

Abstract
Objectives
Caffeine (CAF) and sedative/anesthetic drugs (SADs) are often coadministered to premature infants in the neonatal intensive care unit (NICU). While SAD neurotoxicity in the developing brain is well established, it is not fully clear whether CAF interacts with SADs and whether this interaction is detrimental. Using a mouse model of prematurity, we hypothesized that CAF would increase apoptotic neurotoxicity when coadministered with SADs.
Methods
Postnatal day 3 mice were treated with vehicle or 80 mg/kg CAF prior to challenge with 6 mg/kg midazolam, 40 mg/kg ketamine, or 40 μg/kg fentanyl. Six hours later, pups were sacrificed for activated caspase 3 (AC3) immunohistochemistry, and number of AC3 positive cells per mm3 throughout neocortex, hippocampus, caudate, thalamus, and colliculi was analyzed.
Results
CAF caused a statistically significant increase in AC3 positive cells when coadministered with midazolam (p = 0.002), ketamine (p = 0.014), or fentanyl (p <0.001). Our composite dataset suggests that the addition of CAF to these SADs has a supra-additive effect, causing more neurotoxicity than expected.
Conclusions
CAF may augment the neurotoxic action of SADs indicated for neonatal sedation/anesthesia in the NICU by triggering widespread apoptosis in the developing brains of premature infants.
Keywords: Caffeine, midazolam, ketamine, fentanyl, apoptosis, premature infant
Introduction
A compelling body of evidence positions sedative/anesthetic drugs (SADs) as apoptotic neurotoxins in the developing brain [1]. Yet, each year, millions of premature infants are administered SADs for procedural sedation and/or surgical anesthesia in the neonatal intensive care unit (NICU), and exposure to SADs is associated with neuropathology and neurodevelopmental abnormalities in this population [2,3]. However, premature infants are administered cocktails of drugs in the NICU, and it is unclear whether these drug interactions are detrimental to brain development. For example, respiratory dysfunction is ubiquitous in preterm infants and, if untreated, can be rapidly fatal. The most common respiratory disorder affecting premature infants is apnea of prematurity, characterized by bradycardia and cessation of breathing for 20 or more seconds [4].
Caffeine (CAF) is the first-line treatment for apnea of prematurity, and infants are administered CAF for days or weeks to improve respiratory drive [5]. In contrast to its therapeutic reputation, animal studies report that high doses of CAF may be neurotoxic to the developing brain [6]. Particularly troubling are reports that CAF may augment neurotoxicity if coadministered with drugs of abuse or SADs used in neonatal medicine. Black and colleagues [7] reported increased cell death in postnatal day (PND) 7 rat pups challenged with a moderate dose of CAF and morphine. In a series of studies, Yuede et al. [8] administered PND4 mouse pups CAF in conjunction with various drugs known to cause apoptotic neurotoxicity: ethanol, PCP, ketamine, diazepam, or isoflurane. Importantly, pairing CAF with each of these agents caused more neurotoxicity than CAF or the apoptogen alone.
To test the hypothesis that CAF + SAD coexposure may be neurotoxic, we administered PND3 mice CAF with or without a clinically relevant dose of midazolam, ketamine, or fentanyl and evaluated brains for evidence of apoptosis 6 h later. The PND3 mouse brain is comparable to that of an extremely premature infant of <28 weeks gestational age [9,10]. Infants of such early gestational age are at high risk of apnea of prematurity and may require days or weeks of CAF therapy to stimulate respiratory drive while being intermittently or chronically exposed to SADs for medical procedures. If CAF adversely interacts with SADs to increase neurotoxicity in the developing brain, then hundreds of thousands of premature neonates may be at risk for debilitating brain injury and long-term neurodevelopmental impairment.
