

Abstract
It is increasingly recognized that B cells have multiple functions that contribute to the pathogenesis of autoimmunity. Specific targeting of B cells might therefore be an appropriate therapeutic intervention. The tumor necrosis factor-like molecule BAFF (BLyS) is a key B cell survival factor and its receptors are expressed on most peripheral B cells. Several different BAFF antagonists are under development and in early clinical trials. We review here the rationale for BAFF blockade, and its predicted mechanism of action in autoimmune diseases.
Keywords: autoimmune diseases, B cells, BAFF (BLyS), co-stimulation, therapy
Introduction
B cells are important in the pathogenesis of autoimmune disease. They produce autoantibodies that mediate tissue injury, they function as antigen-presenting cells that present epitopes of self-antigen to autoreactive T cells, and they produce soluble mediators involved in the organization of lymphoid tissues and in the initiation and perpetuation of inflammatory processes [1]. In some autoimmune diseases, B cells migrate to inflamed sites, where they act as local effector cells [2,3]. Because autoreactive B cells have a role in both the inductive and effector arms of autoimmune disease, there is considerable interest in B cell depletion or modulation as a therapeutic strategy.
BAFF, APRIL and their receptors
The B cell survival factor BAFF (BLyS; TNFSF13b), a member of the TNF family, is expressed on the surface of monocytes, dendritic cells [4,5], neutrophils [6], stromal cells [7] and activated T cells [8], and in the serum as a biologically active homotrimer [9]. BAFF-deficient mice are profoundly deficient in B cells, whereas BAFF transgenic mice have increased B cell numbers and develop a lupus-like syndrome [10]. Thus, levels of BAFF must be tightly regulated to maintain B cell survival without triggering autoimmunity.
B cells express three different BAFF receptors (transmembrane activator and calcium modulator ligand interactor [TACI; TNFRSF13b], BCMA [B cell maturation antigen; TNFRSF17] and BAFF-R [BAFF receptor; TNFRSF13c]) at various times during their differentiation (Figs 1 and 2). BCMA is expressed on transitional type 1 (T1) cells [11] and on plasma cells [12,13], whereas TACI and BAFF-R are expressed on transitional type 2/3 and mature B cells [11]. BAFF-R is upregulated by B cell receptor (BCR) ligation on mature B cells [11] and is expressed on resting memory B cells [12]. BAFF-R mediates most BAFF-dependent functions in the naive B cell population [11], whereas BCMA is needed for the optimal generation of long-lived plasma cells [13]. TACI has mixed positive and negative B cell regulatory functions; TACI-deficient mice have decreased serum IgM and decreased IgM responses to T-independent antigens, yet they have increased B cell numbers and develop an autoimmune phenotype [14]. Engagement of TACI on B cells results in a decreased proliferative response to lipopolysaccharide or anti-CD40L stimulation and an increase in apoptosis [14], but the signaling pathways that mediate this effect have not yet been elucidated. In addition, TACI might act as a sink for BAFF and prevent its binding to BAFF-R.
Figure 1.
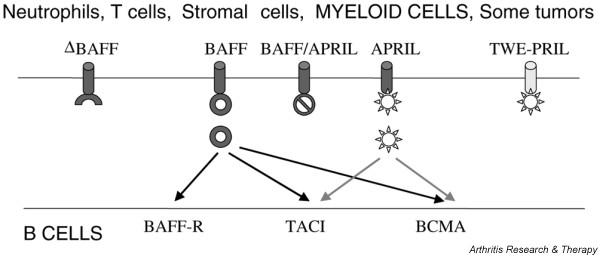
Interactions of BAFF and its homologs with the three BAFF receptors. Sites of action of potential blockers are described in Table 1. APRIL, a proliferation-inducing ligand; BAFF-R, BAFF receptor; BCMA, B cell maturation antigen; ΔBAFF, alternatively spliced form of BAFF that does not bind to BAFF receptors; TACI, transmembrane activator and calcium modulator ligand interactor; TWE-PRIL, a fusion protein of TWEAK (TNFSF12) and APRIL.
