL cells circulating in the PB are added to the population per day (referred to a “newly born” cells) and identified distinct CLL subpopulations that contain variable fractions of these newly born cells.10-13 However, the anatomical compartment where active CLL cell proliferation occurs remains unknown. Proliferative or newly born CLL cells have been detected in PB, BM and LN, albeit of different clone sizes and with the use of different methodologies.10-13 We recently showed that gene expression profiles of CLL cells in LNs are similar to those of activated, proliferating B-cells, while gene expression profiles of CLL cells present in the PB are similar to those of resting memory B-cells.14, 15 We, therefore, hypothesized that the LN will be a critical site for CLL proliferation and progression.
Two cell surface membrane molecules have been particularly useful in identifying functionally different populations of CLL cells in the PB. These are the chemokine C-X-C motif receptor 4 (CXCR4), a chemokine receptor known to regulate cell trafficking, and CD5, a cell surface molecule expressed on normal T-cells, on a fraction of normal B-lymphocytes, especially upon activation, and, characteristically, on CLL B-cells. Using the reciprocal densities of these two molecules on the surface of CLL cells obtained from the PB of patients who consumed 2H2O, the CXCR4dimCD5bright fraction was identified as the population with the highest proportion of 2H-labelled cells and has, therefore, been referred to as the proliferative subset.16 Based on this data, we hypothesized that the CXCR4dimCD5bright population contains the cells that recently emigrated from the LNs into the circulating blood; however, the proliferative fraction of CLL cells in the LN remains to be characterized.
Here we sought to directly compare cellular growth rates of CLL cells collected simultaneously from patient matched PB, LNs, and BM using the 2H in vivo labeling method and concurrent analysis of all three compartments. We now show conclusively that the proportion of newly born CLL cells is highest in the LN, compared to the PB and BM. Further, we directly demonstrate that the proliferative fraction of the clone is contained in the CXCR4dimCD5bright population in all three compartments.
Materials and methods
Study Design
This study was designed as an auxiliary study to complement work on CLL within the Hematology Branch at the National Heart, Lung, and Blood Institute (Bethesda, MD, USA) and was registered as NCT01117142 with clinicaltrials.gov. The institutional review board approved these studies. Inclusion criteria were a diagnosis of CLL/SLL, age ≥18 years of age, absolute neutrophil count ≥1,000/μL, platelet count ≥50,000/μL. Patients using agents described to affect CLL cell biology or proliferation (including phosphodiesterase inhibitors, prednisone, cyclosporine, and green tea extract) were excluded. Lymph node biopsies were only obtained from patients with enlarged superficial lymph nodes. All study participants gave written informed consent. Diagnoses were made by established criteria.17
2H2O administration and sample collection protocol
Patients were instructed to drink 42 ml (46 gm) of 100% 2H2O (Sigma Aldrich, St. Louis, MO, USA) in the morning, noon, and evening for the first four days. Afterwards, patients drank 42 ml daily in the morning until they completed a total of four weeks. The goal was to achieve enrichment of 1-1.5% 2H2O of total body water. Consumption of 2H2O commenced on Day 1. A baseline PB sample was obtained prior to 2H2O ingestion. Patients returned at scheduled intervals during and following the labeling period to donate blood for cell and serum analyses. BM and LN biopsies were performed on Day 13 ± 2. A labeling period of 2 weeks was chosen to provide sufficient time for equilibration of 2H2O in total body water and incorporation of 2H2O into DNA of proliferating cells. Saliva samples were collected to determine total body water enrichment. In addition, patients kept a daily log of 2H2O intake which was discussed with each patient to assure compliance. No major unexpected side effects were encountered, although two patients reported a transient sense of lightheadedness and one patient reported a change in taste during the loading phase. These symptoms resolved spontaneously.
Cell preparation
PB mononuclear cells (PBMC) were isolated by density gradient centrifugation with Lymphocyte Separation Medium (MP Biomedicals, Santa Ana, CA, USA) and used fresh or after thawing frozen aliquots that had been cryopreserved in liquid nitrogen in 10% dimethyl sulfoxide and 90% fetal calf serum (Sigma Aldrich). Single cell suspensions from LN biopsies and BM aspirates were handled similarly.14 CLL cells (>98% of CD19+ cells were CLL cells) were isolated using positive selection (CD19+ magnetic beads, Miltenyi, Auburn, CA, USA) and snap frozen.
Sorting into subpopulations with differential CXCR4 and CD5 expression and Ki67 staining
Isolation of cell fractions on the basis of expression of CXCR4 and CD5 was carried out as previously described.16 Flow cytometry reagents were purchased from BD Bioscience, San Jose, CA, USA. Briefly, cells were incubated with murine anti-human mAbs: CD5-FITC (cat# 555352), CD19-APC (cat# 555415) and CXCR4-PE (cat #555974). After gating on CD19+CD5+ events, cells were sorted with a BD FACSAria on the basis of intensity of CXCR4 and CD5 expression. Isolated cell pellets were stored at −80°C until analysis.
Ki67 expression was assessed in CLL cells stained with CD19-PE-Cy5 (cat# 555414) and CD5-PE-Cy7 (cat# 348790).18 After surface staining, cells were washed and suspended in 4% paraformaldehyde (on ice, 1h), washed, resuspended in 70% ethanol (at –20°C, 2h), washed, incubated with anti-Ki67-FITC (cat# 556026, BD Biosciences) for 20min at room temperature, washed, and analyzed on a BD FACSCanto II flow cytometer. Data were analyzed using FlowJo software (Tree Star Inc.).
Measurement of body 2H2O enrichment and 2H incorporation into cellular DNA
2H2O enrichment in plasma was determined at KineMed Inc. (Emeryville, CA, USA), using variations of two methods described previously.19 Analysis of deuterium incorporation into DNA was determined using methods previously described.20, 21 Briefly, cells were disrupted by incubation with proteinase K and filtered to remove cell debris. DNA retained on the filter was hydrolyzed. Clean up, derivatization, and isotope ratio mass spectrometry (IRMS) was carried out as previously detailed.21 Sorted subpopulations with differential CXCR4 and CD5 membrane densities were analyzed by gas-chromatography/mass spectrometry (GC/MS) as previously described.16
Body 2H2O enrichments to estimate precursor enrichment
Body water enrichment data were evaluated using a single compartment model in which the fit parameters were the subject's total body water (TBW) volume and body water turnover. In this model, TBW is considered a single, homogeneous, well-mixed pool at steady-state, into which heavy water is introduced in known amounts and at known intervals. The model parameters, TBW and body water turnover (kw), were determined by least squares fit to the measured body water enrichment data obtained throughout the study. These data were analyzed with SAAM II software (Simulation, Analysis, and Modeling; SAAM Institute, University of Washington, Seattle, WA, USA). In this way, a continuous time course of body water enrichment was obtained for each subject. The average heavy water exposure during the labeling period relates to the amount of isotope that could be incorporated into newly synthesized DNA. To calculate average body water exposure for each sample timepoint, the body water enrichment time course modeled as above was integrated up to that timepoint (using the trapezoid rule) and represents the time averaged precursor enrichment (p) at each timepoint.