Methods
Animals
Litters of PND3 ICR mouse pups were used for all experiments, and littermates were randomly assigned to receive either vehicle (Control), CAF alone (CAF), SAD alone (SAD), or CAF paired with a SAD (CAF + SAD). Independent sets of litters were used for each SAD tested, and day of birth was defined as PND0. Only healthy pups with a full belly of milk visible through the abdominal wall were included, and all groups had n ≥ 5 pups. Drugs were delivered intraperitoneally, and after initial injections, pups were maintained separate from the mother in a V1200 Mediheat Veterinarian Recovery Chamber (Harvard Apparatus, Holliston, MA) at 30 °C. Since midazolam, ketamine, and fentanyl are short-acting SADs, pups in the SAD and CAF + SAD groups received a booster dose of their respective SAD 3 h later to maintain levels of drug, while pups in the control and CAF groups were administered vehicle. All procedures were approved by the Washington University in St. Louis Animal Studies Committee.
Caffeine
To treat apnea of prematurity, CAF is administered as a bolus (20 mg/kg) supplemented by daily maintenance doses (5–10 mg/kg/day) until respiratory drive improves [5]. Since our aim was to study acute neurotoxicity, we used a single 80 mg/kg dose of CAF (Sigma-Aldrich, St. Louis, MO) to model the bolus phase of this regimen. Prior studies have established that this dose produces blood levels of 38 μg/ml in infant mice [8], which is well within the therapeutic range of 5 to 50 μg/ml for apnea of prematurity in preterm infants [11].
Experiment 1: CAF + midazolam
For experiment 1, we tested the apoptogenic action of CAF and midazolam. Midazolam is a GABA mimetic of the benzodiazepine class used for procedural sedation or as a presedative before general anesthesia. Littermates received vehicle or CAF with or without 6 mg/kg midazolam (Abbott Laboratories, Chicago, IL). Since the sedating dose of midazolam is far higher in mice (40 mg/kg) [12] than in human neonates (up to 0.3 mg/kg/h) [13], the 6 mg/kg dose translates into a 0.02 mg/kg bolus in premature infants and one that is likely to be subsedating.
Experiment 2: CAF + ketamine
Ketamine is a noncompetitive antagonist of glutamate NMDA receptors. A potent SAD with rapid onset, ketamine maintains airway reflexes while minimizing respiratory depression and is indicated for brief, painful procedures. Guidelines for induction and maintenance of ketamine anesthesia in neonates suggest slow infusion of up to 1.2 mg/kg/h [14]. However, the ED50 of ketamine to induce anesthesia in mice is 80 mg/kg [15]. In experiment 2, we injected littermates with vehicle or 40 mg/kg ketamine (Letco Medical, Decatur, AL) with or without CAF. The 40 mg/kg ketamine dose approximates a 0.6 mg/kg bolus in human neonates, which is within the range for anesthesia induction in the NICU.
Experiment 3: CAF + fentanyl
Fentanyl is a μ opioid receptor agonist and a first-line general anesthetic in neonatal and pediatric medicine [13]. Fentanyl anesthesia is accomplished by infusion of up to 4 μg/kg/h [12], which is 15 times lower than the dose indicated for anesthesia in mice (60 μg/kg) [16]. For experiment 3, the chosen dose of 40 μg/kg fentanyl (Hospira Inc., Lake Forest, IL) is equivalent to a 2.7 μg/kg bolus in human infants.
Histopathology and quantitative evaluation of neurotoxicity
Six hours after initial injections, animals were deeply anesthetized with sodium pentobarbital and transcardially perfused with 4% paraformaldehyde (PFA) with Tris buffer. Brains were harvested and postfixed in 4% PFA for 24 h at 4 °C, embedded in 3% agar, and serially sectioned in the sagittal plane at 75 μm on a vibratome. Every 8th section was immunolabeled for activated caspase 3 (AC3) as previously described [17]. Commitment to cell death occurs prior to cell surface expression of AC3, making AC3 a reliable marker of apoptosis.
Brain regions maximally affected by neuroapoptogens include subiculum and hippocampal formation, caudate putamen, retrosplenial cortex, thalamus, superior and inferior colliculi, and all major divisions of neocortex [8]. Serial sectioning yielded 3 to 4 sections per animal encompassing these regions, which were outlined as a single structure using Stereo Investigator software (MicroBrightField, Inc., Colchester, VT) on a Pentium III PC connected to a Prior Optiscan motorized stage (ES103 XYZ system, Prior Scientific Inc., Rockland, MA) with a Nikon Labophot-2 microscope. Thus, apoptotic profile counts are the mean density per mm3 across all regions combined. The population estimator function of Stereo Investigator was used to mark AC3-positive profiles with visible processes and those with pyknotic soma to ensure that no profile would be missed or counted twice. Estimated population of AC3-positive cells from each brain was divided by tissue volume to obtain density of AC3-positive profiles per mm3. All counts were performed by an observer blind to treatment.