Figure 2.
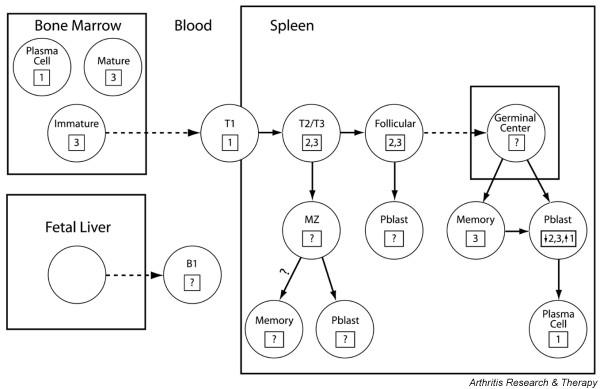
Stages of B cell development and expression of BAFF receptors. The BAFF receptor expressed is shown in the box (1, B cell maturation antigen [BCMA]; 2, transmembrane activator and calcium modulator ligand interactor [TACI]; 3, BAFF receptor [BAFF-R]). A broken line indicates stages of differentiation that can occur independently of BAFF. The necessity of BAFF for the survival of established memory cells or of long-lived plasma cells is not yet certain.
TACI and BCMA also bind APRIL (for 'a proliferation-inducing ligand'), a molecule homologous to BAFF, which is not necessary for normal B cell development [15] but induces B cell proliferation, class switching and survival [12,16]. To further complicate matters, APRIL and BAFF can form heterotrimers [17] and the extracellular domain of APRIL can form a hybrid molecule with the intracellular domain of TWEAK (TWE-PRIL; TNFSF12) as a result of alternative splicing [18]. The physiologic role of these mixed molecules remains to be defined. Finally, ΔBAFF is an alternatively spliced form of BAFF that does not bind to BAFF receptors. When ΔBAFF is co-expressed with BAFF, it acts in a dominant negative fashion both because heterotrimers of ΔBAFF/BAFF are not functional and because their formation results in intracellular retention of BAFF [19] (Fig. 1).
Function of BAFF and APRIL
BAFF prolongs B cell survival by regulating the expression of Bcl-2 gene family members [20]. BAFF might also enhance signaling through the BCR by upregulating expression of the BCR co-receptors CD21 and CD19 and by potentiating BCR-mediated phosphorylation of CD19 [21,22]. Recent studies suggest that the levels of BAFF can influence the selection and differentiation of autoreactive B cells in the periphery [23,24]; this should be a fruitful area for further investigation.
Germinal centers are shorter-lived in BAFF-deficient mice than in wild-type mice. Nevertheless, somatic mutation and class switching occur in these attenuated germinal centers and the antibody response to T-dependent antigens is of lower titer but not of lower affinity [25]. BAFF and APRIL prolong the survival but do not induce the proliferation of memory B cells in vitro [12]. However, our own data suggest that memory cells survive in vivo in the presence of BAFF blockade [26]. BAFF and APRIL enhance the survival of plasmablasts, but there are still conflicting data about whether they are required for the survival of long-lived plasma cells [12,13]. Finally, the B1 compartment seems to be independent of both BAFF and APRIL [11,27,28].
The role of BAFF and APRIL in T cell co-stimulation is controversial [26,29]. BAFF enhances T cell proliferation in vitro [8] but T cell numbers are normal in BAFF-deficient mice [27]. T cells are expanded twofold in number and seem activated in BAFF transgenic mice [10], but this might be secondary to B cell expansion rather than a direct effect on T cells. T cells from TACI-deficient mice hyperproliferate in response to anti-CD3 and BAFF, suggesting that TACI might in fact negatively regulate T cell function [14]. In contrast, APRIL seems to enhance T cell proliferation [30], perhaps through a different receptor.