2H enrichments in DNA and calculation of fractional DNA synthesis
In order to calculate the newly born CLL cell fraction for each timepoint from GC/P/IRMS data, atom percent excess (APE) was first calculated from measured 2H–1H:1H–1H abundance ratios in the GC peaks corresponding to the dR derivative. The maximal or asymptotic amount of 2H incorporation (APE*) for fully turned-over (100% new) DNA was calculated as: APE* = p × 5.5/59, where p is the average precursor enrichment (as above) and the numerical conversion ratio corresponds to the number of hydrogen atoms in dR accessible to 2H2O labeling, divided by the number of hydrogen atoms in the derivative. Fractional DNA synthesis (F) was then calculated using the ratio of the measured APE to the APE*.
For the DNA samples analyzed for isotopic enrichment by GC/MS, F was calculated from the deuterium enrichment in dR (EM1) and the time-averaged p using a mathematical relationship previously described (F = EM1/EM1*, where EM1* = 3.025 × p+0.0014).21 A daily rate of appearance of newly born CLL cells (k) was obtained by fitting F to the exponential rise-to-plateau equation [F = 1-exp (-k t)].
Statistics
To compare measurements in patient samples from PB, LN, and BM either across time or compartment, a paired Student's t-test was used unless otherwise stated (Prism5, GraphPad, La Jolla, CA, USA). Values of P<0.05 were considered statistically significant.
Results
2H incorporation into the DNA of CLL cells is higher in LN compared to PB and BM
Fifteen patients ingested 2H2O for 2-4 weeks; 12 had CLL and three SLL. Collectively, we will refer to the tumor cells as CLL cells. The demographic and baseline disease characteristics of the patients are summarized in Table 1. One patient was previously treated; the other 14 patients were treatment naïve, eight had stable disease, and seven had active or progressive disease at the time of 2H2O labeling. Seven patients had active disease and initiated treatment within a median of 1.6 months from starting 2H2O intake. The content of 2H2O in total body water was measured weekly. Enrichment of 2H2O in the desired range of 1 to 1.5% of total body water was reached by day 7 and maintained throughout the labeling period (Figure 1a). Excisional LN biopsies and/or BM aspirates were obtained on day 13. PB was sampled at baseline and on day 13 and when possible on day 28 and day 35. CLL cells were purified using positive selection, and 2H incorporation into the cellular DNA was measured by mass spectrometry. Based on the level of 2H incorporation into the DNA of CLL cells, the newly born CLL cell fraction, i.e. cells generated during the labeling period, was determined for each sample and timepoint. The newly born CLL cell fraction differed significantly between the three anatomic sites (P<0.01, by repeated measures ANOVA). At day 13, the fraction of newly born CLL cells was largest in the LN at 33% followed by 23% in PB and 10% in BM (Figure 1b). Thus, in some patients up to one third of the tumor burden in the LN at day 13 consisted of cells born within the preceding two weeks.
Table 1.
Patient demographics and disease characteristics
| Patient | Disease | Age(y) | Gender | Stage1 | Time From Diagnosis (years) | Time To Treatment (months)2 | Disease Status3 | LDT4 (months) | IGHV | VH Gene | CD38 (%) | Cytogenetic Status5 (%) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| HW01 | CLL | 48 | F | 3 | 4 | 51 | A | 9.19 | M | VH4-34 | 2 | 13q- (93%) |
| HW02 | SLL | 59 | F | 4 | 3 | 40 | A | ND | U | VH3-09 | 39 | +12 (49%) |
| HW03 | CLL | 77 | M | 2 | 11 | 143 | S | 19.7 | U | VH1-69 | 64 | no abnormality |
| HW04 | CLL | 56 | F | 3 | 1 | 14 | A | 3.34 | U | VH4-39 | 82 | +12 (64%) |
| HW05 | CLL | 61 | M | 1 | 3 | 86 | S | ND | M | VH1-18 JH4b | 2 | no abnormality |
| HW06 | CLL | 57 | F | 3 | 2 | 38 | S | 13.55 | U | VH1-eJH3b | 43 | 13q- (8%) |
| HW07 | SLL | 75 | M | 4 | 5 | 63 | S | ND | M | VH7-04.1 | 71 | +12 (56%), 13q- (45%) |
| HW08 | CLL | 47 | M | 3 | 3 | 39 | A | 20.82 | M | VH3-74 | 8 | 17p- (16%), +12 (17%), 13q- (19%) |
| HW09 | CLL | 73 | M | 2 | 1 | 31 | S | 11.79 | M | VH3-23 | 0.8 | 11q- (9%), 13q- (60%) |
| HW11 | CLL | 55 | F | 1 | 4 | 79 | S | 24 | M | VH3-07 | 2 | 13q- (60%) |
| HW12 | SLL | 73 | F | 4 | 5 | 92 | S | ND | M | VH4-34 | ND | +12 (8%) |
| HW13 | CLL | 67 | M | 2 | 5 | 84 | S | 19.67 | U | VH3-21 | 3 | 13q- (73%) |
| HW15 | CLL | 63 | M | 1 | 1 | 16 | A | 4.86 | U | VH1-eJ6b | 81 | +12 (62%), 13q- (7%) |
| HW18 | CLL | 33 | M | 2 | 1 | 11 | A | ND | U | VH1-69 | 0.7 | p53 mutation* |
| HW20 | CLL | 69 | F | 2 | 1 | 8 | A | 2.74 | U | VH3-30 | 0.9 | 17p- (43%) |
F, Female; M, Male; Mu, Mutated; U, Unmutated; ND, Not done
Rai Staging for patients with CLL and Ann Arbor Staging System for patients with SLL
Time to treatment is calculated from time of diagnosis to initiation of treatment or last follow-up for patients with stable disease where treatment indications have not been met.