Data were analyzed via one-way analysis of covariance (ANCOVA) with litter as a covariate to control for inter-litter variability in background levels of physiologic apoptosis. Positive results were followed by Fisher’s LSD post hoc to determine differences between treatments, and Cohen’s d was calculated as a measure of effect size. Data were analyzed using Prism 5.0 (GraphPad Software, Inc., San Diego, CA) and SPSS 22 (IBM, Armonk, NY) and are expressed as mean ± SEM. A priori significance threshold was set at α = 0.05. Thus, p values less than 0.05 were considered statistically significant.
Results
General observations
As expected, our doses of midazolam, ketamine, and fentanyl were subsedative/anesthetic doses. Animals were ambulatory and responded vigorously to tail and toe pinch throughout the duration of experiments. No pups displayed signs of respiratory impairment or skin discoloration, suggesting that animals were not hypoxic.
Caffeine + midazolam
When 80 mg/kg CAF was administered alone or in combination with a 6 mg/kg dose of midazolam, there was a significant effect of treatment, F(3, 19) = 8.10, p = 0.001. Post hoc analysis revealed that the CAF + midazolam group had a higher mean density of AC3-positive profiles per mm3 than either the CAF (p = 0.01, Cohen’s d = 2.72) or midazolam (p = 0.002, Cohen’s d = 2.56) groups (Figure 1(A)). These data suggest that the CAF + midazolam cocktail increases neurotoxicity in the immature mouse brain in substrates critical for normal brain function.
Figure 1. Apoptogenic action of CAF with NICU SADs.
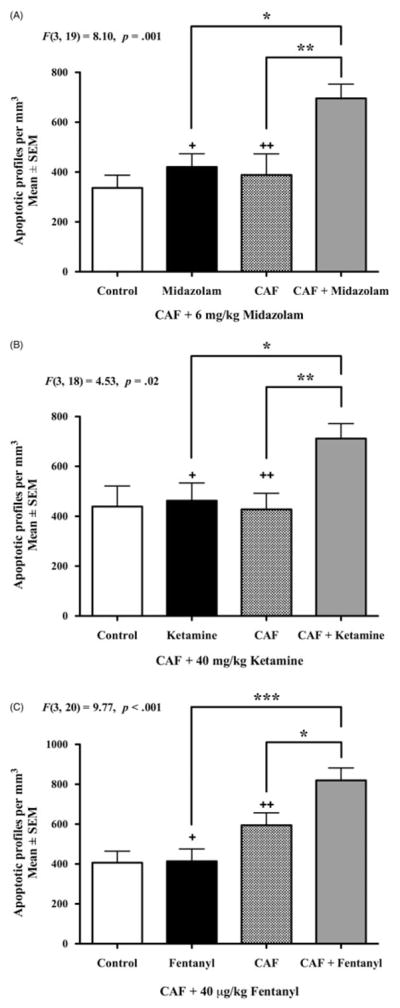
Six hours after drug administration, PND3 brains were assessed for apoptotic neurotoxicity via AC3 immunolabeling (n ≥5 per group). Cell counts for the control groups represent physiologic apoptosis, that is, the background rate of natural cell death in the PND3 brain. Panel A displays the mean density counts of apoptotic profiles after challenge with CAF + 6 mg/kg midazolam. A single exposure to midazolam or CAF caused no increase in apoptosis compared to controls. However, when CAF was combined with midazolam there was a statistically significant increase in apoptosis compared to either alone. Panel B demonstrates the neurotoxic reaction of CAF +40 mg/kg ketamine. The CAF + ketamine cocktail caused a significant increase in apoptotic profiles per mm3 compared to CAF alone or ketamine alone. The apoptotic neurotoxicity of CAF + 40 μg/kg fentanyl is illustrated in Panel C. CAF + fentanyl coexposure led to a statistically significant increase in mean number of apoptotic cells per mm3 versus the CAF only and fentanyl only groups. Panel A: +Cohen’s d = 2.56, ++Cohen’s d = 2.72 compared to CAF + midazolam. Panel B: +Cohen’s d = 1.45, ++Cohen’s d = 1.75 compared to CAF + ketamine. Panel C: +Cohen’s d = 2.47, ++Cohen’s d = 1.23 compared to CAF +Fentanyl *p <0.05, **p <0.01, ***p <0.001.