BAFF antagonists for autoimmune diseases
BAFF and APRIL levels are increased in patients with systemic lupus erythematosus (SLE) [17] and correlate with titers of autoantibodies [31] and disease activity scores [32]. Both BAFF and APRIL are found in the synovial fluid of patients with rheumatoid arthritis, in whom they might prolong the survival of pathogenic B cells [33]. BAFF is also expressed in salivary glands of patients with Sjögren's syndrome [34] and in the central nervous system of mice with experimental autoimmune encephalomyelitis [35]. These findings, together with the observation that BAFF receptors are expressed on nearly all B cells [12,13], suggest that BAFF antagonism might be a useful therapeutic strategy for autoimmune diseases in which B cells have a pathogenic role [36]. Pre-clinical data in mice show that BAFF antagonists substantially delay the onset of disease in SLE-prone NZB/W mice [26,28,37] and prevent collagen-induced arthritis in DBA1 mice [29].
Two classes of human BAFF antagonist are being developed (Table 1). The first is a human antibody (anti-BLyS; LymphoStat-B) that binds soluble BAFF and prevents its interaction with all three receptors with equivalent potency [38]. It is currently in Phase 2 clinical trial for human SLE and rheumatoid arthritis. The second class of BAFF antagonist is a fusion protein of one of the BAFF receptors with immunoglobulin. Both TACI-Ig and BAFF-R-Ig are under development. These might have different properties, because TACI-Ig blocks the action of both BAFF and APRIL, whereas BAFF-R-Ig blocks only BAFF. LymphoStat-B might differ in its effects from BAFF-R-Ig because it does not block membrane-bound BAFF [38]. Human BCMA-Ig binds both BAFF and APRIL but it is sevenfold to eightfold less potent than BAFF-R-Ig and might therefore not be a useful therapeutic agent [39]. A BCMA-Ig mutant that blocks only APRIL has been generated and might be useful for further dissecting the function of APRIL in autoimmunity [40]. Finally, a 26 amino acid domain of BAFF-R that forms a β-turn structure has been found to bind BAFF with the same affinity as full-length BAFF-R. A six amino acid peptide derived from this domain is sufficient to block NF-κB2/p52 induction by BAFF in primary B cells. TACI and BCMA have homologous sequences in this region, so peptide antagonists might be derived for all three receptors [37].
Table 1.
List of current and potential BAFF antagonists
| Blocks BAFF | |||||
| Therapeutic agent | Soluble | Membrane | Blocks APRIL | Current status | Reference |
| LymphoStat-B (Human Genome Sciences) | + | - | - | Phase II clinical trialsa | [38] |
| BAFF-R-Ig (Biogen Idec) | + | + | - | In development | [39] |
| TACI-Ig (ZymoGenetics Serono) | + | + | + | Phase I clinical trials | [28] |
| BCMA-Ig | +b | +/- | + | - | [39] |
| Mutated BCMA-Ig | - | - | + | - | [40] |
| MiniBR3 (BAFF-R) | + | + | - | Pre-clinical | [37] |
| Peptidomimetics of: | |||||
| BAFF-R | + | + | - | Pre-clinical | [37] |
| TACI | + | + | + | Hypothetical | [37] |
| BCMA | + | + | + | Hypothetical | [37] |
aSystemic lupus erythematosus and rheumatoid arthritis. bLow affinity for BAFF. BAFF-R, BAFF receptor; BCMA, B cell maturation antigen; TACI, transmembrane activator and calcium modulator ligand interactor.
Mechanism of action of BAFF blockade
BAFF blockade mediates profound effects on B cells in mice. Within 1–2 weeks of administration of TACI-Ig or BAFF-R-Ig to normal mice, the spleen and lymph nodes decrease in size and the B cell frequency decreases by 50%. The decrease in T2, marginal-zone and follicular B cells is relatively greater than the decrease in T1 cells, but B1 cells are unaffected ([28] and M Ramanujam, unpublished data). Germinal center formation is impaired [25], as is the generation of memory responses [11]. However, established memory cells can survive a short period of BAFF blockade [26]. One study has reported a decrease in the frequency of antigen-specific bone marrow plasma cells after TACI-Ig treatment [13]. Recovery of B cells takes several months after the cessation of treatment.