Disease status by IWCLL guidelines; A, Active disease, treatment indicated; S, Stable disease, no treatment indicated.
Lymphocyte doubling time. LDT is not applicable for SLL patients and not available for HW05 who had no change in ALC during the observation period and HW18 as there were only two time points available.
Cytogenetic status was determined by FISH on peripheral blood cells
This patient was included in the analyses for 17p-
Figure 1. 2H2O enrichment in body water and 2H incorporation into cellular DNA to label “newly born” CLL cells.
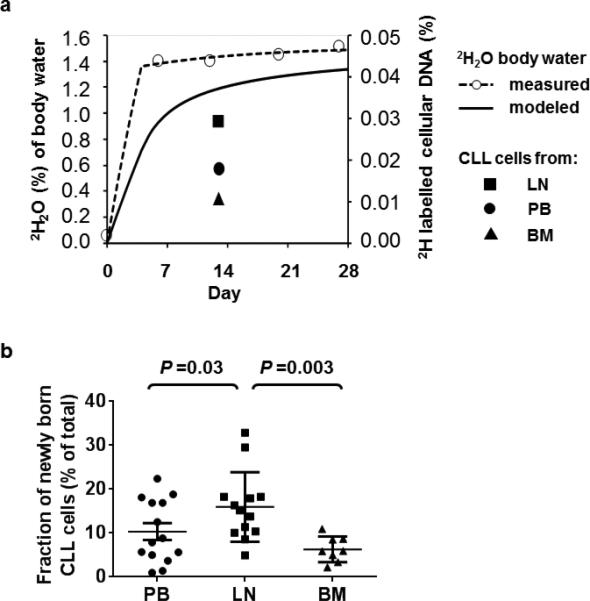
(a) Heavy water enrichment in body water (left y-axis) and the fraction of 2H labelled cellular DNA (right y-axis) in CLL cells obtained from three anatomic compartments (LN, lymph node, PB, peripheral blood, BM, bone marrow) in a representative patient (HW04) is shown. 2H2O was consumed for 28 days. On days 7, 14, 21, and 28 saliva and/or serum samples were collected to determine 2H exposure. These time points (open circles, dashed line) were used to calculate the average 2H exposure per day (solid line). The goal range of 1 to 1.5% 2H2O in body water was achieved by day 7. (b) The fraction of newly born CLL cells on day 13 is shown for PB (circle), LN (square), and BM (triangle). Plot depicts the mean ± standard error of the mean. Comparisons between different anatomic sites were done by repeated measures ANOVA.
In a previous study the “birth rate” of cells was estimated from the fraction of newly born CLL cells present in PB samples.10 Here we concurrently measured the fraction of newly born CLL cells in three different anatomic compartments. Because the fraction of newly born CLL cells at any one site is determined by the birth rate at the given site and the rate and direction of exchange of newly born cells between compartments we refer to k as the % newly born cells added to the total population in any given compartment per day. The median value of k in the PB was 0.56 (range 0.08 – 2.11), in the LN 1.27 (0.42 – 3.32), and in the BM 0.51 (range 0.18 – 0.96). Across different patients and compartments k was highly variable (Table 2). In twelve patients PB and LN samples were obtained concurrently and k was significantly higher in LN than PB in all except on patient (P=0.03; Figure 2a). In eight patients PB and BM samples were obtained concurrently; k was not significantly different across all eight samples (P=0.2; Figure 2b) and in four of eight patients, k was actually lower in the BM than in PB. In five of six patients with concurrent BM and LN samples k was higher in LN than BM but the overall difference did not reach significance (P=0.07; Figure 2c).
Table 2.
Rate of appearance of new cells k (%/day)
| Patient | Disease | Day 13* ALC (×103/μL) | 13* PB k | 13* LN k | 13* BM k | 28* PB k | 35* PB k |
|---|---|---|---|---|---|---|---|
| HW01 | CLL | 243 | 0.42 | 1.65 | 0.42 | 0.42 | 0.38 |
| HW02 | SLL | 3 | - | 1.27 | - | - | - |
| HW03 | CLL | 37 | 0.62 | - | 0.23 | 0.79 | 0.79 |
| HW04 | CLL | 163 | 1.66 | 2.92 | 0.96 | 1.69 | - |
| HW05 | CLL | 20 | 0.08 | - | 0.18 | 0.11 | 0.11 |
| HW06 | CLL | 142 | 0.48 | 1.24 | - | 0.53 | - |
| HW07 | SLL | 2 | 2.11 | 0.91 | 0.50 | 2.04 | - |
| HW08 | CLL | 116 | 1.12 | 1.37 | 0.76 | 0.97 | - |
| HW09 | CLL | 75 | 0.34 | 0.82 | - | 0.49 | 0.47 |
| HW11 | CLL | 30 | 0.12 | 0.42 | 0.51 | 0.14 | 0.14 |
| HW12 | SLL | 4 | 0.49 | 0.88 | 0.74 | 0.36 | 0.31 |
| HW13 | CLL | 13 | 0.68 | 1.00 | - | 0.64 | 0.52 |
| HW15 | CLL | 23 | 1.42 | 1.56 | - | 0.92 | 1.36 |
| HW18 | CLL | 33 | 0.5 | 1.44 | - | - | - |
| HW20 | CLL | 101 | 1.74 | 3.32 | - | - | - |
±2 days; ALC, Absolute lymphocyte count;
PB, Peripheral blood; BM, Bone marrow; LN, Lymph node
6 patients provided PB, LN, and BM; 6 patients provided PB and LN; 2 patients provided PB and BM and in one patient only the LN sample could be analyzed.
Figure 2. Growth rates of CLL cells are higher in the LN than in PB or BM.