Caffeine + ketamine
Similar to the CAF + midazolam study, we found a significant effect of treatment in the CAF + ketamine study, F(3, 18) = 4.53, p = 0.02 (Figure 1(B)). In the brains of animals challenged with 80 mg/kg CAF +40 mg/kg ketamine, we observed a statistically significant increase in AC3-positive profiles per mm3 compared to CAF alone (p = 0.004, Cohen’s d = 1.75) or ketamine alone (p = 0.014, Cohen’s d = 1.45). Thus, doses of CAF + ketamine consistent with neonatal medical protocols caused a widespread and robust neurotoxic reaction.
Caffeine + fentanyl
Statistical analysis also revealed an overall significant effect of treatment in the CAF + fentanyl experiment F(3, 20) = 9.77, p <0.001. When groups were compared via post hoc analysis, 80 mg/kg CAF +40 μg/kg fentanyl co-exposure group exhibited more AC3-positive profiles per mm3 than their CAF (p = 0.018, Cohen’s d = 1.23) or fentanyl (p <0.001, Cohen’s d = 2.47) treated cohorts (Figure 1(C)). Results from this experiment suggest that clinically relevant doses of CAF + fentanyl may be neurotoxic to the developing mammalian brain.
Overview of CAF + SAD neurotoxicity
Figure 2(A) provides an overview of the apoptotic actions of CAF + SADs. We generated a composite dataset by combining density counts from all three experiments into four groups: Control, SAD, CAF, and CAF + SAD. There was a significant effect of treatment driven by the combination of CAF with our primary SADs, F(3, 67) = 19.20, p <0.001. CAF + SAD coexposure produced a massive increase in apoptosis compared to both CAF alone (p <0.001, Cohen’s d = 1.66) and SAD alone (p <0.001, Cohen’s d = 2.10). In Figure 2(B), the composite dataset has been adjusted by subtracting out control density counts that represent physiologic apoptosis, the natural rate of cell death expected in the PND3 mouse brain. The remaining values reflect cell death attributable solely to drug exposure, that is, the amount of apoptosis caused by each treatment. Exposure to SAD added 38 AC3-positive profiles per mm3, while exposure to CAF added 59 AC3-positive profiles per mm3. The dashed line represents the number of apoptotic cells expected by simply adding together SAD apoptosis and CAF apoptosis (38 + 59 = 97). The mean number of AC3-positive profiles in the CAF + SAD group was much greater than predicted, resulting in an additional 245 apoptotic cells per mm3.
Figure 2.

Overview of the apoptogenic action of CAF. To provide a general overview of the pro-apoptotic action CAF when combined with SADs, we generated a composite dataset (Panel A) of the density counts for all animals exposed to SADs (midazolam, ketamine, or fentanyl), CAF, or CAF + SADs at PND3. In this composite dataset, the addition of CAF to the SADs tested caused significantly more apoptotic neurotoxicity than either CAF or SAD alone. Panel B displays the composite dataset from Panel A adjusted to reflect cell death attributable to drug exposure. By subtracting out the control value representing the natural background rate of physiologic apoptosis, the remaining values reflect the amount of apoptotic cell death attributable to drug exposure. Exposure to SADs added 38 AC3-positive profiles per mm3, while exposure to CAF added 59 AC3-positive profiles per mm3. The dashed line is the number of apoptotic cells that would be expected by simply adding together SAD apoptosis and CAF apoptosis (38 + 59 = 97). However, the mean number of AC3-positive profiles in the CAF + SAD group was much greater than predicted, resulting in an additional 245 apoptotic cells per mm3. Our data suggest that CAF may have supra-additive properties when combined with SADs. Panel A: +Cohen’s d= 2.10, ++Cohen’s d = 1.66 compared to CAF + SADs ***p <0.001.