We have found that in normal mice TACI-Ig induces a reversible decrease in the serum levels of IgM and IgG1 and impairs primary IgM immune responses to a T-dependent antigen, but BAFF-R-Ig has little effect on serum immunoglobulin levels. In SLE-prone NZB/W mice, which have a marked polyclonal increase in serum IgM, TACI-Ig, but not BAFF-R-Ig, normalizes serum IgM levels for months after a short treatment course. Nevertheless, both agents delay disease onset. BAFF blockade has little effect on total serum IgG in NZB/W mice. In the NZB/W SLE model, both fusion proteins have only modest effects on the emergence of IgG anti-double-stranded DNA antibodies but they induce B cell depletion and a marked delay in the expansion of activated and memory T cells. The addition of a short course of cytotoxic T lymphocyte-associated antigen 4 (CTLA4)Ig to TACI-Ig results in a decrease in the serum levels of IgG and of autoantibodies, perhaps because T cell-derived cytokines augment the effect of BAFF on plasma cell survival [12]. Despite some differences in the immunologic effects of TACI-Ig and BAFF-R-Ig, both reagents delay disease onset and death by 4–5 months when given before the emergence of nephritis in NZB/W F1 mice, and the combination of TACI-Ig and CTLA4Ig can even reverse established nephritis in this model ([26] and M Ramanujam and A Davidson, unpublished data). In the collagen-induced arthritis model, TACI-Ig given just before disease onset inhibits both anti-collagen antibodies and T cell proliferative and cytokine responses to collagen and markedly attenuates disease [29].
The effect of a 4-week course of anti-BLyS in primates is similar to that of BAFF-R-Ig, with a decrease in B cells in secondary lymphoid organs but no effect on serum Ig [38]. A Phase 1 study of anti-BLyS in patients with SLE showed that the agent has a half-life of 13–17 days, that it reduces the number of circulating B cells and that, in some patients, serum levels of anti-DNA antibodies decreased after treatment [41].
The extensive information available about BAFF and APRIL allows us to make several predictions about the effects of BAFF blockade in autoimmune disease. The first is that BAFF blockade will deplete B2 cells but not B1 cells. B2 cells are bone marrow-derived B cells that differentiate into either marginal-zone or follicular cells in a BAFF-dependent manner. B1 cells might constitute a separate self-renewing lineage that derives from the fetal liver, is located predominantly in the peritoneal cavity and does not seem to depend on BAFF for survival [42,43] (Fig. 2). Thus, autoimmune diseases in which B1 cells have a dominant role might be resistant to BAFF blockade. Second, diseases in which BCMA-expressing plasma cells produce pathogenic antibodies might be more sensitive to blockade with TACI-Ig than with selective BAFF blockers because APRIL can support the survival of these cells. TACI-Ig might also be more effective for diseases in which short-lived extrafollicular plasma cells produce IgM autoantibodies. In contrast, if autoantibody-producing plasmablasts are continuously newly formed from memory cells or naive cells that predominantly express BAFF-R, blockade of BAFF alone should be effective. Third, the immunoglobulin repertoire of newly emerging B cells may be altered by BAFF blockade because the survival of transitional B cells will be decreased and because of possible alterations in the strength of the BCR signal, but the repertoire of established memory cells is unlikely to be altered. Fourth, depletion of B cells by blockade of BAFF should have indirect effects on other cell types and on inflammatory mediators, some of which might improve disease activity independently of effects on autoantibody production. These include decreased antigen presentation to T cells, decreased epitope spreading, decreased cytokine secretion, decreased immune complex formation and decreased infiltration of target organs. Finally, BAFF blockade might synergize with agents that block T cell activation (Fig. 3). It is important to explore these hypotheses in autoimmune individuals and in rodent auto-immunity models because intrinsic B cell hyperreactivity, the provision of excessive T cell help and the presence of inflammatory mediators might alter the normal dependence of B cells on BAFF or APRIL and thus the response to blockade.