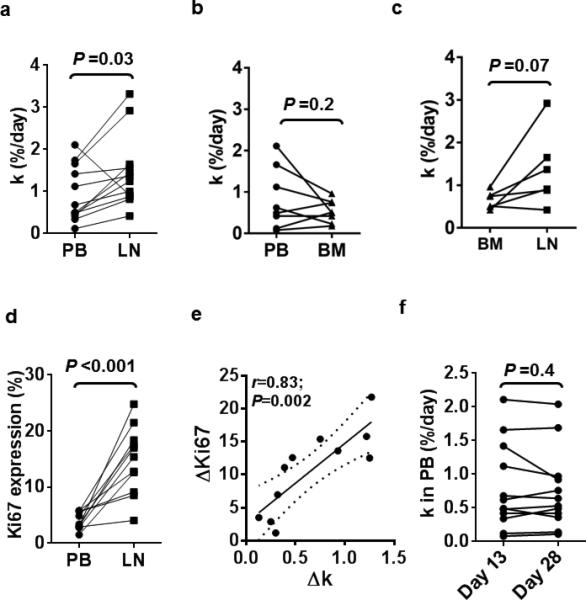
(a-c) The population growth rate k, defined as the % newly born cells added to a population per day is compared between concurrently obtained samples from PB and LN (n=12) (a), PB and BM (n=8) (b) and BM and LN (n=6) (c). (d-e) Expression of the proliferation marker Ki67 is higher in LN than in PB and correlated to growth rate. (d) Ki67 expression measured by flow cytometry in CLL cells obtained on day 13 from PB and LN (n=11). (e) The correlation between the change in Ki67 expression and the population growth rate k in the LN compared to PB is shown (n=11). Solid line represents linear regression fit and dotted lines represent 95% confidence intervals. P-value and r-value were calculated by Pearson's correlation. (f) The population growth rate k estimated from analysis of PB on day 13 and day 28 (n=12). Paired data (samples from the same patient) are connected by a line; statistical comparison by paired Student t test.
To estimate cell proliferation we stained cells from PB and LN for Ki67 using flow cytometry (Figure 2d). The median fraction of Ki67 expressing CLL cells in the LN was 15.4% (range 4.1 - 24.8) compared with 3.4% (range 1.5 - 5.8) in the PB. The measured increase in cell proliferation in the LN between the two assays, in one detected as Ki67 expression and in the other as more 2H2O incorporation into cellular DNA, showed a strong positive correlation (r=0.83; P=0.002; Figure 2e). Interestingly, while k in CLL cells in the LN on average doubled compared to PB (Figure 2a), the fraction of Ki67 positive cells in LN quadrupled (Figure 2d). While Ki67 is transiently expressed in cells entering the cell cycle and in recently divided cells, 2H2O permanently labels cells that divided at any timepoint during the labeling period. Therefore, the difference in the estimated fraction of dividing cells is consistent with active cell proliferation in LN, from where newly formed cells emigrate into the PB.
In patients who completed four weeks of 2H2O ingestion, we obtained additional PB samples on day 28 and day 35 (Table 2). Consistent with continuous cell proliferation and 2H2O incorporation into cellular DNA, the fraction of newly born cells increased from day 13 to day 28 (data not shown). However, the rate at which newly formed cells appeared in the PB was constant over time, with k = 0.51, 0.48, and 0.52 %/day on days 13, 28, and 35, respectively. Further, in individual patients, there was very little variability of k on day 13 compared to day 28 (Figure 2f). Thus, the birth rate of CLL cells, while highly variable between different patients, appears to be quite steady in the individual patient, at least over the timeframe studied.
The fraction of newly born cells (k) is highest in the CXCR4dim/CD5bright subset
Previous cell kinetic studies using 2H2O labeling showed that a CXCR4dim/CD5bright subpopulation in the PB contained a larger fraction of newly formed cells than the bulk of the CLL cells.16, 22 These CXCR4dim/CD5bright cells are thought to be recent emigrants from tissue sites, in particular from LNs. Here, we extended this analysis to LN and BM samples. CLL cells from PB, LN, and BM were sorted into three subpopulations as previously described:16, 22 the CXCR4dim/CD5bright (db) “proliferative”, the CXCR4int/CD5int (ii) “bulk”, and the CXCR4bright/CD5dim (bd) resting/re-entry subsets (Figure 3a). The CXCR4dim/CD5bright subset had the highest mean k in all three compartments and contained at least 3-times as many newly born cells than any of the other fractions within the same anatomic site (P<0.0001 in PB and LN; Table 3, Figure 3b). Interestingly, k in the CXCR4dim/CD5bright subset from LN and PB was quite comparable at 3.6 and 3.2%/day, respectively. At this rate, the whole proliferative fraction (CXCR4dim/CD5bright) is being replaced with newly formed cells every month. In contrast, only between 0.5 to 1% newly formed cells are added each day to the CXCR4int/CD5int and the CXCR4bright/CD5dim subsets that therefore contain mostly older cells (estimated greater than 70% of cells older than 1 month). Compared to LN and PB, k for each subset was the lowest in the BM.
Figure 3. Newly divided CLL cells are enriched in the CXCR4dim/CD5bright population.
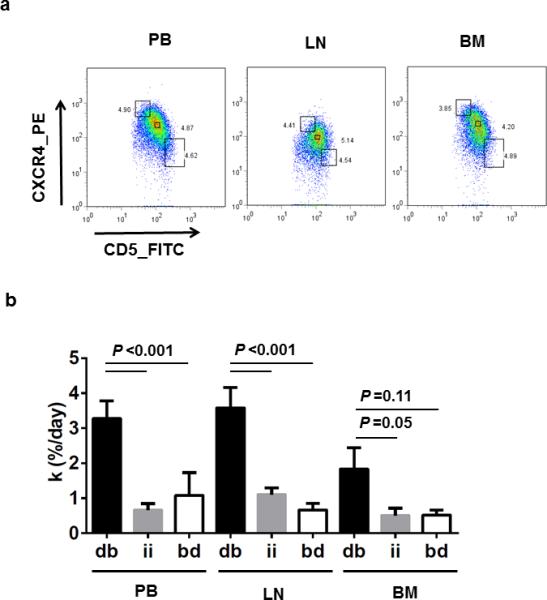
(a) Dot plots of CD19+CD5+ CLL cells collected from PB, LN, and BM on the basis of reciprocal densities of CXCR4 and CD5 surface membrane levels. Flow gates were set to include ~3 to 5% of total cells in each fraction; from top to bottom: CXCR4bright/CD5dim (bd), CXCR4int/CD5int (ii) and CXCR4dim/CD5bright (db). (b) Mean (± standard error of the mean) of the fraction of newly born CLL cells (k) for each of the three flow cytometry sorted fractions (as shown in panel a; db, ii, and bd) for each of the three anatomic compartments (PB (n=11), LN (n=13), and BM (n=4)). Results are detailed for each patient in Table 3. Paired T test was used to compare k between cellular fractions within each anatomic compartment.