Figure 3 is representative images of neural substrates critical to learning and memory and information processing: hippocampus (Figure 3(A)), thalamus (Figure 3(B)), and retrosplenial cortex (Figure 3(C)). Qualitative evaluation suggests that CAF + SAD coexposure may accelerate apoptosis since many cells have morphological features consistent with an advanced stage of degeneration. Six hours after drug challenge, AC3-positive cortical neurons typically are intact, with well-defined soma and dendritic arborization. However, in CAF + SAD brains, many cerebrocortical neurons were pyknotic and embedded in a field of cellular debris. Many others exhibited extensive dendritic fragmentation and discontinuous, beaded axons characteristic of late stage apoptosis (8–10-h postchallenge) [18].
Figure 3.

Representative images of CAF + SAD neurotoxicity in the PND3 mouse brain. The addition of CAF to SADs caused apoptotic neurotoxicity in every region examined. The above panels are representative images of select neural substrates key to learn and memory and information processing: hippocampus, thalamus, and retrosplenial cortex. Each CAF + SAD pairing produced a unique pattern of cell death in those regions, and generally, many cells were in an advanced state of degeneration (white arrows) characteristic of late-stage apoptosis. Note the diverse morphology of AC3-positive cells, suggesting that a specific cell type is not preferentially vulnerable to CAF + SAD neurotoxicity. Panel A illustrates apoptotic neurotoxicity in the hippocampus after CAF + SAD challenge. Numerous apoptotic profiles are visible and scattered throughout the CA2 and CA3 hippocampal fields. Panel B focuses on anterior thalamus and neighboring dorsal and ventral thalamic nuclei. In control animals, few apoptotic cells were observed in these regions. The CAF + SAD cocktail substantially increased the density of AC3-positive profiles, many of which possess a medium-sized stellate morphology. Panel C represents detailed views of neuroapoptosis in retrosplenial cortex. These images reveal that CAF combined with midazolam, ketamine, or fentanyl caused a robust apoptotic reaction that involved small pyramidal neurons in superficial cortical layers and larger projection neurons in deeper cortical layers.
Discussion
In a series of studies, we report that CAF has pro-apoptotic properties when coadministered with SADs frequently employed in neonatal medicine. Using a model of prematurity, we challenged PND3 mouse pups with CAF + midazolam, CAF + ketamine, or CAF + fentanyl and assessed neurotoxicity via AC3 immunolabeling. When paired with these agents, CAF significantly increased apoptosis compared to CAF and each SAD alone. By subtracting out physiologic (control) apoptosis, our composite dataset (Figure 2(B)) provides a clear overview of cell death attributed to SADs, CAF, and CAF + SADs. A reasonable expectation was that CAF + SAD neurotoxicity would be additive (CAF apoptosis + SAD apoptosis = CAF + SAD apoptosis). Yet, our data suggest otherwise; CAF may have a supra-additive, pro-apoptotic effect when combined with SADs.
An important design consideration was to model a NICU scenario in which a premature infant is exposed to a bolus of CAF for apnea of prematurity in conjunction with an SAD for procedural sedation or general anesthesia. Appropriately, we scaled all doses to account for metabolic differences between rodents and humans. Thus, the doses of CAF, midazolam, ketamine, and fentanyl are consistent with standard medical protocols and considered safe and effective for infants in the NICU [13,14]. In practice, premature infants are often treated with CAF and SADs on a chronic or repetitive basis [5]. While it is unknown how such a regimen affects brain development, our data suggest that acute exposure to a CAF + SAD cocktail triggers neurotoxicity in substrates critical for normal behavior and cognition, and it is plausible that prolonged durations delete a much larger number of cells that could lead to debilitating neurological outcomes.
Another unknown is the mechanism(s) by which CAF exacerbates neurotoxicity when combined with SADs. CAF is a potent adenosine receptor antagonist, and the ability of CAF to stimulate respiration and reduce spells of apnea is mediated by adenosine A2A receptor subtype antagonism. A2A is excitatory and expressed densely on GABAergic neurons that, upon adenosine binding, exert inhibitory control over glutamate neurons in medulla [19]. By antagonizing A2A receptors, CAF disinhibits glutamate neurons, resulting in a net increase in excitatory neurotransmission in medulla and enhanced respiratory drive.