Table 3.
Rate of appearance of new cells k (%/day) for sorted fractions
| ID | PB Day 13* k | LN Day 13* k | BM Day 13* k | ||||||
|---|---|---|---|---|---|---|---|---|---|
| db | ii | bd | db | ii | bd | db | ii | bd | |
| HW01 | 3.57 | 0.69 | 0.48 | 3.60 | 1.31 | 0.59 | 1.12 | −0.05 | 0.13 |
| HW02 | - | - | - | 6.11 | 1.53 | 0.68 | - | - | - |
| HW03 | - | - | - | - | - | - | - | - | - |
| HW04 | 3.34 | 1.79 | 0.76 | 5.89 | 1.92 | 0.83 | 0.61 | 0.31 | 0.77 |
| HW05 | - | - | - | - | - | - | - | - | - |
| HW06 | 2.93 | 0.35 | 0.06 | 1.68 | 1.02 | 0.64 | - | - | - |
| HW07 | - | - | - | 2.33 | 0.38 | 0.13 | - | - | - |
| HW08 | 3.27 | 0.37 | 0.21 | 4.25 | 0.87 | −0.01 | 2.33 | 0.79 | 0.57 |
| HW09 | 1.80 | 0.25 | 0.34 | 2.23 | 0.86 | 0.20 | - | - | - |
| HW11 | 2.04 | 0.22 | 0.43 | 0.40 | 0.37 | 1.74 | - | - | - |
| HW12 | 1.01 | −0.07 | −0.20 | 1.65 | 0.27 | 0.30 | - | - | - |
| HW13 | 3.34 | 0.22 | 0.05 | 3.05 | 1.36 | 0.22 | - | - | - |
| HW15 | 7.29 | 1.13 | 7.36 | 7.91 | 0.30 | 0.21 | - | - | - |
| HW18 | 2.74 | 0.53 | 1.85 | 4.43 | 1.64 | 0.86 | - | - | - |
| HW20 | 4.70 | 1.76 | 0.54 | 3.02 | 2.54 | 2.31 | 3.29 | 0.91 | 0.64 |
ID, Identification
±2 days; PB, Peripheral blood; BM, Bone marrow;
LN, Lymph node; Sorted fractions analyzed for each compartment: CXCR4dim/CD5bright (db); CXCR4intermediate/CD5intermediate (ii); CXCR4bright/CD5dim (bd).
Birth rate of CLL cells and disease characteristics
Next, we correlated the growth rate of the LN populations with clinical characteristics and prognostic markers. Of the 13 patients who underwent LN sampling, seven had active disease and six had stable disease according to IWCLL criteria (Table 1, Figure 4).17 Patients with active disease had the highest growth rate, on average 1.6% of the LN clone per day, which was more than twice the rate in patients with stable disease (Figure 5a, P=0.001), while the growth rate of the PB and BM was not statistically different between patients with active and stable disease. There was an inverse correlation of the growth rate in LN populations with lymphocyte doubling time (ρ=−0.83; P=0.008; Figure 5b) and the time to first treatment (r=−0.685; P=0.01, Figure 5c), consistent with increased proliferation in the LN as a driver of active disease and the requirement for early treatment. There was also a trend towards higher growth rate in patients with IGHV unmutated CLL (Ig-unmutated) compared to patients with IGHV mutated (Ig-mutated) disease (P=0.07; Figure 5d). CCL3 and CCL4 are chemokines secreted by activated CLL cells in the LN and high CCL3 serum concentrations have been associated with more rapid disease progression.14, 23 We therefore measured CCL3 and CCL4 serum levels in our patients. While there was a trend for higher CCL3 serum levels in patients with Ig-unmutated CLL there was no significant correlation between k in LN and CCL3 (r= 0.36; P= 0.23) or CCL4 (r= 0.295; P= 0.32) serum levels (Supplemental Data).
Figure 4. Rate of addition of new cells to the LN population and clinical characteristics of the patients.
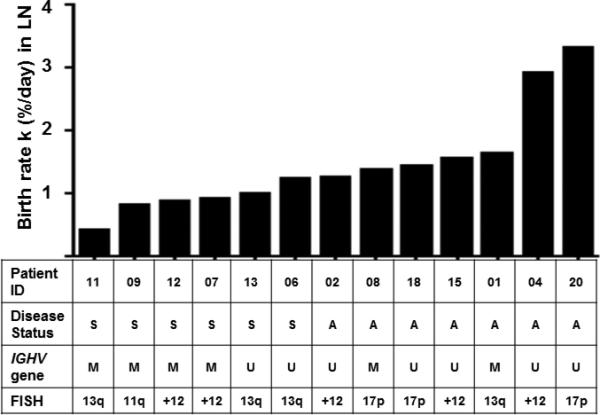
Bars depict k in the LN of the individual patients. Patients are arranged from left to right by increasing k values. Disease status (S, stable disease; A, active disease); IGHV mutation status (M, mutated; U, unmutated); and FISH cytogenetics (deletions 13q, 11q, 17p, and trisomy 12 according to the Dohner hierarchical classification).
Figure 5. CLL cell birth rates in the LN and clinical disease characteristics.
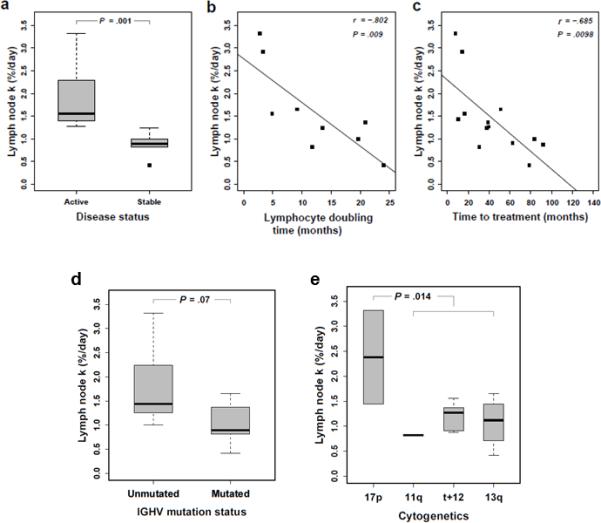
(a) Appearance of newly born cells (k) in the LN of patients stratified by disease status depicted in Box and Whisker plots; median (bold bar, 1.6 for active (n=7) and 0.9 for stable (n=8) disease). Pearson correlation of k in LN and (b) lymphocyte doubling time (n=9 pairs) and (c) time to first treatment (n=13 pairs). (d) k in the LN of patients stratified by IGHV gene mutation status depicted in Box and Whisker plots; median (bold bar, 1.4 for IGHV unmutated (n=8) and 0.9 for IGHV mutated (n=6) CLL). (e) Box and Whisker plots of k in the LN of patients stratified by cytogenetic aberrations, 17p- (n=2), 11q- (n=1), +12 (n=6), 13q- (n=4). All statistical comparisons by Mann-Whitney U test.