Cell death is a well-characterized outcome of runaway glutamate signaling, but our results argue against excitotoxicity as the underlying culprit of CAF-enhanced neurotoxicity. The reaction we observed after CAF + SAD challenge culminates with expression of the executioner caspase AC3, thus fundamentally the reaction is apoptotic programed cell death. This toxicity occurred rapidly, was found in all major brain regions examined, and had no preference for cell type based on the morphological features of AC3 positive cells. Intriguingly, the magnitude of neurotoxicity was similar regardless of whether CAF was paired with a GABA agonist (midazolam), NMDA antagonist (ketamine), or μ opioid agonist (fentanyl).
Dysregulation of intracellular Ca2+ homeostasis is a plausible mechanism for the relatively indiscriminate neurotoxicity that we observed in various brain regions and cell types. Intracellular Ca2+ overload damages mitochondrial membranes, increases expression of pro-apoptotic proteins, and, ultimately, initiates the apoptotic cascade [20]. Ethanol, which possesses both GABA agonist and NMDA antagonist properties, is a potent enhancer of Ca2+ [21] and causes a particularly devastating apoptotic reaction in fetal and neonatal brain. Importantly, the addition of CAF to ethanol-exposed neurons potentiates both intracellular Ca2+ [21] and neuroapoptosis [8,21]. Furthermore, ketamine [22] and propofol [23] raise Ca2+ in immature neurons with a concomitant decrease in cell viability. Similar to ethanol, CAF and SADs may have a deleterious, additive effect by overloading cells with Ca2+, thereby rapidly initiating neuroapoptosis.
Ca2+ is also an important second messenger regulating molecular effectors (e.g., BDNF) and signaling pathways (e.g., Akt) critical for neuronal survival and function. Notably, exposure of immature brain cells to CAF or SADs reduces mRNA and protein levels of BDNF [6,24–26], and altered BDNF expression by CAF or SADs has been implicated in neuroapoptosis [27] and long-term neurodevelopmental impairment [25,28]. In addition, CAF [29] and SADs [27,28] have been shown to downregulate activity of the pro-survival Akt pathway. Thus, CAF + SADs likely cause overlapping disturbances in intracellular Ca2+ and molecular survival signals, and this detrimental interaction tags a failed cell for immediate apoptotic deletion.
Conclusions
Over the last two decades, CAF use in the NICU has surged due to its low cost and lack of adverse side effects [30]. CAF is the first-line treatment for apnea of prematurity and considered safe enough that researchers and clinicians alike question the need to monitor blood levels of CAF in premature infants [11]. Despite its therapeutic reputation, CAF may interact unfavorably with SADs to trigger a particularly aggressive apoptotic response in the infant brain. There are now several clinical studies documenting that exposure of premature infants to SADs is associated with increased risk of neuropathology and neurodevelopmental abnormalities [2,3]. A compelling question is whether CAF co-administration with SADs partially contributes to these adverse outcomes, and retrospective clinical studies may provide a preliminary answer to this question. Perhaps the most immediate clinical action is to use the lowest doses of CAF necessary to stimulate respiratory drive, while diligently monitoring and recording blood levels of CAF in NICU patients.
Acknowledgments
Funding
This work was supported by: K.K.N. NIH grants MH083046, HD052664, HD052664S, and the Intellectual and Developmental Disabilities Research Center at Washington University in St. Louis (NIH/NICHD U54-HD087011).
Footnotes
Disclosure statement
The authors report no conflicts of interest. The authors alone are responsible for the content and writing of this article.
ORCID
Omar Hoseá Cabrera, http://orcid.org/0000-0002-9501-6720
George Townsend Taylor, http://orcid.org/0000-0002-0102-3787
Kevin Kiyoshi Noguchi, http://orcid.org/0000-0001-5307-161X
References
- 1.Jevtovic-Todorovic V, Absalom AR, Blomgren K, et al. Anaesthetic neurotoxicity and neuroplasticity: an expert group report and statement based on the BJA Salzburg Seminar. Br J Anaesth. 2013;111:143–51. doi: 10.1093/bja/aet177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Filan PM, Hunt RW, Anderson PJ, et al. Neurologic outcomes in very preterm infants undergoing surgery. J Pediatr. 2012;160:409–14. doi: 10.1016/j.jpeds.2011.09.009. [DOI] [PubMed] [Google Scholar]
- 3.Morriss FH, Jr, Saha S, Bell EF, et al. Eunice Kennedy Shriver National Institute of Child H, Human Development Neonatal Research N. Surgery and neurodevelopmental outcome of very low-birth-weight infants. JAMA Pediatr. 2014;168:746–54. doi: 10.1001/jamapediatrics.2014.307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Henderson-Smart DJ, Steer P. Methylxanthine treatment for apnea in preterm infants. Cochrane Database Syst Rev. 2001:CD000140. doi: 10.1002/14651858.CD000140. [DOI] [PubMed] [Google Scholar]
- 5.Schmidt B, Roberts RS, Davis P, et al. Caffeine for Apnea of Prematurity Trial G. Caffeine therapy for apnea of prematurity. N Engl J Med. 2006;354:2112–21. doi: 10.1056/NEJMoa054065. [DOI] [PubMed] [Google Scholar]
- 6.Kang SH, Lee YA, Won SJ, et al. Caffeine-induced neuronal death in neonatal rat brain and cortical cell cultures. Neuroreport. 2002;13:1945–50. doi: 10.1097/00001756-200210280-00023. [DOI] [PubMed] [Google Scholar]
- 7.Black AM, Pandya S, Clark D, et al. Effect of caffeine and morphine on the developing pre-mature brain. Brain Res. 2008;1219:136–42. doi: 10.1016/j.brainres.2008.04.066. [DOI] [PubMed] [Google Scholar]
- 8.Yuede CM, Olney JW, Creeley CE. Developmental neurotoxicity of alcohol and anesthetic drugs is augmented by co-exposure to caffeine. Brain Sci. 2013;3:1128–52. doi: 10.3390/brainsci3031128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Blencowe H, Cousens S, Oestergaard MZ, et al. National, regional, and worldwide estimates of preterm birth rates in the year 2010 with time trends since 1990 for selected countries: a systematic analysis and implications. The Lancet. 2012;379:2162–72. doi: 10.1016/S0140-6736(12)60820-4. [DOI] [PubMed] [Google Scholar]
- 10.Workman AD, Charvet CJ, Clancy B, et al. Modeling transformations of neurodevelopmental sequences across mammalian species. J Neurosci. 2013;33:7368–83. doi: 10.1523/JNEUROSCI.5746-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Natarajan G, Botica ML, Thomas R, Aranda JV. Therapeutic drug monitoring for caffeine in preterm neonates: an unnecessary exercise? Pediatrics. 2007;119:936–40. doi: 10.1542/peds.2006-2986. [DOI] [PubMed] [Google Scholar]
- 12.Ben-Shlomo I, Rosenbaum A, Hadash O, Katz Y. Intravenous midazolam significantly enhances the lethal effect of thiopental but not that of ketamine in mice. Pharmacol Res. 2001;44:509–12. doi: 10.1006/phrs.2001.0900. [DOI] [PubMed] [Google Scholar]
- 13.Anand KJ, Barton BA, McIntosh N, et al. Analgesia and sedation in preterm neonates who require ventilatory support: results from the NOPAIN trial. Neonatal outcome and prolonged analgesia in neonates. Arch Pediatr Adolesc Med. 1999;153:331–8. doi: 10.1001/archpedi.153.4.331. [DOI] [PubMed] [Google Scholar]
- 14.Lexicomp. Pediatric & Neonatal Lexi-Drugs. Hudson (OH): Lexi-Comp, Inc; 2016. [Google Scholar]
- 15.Green CJ, Knight J, Precious S, Simpkin S. Ketamine alone and combined with diazepam or xylazine in laboratory animals: a 10 year experience. Lab Anim. 1981;15:163–70. doi: 10.1258/002367781780959107. [DOI] [PubMed] [Google Scholar]