Four types of cytogenetic abnormalities routinely assessed by FISH are associated with clinically meaningful differences in survival. In order of associated shortest to longest median survival time, these are 17p deletion, 11q deletion, trisomy 12, and 13q deletion.24 The presence of 17p deletion was associated with a higher growth rate of the LN population compared to 11q deletion, trisomy 12, and 13q deletion (P=0.01; Figure 5e). The three patients with 17p deletion had higher than median growth rates and active disease, while patients with trisomy 12 or 13q deletion had variable growth rates and some of these patients had stable disease. There was no correlation between growth rates and CD38 expression, age, or gender. The growth rate in the LN for patients with SLL was virtually the same as in patients with CLL.
Discussion
In this report, we identify that the major anatomic site of the proliferating cells in a CLL clone is the LN, and that the primary proliferative fraction of these cells expresses a CXCR4dim/CD5bright phenotype. This is consistent with gene expression profiling studies showing that the LN compared to PB and BM is the site for upregulation of gene signatures associated with B-cell receptor activation and proliferation of CLL cells.14 The higher growth rate of CLL cells in the LN compared to PB and BM measured by in vivo labeling of newborn cells with 2H is further supported by the finding that expression of the cell cycle marker Ki67 is highest in CLL cells in the LN. In fact, the average Ki67 positive fraction in LN was five times the fraction in the PB supporting the view that active clonal proliferation occurs primarily in the LN and this is the site from which newborn cells enter the PB. While we cannot estimate the recirculation capacities of CLL cells, the migration from the LN to PB appears to follow first order kinetics and is dependent on the rate of appearance of new cells (k) in the LN.
This is the first report using 2H in vivo labeling to identify the growth rate of CLL populations concurrently in all three major anatomic compartments. The highest rates of cell growth and proliferation were found in the LN compared to BM and PB. The presence of CLL cells with increased birth rates in the PB and BM is consistent with the finding of similar, but less pronounced expression of gene signatures associated with CLL cell activation and proliferation in the BM compared to the LN and suggests that although the BM is predominately inhabited by CLL cells in a resting state, a small fraction of recently born cells is also found here that may be made up of recent emigrants from LN or of cells having divided in situ. Due to the technical limitations of BM aspiration, representation of all BM resident cells in the aspirate cannot be ensured, and we cannot rule out that BM cells bound more tightly to the microenvironment and, therefore, not represented in the aspirate have a higher birth rate. However, our finding of low CLL cell turnover in the BM is in line with previous work by van Gent and colleagues.13 Of note, k in the PB of HW07, the only patient showing a higher k in PB than LN, was nearly identical on day 13 and day 28. While we cannot provide a definitive explanation for the larger fraction of newly born cells in the PB compared to LN, it is noteworthy that this was a patient with predominantly nodal presentation and an ALC of only 2,000 cells per μl in the blood. It is, possible that the enrichment of newly born cells in the PB may be due to different trafficking and/or homing properties of subpopulations in this patient. With disease progression and/or after having received multiple treatments, the cellular kinetics that we observed may be altered. This work, therefore, does not exclude the possibility that CLL cells may proliferate at a higher rate in the PB or BM in more aggressive disease or relapsed/refractory disease.
Other studies determined cell proliferation in CLL cells using 2H in vivo labeling. The majority of these studies analyzed bulk populations in the PB and estimated the CLL cell growth rate at 0.1 to 1% per day.10, 11, 13 Most recently, a proliferative fraction of CLL cells primarily among CXCR4dim/CD5bright cells was identified in PB.16 Our results confirm the presence of this proliferative population in the PB and extend the first observation by identifying the presence of phenotypically similar populations in the LN and BM that also have a higher growth rate than the other subpopulations in the same compartment (Figure 3). CXCR4 is dynamically regulated in response to antigen stimulation and CXCL12 ligation and thus identifies cells that have recently been exposed to CXCL12 within the tissue compartment or have recently been stimulated.14, 16, 25-27 Notably, the % newly born cells appearing in the CXCR4dimCD5bright populations in PB and LN was not statistically significantly different, albeit still somewhat higher in the LN than in the PB. The similar size of the fraction of newly born cells among the proliferative fraction in PB and LN is not unexpected, based on current thinking. In the prevailing model of CLL cell proliferation,16 the CXCR4brightCD5dim fraction of cells in a resting state. When these cells are activated, CD5 is upregulated and CXCR4 is internalized. These cells, now bearing a CXCR4dim/CD5bright phenotype are released from the LN and migrate into the PB. Eventually these cells become more quiescent leading to downregulation of CD5 and renewed surface expression of CXCR4 that could increase the likelihood of a return to the LN. In line with this view, it has previously been shown that CLL cells expressing activation-induced deaminase (AID) are almost exclusively found in the CXCR4dimCD5bright population; further suggesting these cells represent the proliferative subclone.28
Our observation that the CXCR4dim/CD5bright population contains the highest proportion of newly born cells in any compartment suggests that the interchange between the immunophenotypically defined subpopulations, for example from proliferative to resting/re-entry populations, is quite slow. On the other hand, the minimal difference in the proportion of newly born cells between the CXCR4intCD5int and the CXCR4brightCD5dim subpopulations, would be consistent with rapid equilibration of cells between these two populations. Alternatively, the CXCR4dimCD5bright phenotype could identify a subclonal population that is distinct in its life-cycle from other subpopulations and that could disproportionately contribute to clonal expansion and more fluidly traffic between compartments. Studies are ongoing to further dissect the transcriptomic and functional characteristics of the different subpopulations.
The more progressive disease course associated with the absence of somatic mutations in the IGHV gene, referred to as Ig-unmutated CLL, compared to Ig-mutated CLL is well established.1, 4 The exploratory analyses of CLL growth rates in patients stratified by IGHV gene mutations status measured higher CLL growth rates in the LNs of patients with Ig-unmutated CLL than in patients with Ig-mutated CLL (Figure 4c). Further, high growth rates in the LN were associated with active disease, faster lymphocyte doubling time, and shorter time to first treatment than in CLL with low growth rates. These data are consistent with the prior observation of increased BCR signaling in the LN of Ig-unmutated compared to Ig-mutated CLL and the shorter time to treatment in patients with higher tumor proliferation in the LN and the presence of more aggressive disease in the PB.10, 14 The 15 patients studied here overall reflect the heterogeneity of the CLL patient population quite well, including low to high risk Rai stage and representing both IGHV subtypes equally. At study entry, the time from diagnosis ranged from 1 to 11 years and 7 (47%) patients had active disease requiring treatment. While limited by a small sample size, our findings are statistically sound and consistent with previous gene profiling studies that utilized greater numbers of patients.
Our results identify the LN as the primary anatomical site of CLL cell proliferation from which newly born cells are released into other compartments. Our data advance the understanding of CLL cell trafficking between the LN and the periphery. Further, the subpopulation of CXCR4dim/CD5bright cells contains the proliferative fraction of CLL cells in all three anatomic compartments, identifying this population as a valuable target for novel treatment strategies.
Supplementary Material
Acknowledgments
The authors are grateful to all patients who participated in this study. We thank Ajunae Wells for assistance in the clinic, the NIH Surgery Branch, Judy Starling of the NIH Clinical Center Pharmacy, and Theresa Davies-Hill for preparing lymph node single cell suspensions. In addition, we thank Keyvan Keyvanfar for assistance with flow cytometry. This work was supported by the Intramural Research Program of the National, Heart, Lung and Blood Institute and the National Cancer Institute, the National Institutes of Health and an RO1 grant from the National Cancer Institute, NIH to N.C. C.U.N. was supported by the Danish Cancer Society.
Research support: This research was supported by the Intramural Research Program of the National, Heart, Lung and Blood Institute and the National Cancer Institute. C.U.N. received support from the Danish Cancer Society. Research support was also provided by an RO1 grant from the NIH National Cancer Institute (CA081554) to NC.
Footnotes
Disclosure of Conflicts of Interests:
C.E. and M.G. are employees of Kine Med Inc. C.U.N. reports consultancy for Roche, Janssen, Gilead, and receipt of travel grants from Roche, Gilead and Novartis The remaining authors declare no competing financial interests.
Authorship Contributions
Designed the study: T.M.H., S.-S.C., N.S., C.E., X.T., T.E.H., N.C., A.W.
Cared for patients: T.M.H., J.V., T.E.H., G.E.M., G.A., S.S., M.F., A.W.
Collected data: T.M.H., S.-S.C., N.S., J.V., C.E., S.S., D.C.A., M.S-S., C.M.Y., C.U.N., S.E.M.H., N.C., A.W.
Analyzed data: T.M.H., S.-S.C., N.S., C.E., M.G., X.T., D.C.A., M.S.-S., C.M.Y., C.U.N., N.C., A.W.
Wrote manuscript: T.M.H., S.-S.C., C.E., N.C., A.W.
References
- 1.Chiorazzi N, Rai KR, Ferrarini M. Chronic lymphocytic leukemia. N Engl J Med. 2005 Feb 24;352(8):804–815. doi: 10.1056/NEJMra041720. [DOI] [PubMed] [Google Scholar]
- 2.Santos FP, O'Brien S. Small lymphocytic lymphoma and chronic lymphocytic leukemia: are they the same disease? Cancer J. 2012 Sep-Oct;18(5):396–403. doi: 10.1097/PPO.0b013e31826cda2d. [DOI] [PubMed] [Google Scholar]
- 3.Zenz T, Mertens D, Kuppers R, Dohner H, Stilgenbauer S. From pathogenesis to treatment of chronic lymphocytic leukaemia. Nat Rev Cancer. 2010 Jan;10(1):37–50. doi: 10.1038/nrc2764. [DOI] [PubMed] [Google Scholar]
- 4.Sun C, Wiestner A. Prognosis and therapy of chronic lymphocytic leukemia and small lymphocytic lymphoma. Cancer Treat Res. 2015;165:147–175. doi: 10.1007/978-3-319-13150-4_6. [DOI] [PubMed] [Google Scholar]
- 5.Wierda WG, O'Brien S, Wang X, Faderl S, Ferrajoli A, Do KA, et al. Multivariable model for time to first treatment in patients with chronic lymphocytic leukemia. J Clin Oncol. 2011 Nov 1;29(31):4088–4095. doi: 10.1200/JCO.2010.33.9002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chiorazzi N. Implications of new prognostic markers in chronic lymphocytic leukemia. Hematology Am Soc Hematol Educ Program. 2012;2012:76–87. doi: 10.1182/asheducation-2012.1.76. [DOI] [PubMed] [Google Scholar]
- 7.Bulian P, Shanafelt TD, Fegan C, Zucchetto A, Cro L, Nuckel H, et al. CD49d Is the Strongest Flow Cytometry-Based Predictor of Overall Survival in Chronic Lymphocytic Leukemia. J Clin Oncol. 2014 Mar 20;32(9):897–904. doi: 10.1200/JCO.2013.50.8515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rossi D, Rasi S, Spina V, Bruscaggin A, Monti S, Ciardullo C, et al. Integrated mutational and cytogenetic analysis identifies new prognostic subgroups in chronic lymphocytic leukemia. Blood. 2013 Feb 21;121(8):1403–1412. doi: 10.1182/blood-2012-09-458265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Stilgenbauer S, Schnaiter A, Paschka P, Zenz T, Rossi M, Dohner K, et al. Gene mutations and treatment outcome in chronic lymphocytic leukemia: results from the CLL8 trial. Blood. 2014 May 22;123(21):3247–3254. doi: 10.1182/blood-2014-01-546150. [DOI] [PubMed] [Google Scholar]