- 16.Gargiulo S, Greco A, Gramanzini M, et al. Mice anesthesia, analgesia, and care, Part I: anesthetic considerations in preclinical research. ilar J. 2012;53:E55–69. doi: 10.1093/ilar.53.1.55. [DOI] [PubMed] [Google Scholar]
- 17.Cabrera O, Dougherty J, Singh S, et al. Lithium protects against glucocorticoid induced neural progenitor cell apoptosis in the developing cerebellum. Brain Res. 2014;1545:54–63. doi: 10.1016/j.brainres.2013.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Olney JW, Wozniak DF, Jevtovic-Todorovic V, et al. Drug-induced apoptotic neurodegeneration in the developing brain. Brain Pathol. 2002;12:488–98. doi: 10.1111/j.1750-3639.2002.tb00467.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zaidi SI, Jafri A, Martin RJ, Haxhiu MA. Adenosine A2A receptors are expressed by GABAergic neurons of medulla oblongata in developing rat. Brain Res. 2006;1071:42–53. doi: 10.1016/j.brainres.2005.11.077. [DOI] [PubMed] [Google Scholar]
- 20.Orrenius S, Gogvadze V, Zhivotovsky B. Calcium and mitochondria in the regulation of cell death. Biochem Biophys Res Commun. 2015;460:72–81. doi: 10.1016/j.bbrc.2015.01.137. [DOI] [PubMed] [Google Scholar]
- 21.Hirata H, Machado LS, Okuno CS, et al. Apoptotic effect of ethanol is potentiated by caffeine-induced calcium release in rat astrocytes. Neurosci Lett. 2006;393:136–40. doi: 10.1016/j.neulet.2005.09.066. [DOI] [PubMed] [Google Scholar]
- 22.Wang C, Liu F, Patterson TA, et al. Relationship between ketamine-induced developmental neurotoxicity and NMDA receptor-mediated calcium influx in neural stem cell-derived neurons. Neurotoxicology. 2016 doi: 10.1016/j.neuro.2016.04.015. [Epub ahead of print] [DOI] [PubMed]
- 23.Kahraman S, Zup SL, McCarthy MM, Fiskum G. GABAergic mechanism of propofol toxicity in immature neurons. J Neurosurg Anesthesiol. 2008;20:233–40. doi: 10.1097/ANA.0b013e31817ec34d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Massara LD, Osuru HP, Oklopcic A, et al. General anesthesia causes epigenetic histone modulation of c-Fos and brain-derived neurotrophic factor, target genes important for neuronal development in the immature rat hippocampus. Anesthesiology. 2016;24:1311–27. doi: 10.1097/ALN.0000000000001111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pesic V, Milanovic D, Popic J, et al. Neonatal propofol anesthesia modifies activity-dependent processes and induces transient hyperlocomotor response to d-amphetamine during adolescence in rats. Int J Dev Neurosci. 2015;47:266–77. doi: 10.1016/j.ijdevneu.2015.09.009. [DOI] [PubMed] [Google Scholar]
- 26.Mioranzza S, Nunes F, Marques DM, et al. Prenatal caffeine intake differently affects synaptic proteins during fetal brain development. Int J Dev Neurosci. 2014;36:45–52. doi: 10.1016/j.ijdevneu.2014.04.006. [DOI] [PubMed] [Google Scholar]
- 27.Pesic V, Milanovic D, Tanic N, et al. Potential mechanism of cell death in the developing rat brain induced by propofol anesthesia. Int J Dev Neurosci. 2009;27:279–87. doi: 10.1016/j.ijdevneu.2008.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Karen T, Schlager GW, Bendix I, et al. Effect of propofol in the immature rat brain on short- and long-term neurodevelopmental outcome. PLoS One. 2013;8:e64480. doi: 10.1371/journal.pone.0064480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Saiki S, Sasazawa Y, Imamichi Y, et al. Caffeine induces apoptosis by enhancement of autophagy via PI3K/Akt/mTOR/p70S6K inhibition. Autophagy. 2014;7:176–87. doi: 10.4161/auto.7.2.14074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Clark RH, Bloom BT, Spitzer AR, Gerstmann DR. Reported medication use in the neonatal intensive care unit: data from a large national data set. Pediatrics. 2006;117:1979–87. doi: 10.1542/peds.2005-1707. [DOI] [PubMed] [Google Scholar]