- 10.Messmer BT, Messmer D, Allen SL, Kolitz JE, Kudalkar P, Cesar D, et al. In vivo measurements document the dynamic cellular kinetics of chronic lymphocytic leukemia B cells. J Clin Invest. 2005 Mar;115(3):755–764. doi: 10.1172/JCI23409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Defoiche J, Debacq C, Asquith B, Zhang Y, Burny A, Bron D, et al. Reduction of B cell turnover in chronic lymphocytic leukaemia. Br J Haematol. 2008 Oct;143(2):240–247. doi: 10.1111/j.1365-2141.2008.07348.x. [DOI] [PubMed] [Google Scholar]
- 12.Hayes GM, Busch R, Voogt J, Siah IM, Gee TA, Hellerstein MK, et al. Isolation of malignant B cells from patients with chronic lymphocytic leukemia (CLL) for analysis of cell proliferation: Validation of a simplified method suitable for multi-center clinical studies. Leuk Res. 2009 Oct 23; doi: 10.1016/j.leukres.2009.09.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.van Gent R, Kater AP, Otto SA, Jaspers A, Borghans JA, Vrisekoop N, et al. In vivo dynamics of stable chronic lymphocytic leukemia inversely correlate with somatic hypermutation levels and suggest no major leukemic turnover in bone marrow. Cancer Res. 2008 Dec 15;68(24):10137–10144. doi: 10.1158/0008-5472.CAN-08-2325. [DOI] [PubMed] [Google Scholar]
- 14.Herishanu Y, Perez-Galan P, Liu D, Biancotto A, Pittaluga S, Vire B, et al. The lymph node microenvironment promotes B-cell receptor signaling, NF-kappaB activation, and tumor proliferation in chronic lymphocytic leukemia. Blood. 2011 Jan 13;117(2):563–574. doi: 10.1182/blood-2010-05-284984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Klein U, Tu Y, Stolovitzky GA, Mattioli M, Cattoretti G, Husson H, et al. Gene Expression Profiling of B Cell Chronic Lymphocytic Leukemia Reveals a Homogeneous Phenotype Related to Memory B Cells. J Exp Med. 2001;194(11):1625–1638. doi: 10.1084/jem.194.11.1625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Calissano C, Damle RN, Marsilio S, Yan XJ, Yancopoulos S, Hayes G, et al. Intraclonal complexity in chronic lymphocytic leukemia: fractions enriched in recently born/divided and older/quiescent cells. Mol Med. 2011;17(11-12):1374–1382. doi: 10.2119/molmed.2011.00360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hallek M, Cheson BD, Catovsky D, Caligaris-Cappio F, Dighiero G, Dohner H, et al. Guidelines for the diagnosis and treatment of chronic lymphocytic leukemia: a report from the International Workshop on Chronic Lymphocytic Leukemia updating the National Cancer Institute-Working Group 1996 guidelines. Blood. 2008 Jun 15;111(12):5446–5456. doi: 10.1182/blood-2007-06-093906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Saba NS, Liu D, Herman SE, Underbayev C, Tian X, Behrend D, et al. Pathogenic role of B-cell receptor signaling and canonical NF-kappaB activation in mantle cell lymphoma. Blood. 2016 Jul 7;128(1):82–92. doi: 10.1182/blood-2015-11-681460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lis G, Wassenaar LI, Hendry MJ. High-precision laser spectroscopy D/H and 18O/16O measurements of microliter natural water samples. Anal Chem. 2008 Jan 1;80(1):287–293. doi: 10.1021/ac701716q. [DOI] [PubMed] [Google Scholar]
- 20.Busch R, Neese RA, Awada M, Hayes GM, Hellerstein MK. Measurement of cell proliferation by heavy water labeling. Nat Protoc. 2007;2(12):3045–3057. doi: 10.1038/nprot.2007.420. [DOI] [PubMed] [Google Scholar]
- 21.Voogt JN, Awada M, Murphy EJ, Hayes GM, Busch R, Hellerstein MK. Measurement of very low rates of cell proliferation by heavy water labeling of DNA and gas chromatography/pyrolysis/isotope ratio-mass spectrometric analysis. Nat Protoc. 2007;2(12):3058–3062. doi: 10.1038/nprot.2007.421. [DOI] [PubMed] [Google Scholar]
- 22.Calissano C, Damle RN, Hayes G, Murphy EJ, Hellerstein MK, Moreno C, et al. In vivo intraclonal and interclonal kinetic heterogeneity in B-cell chronic lymphocytic leukemia. Blood. 2009 Nov 26;114(23):4832–4842. doi: 10.1182/blood-2009-05-219634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sivina M, Hartmann E, Kipps TJ, Rassenti L, Krupnik D, Lerner S, et al. CCL3 (MIP-1alpha) plasma levels and the risk for disease progression in chronic lymphocytic leukemia. Blood. 2011 Feb 3;117(5):1662–1669. doi: 10.1182/blood-2010-09-307249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dohner H, Stilgenbauer S, Benner A, Leupolt E, Krober A, Bullinger L, et al. Genomic aberrations and survival in chronic lymphocytic leukemia. N Engl J Med. 2000;343(26):1910–1916. doi: 10.1056/NEJM200012283432602. [DOI] [PubMed] [Google Scholar]
- 25.Burger JA, Burger M, Kipps TJ. Chronic lymphocytic leukemia B cells express functional CXCR4 chemokine receptors that mediate spontaneous migration beneath bone marrow stromal cells. Blood. 1999 Dec 1;94(11):3658–3667. [PubMed] [Google Scholar]
- 26.Bennett F, Rawstron A, Plummer M, de Tute R, Moreton P, Jack A, et al. B-cell chronic lymphocytic leukaemia cells show specific changes in membrane protein expression during different stages of cell cycle. Br J Haematol. 2007 Nov;139(4):600–604. doi: 10.1111/j.1365-2141.2007.06790.x. [DOI] [PubMed] [Google Scholar]
- 27.Vlad A, Deglesne PA, Letestu R, Saint-Georges S, Chevallier N, Baran-Marszak F, et al. Down-regulation of CXCR4 and CD62L in chronic lymphocytic leukemia cells is triggered by B-cell receptor ligation and associated with progressive disease. Cancer Res. 2009 Aug 15;69(16):6387–6395. doi: 10.1158/0008-5472.CAN-08-4750. [DOI] [PubMed] [Google Scholar]
- 28.Patten PE, Chu CC, Albesiano E, Damle RN, Yan XJ, Kim D, et al. IGHV-unmutated and IGHV-mutated chronic lymphocytic leukemia cells produce activation-induced deaminase protein with a full range of biologic functions. Blood. 2012 Dec 6;120(24):4802–4811. doi: 10.1182/blood-2012-08-449744. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.