Figure 3.
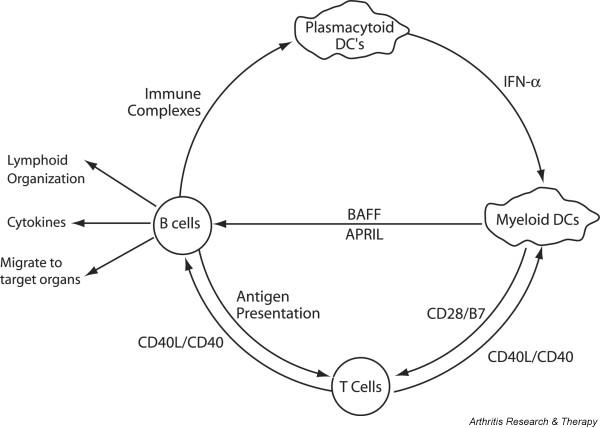
BAFF forms part of an amplification loop that is activated by inflammation. Immune complexes induce interferon-α (IFN-α) secretion from plasmacytoid dendritic cells (DCs) that in turn stimulates myeloid dendritic cells to further activate T and B cells. Activated B cells act as effectors that secrete cytokines and chemokines, present antigen to T cells and migrate to inflamed tissues. Antagonism of T cell activation by dendritic cells or of B cell activation by T cells might synergize with BAFF blockade. APRIL, a proliferation-inducing ligand.
Is BAFF blockade immunosuppressive?
An obvious concern about BAFF blockade is its immunosuppressive potential. TACI-Ig blocks the T-independent response of marginal-zone B cells to bacterial antigens [44] but does not block the response of B1 cells (M Ramanujam, unpublished data). The effect of BAFF-R-Ig on anti-bacterial responses has not yet been reported, but primary IgM responses to both T-dependent and T-independent antigens seem intact in BAFF-R-deficient mice [45]. The effect of BAFF blockade on anti-bacterial responses is clearly of concern for patients with SLE, who are prone to infections with encapsulated organisms. The absence of BAFF or BAFF-R results in diminished, but not absent, IgG responses to T-dependent antigens [25,45]. The physiologic impact of this decrease with regard to protective immune responses remains to be determined. Finally, the effect of BAFF blockade on T cell responses to infectious agents needs to be investigated further.
Conclusions
BAFF is a rational target for therapy of autoimmune diseases, and several different antagonists that may have different properties are being developed. B cell depletion is expected to have multiple direct and indirect effects on the autoimmune response. The potential ability of BAFF blockade to modulate memory B cells and plasmablasts might result in therapeutic benefit for diseases in which pathogenic autoantibodies derive from these cells. Analysis of the effects of BAFF blockade in vivo might yield important insights into the pathogenesis of autoimmune diseases. Much remains to be learned about the appropriate clinical use of this new class of drugs, including its safety profile and its ability to synergize with other conventional or biological immunosuppressive agents.
Competing interests
None declared.
Abbreviations
APRIL = a proliferation-inducing ligand, TNFSF13a; BAFF = B cell-activating factor of the tumor necrosis factor family, TNFSF13b; BAFF-R = BAFF receptor, also called BR-3, TNFRSF13c; BCMA = B cell maturation antigen, TNFRSF17; BCR = B cell receptor; CTLA4 = cytotoxic T lymphocyte-associated antigen 4; SLE = systemic lupus erythematosus; T1 = transitional type 1; TACI = transmembrane activator and calcium modulator ligand interactor, TNFRSF13b; TNF = tumor necrosis factor; TWEAK = TNFSF12.
Acknowledgments
Acknowledgements
This work was supported by grants from the SLE Foundation to MR, and from the NIH to AD (AI47291 and AI31229).
References
- Lipsky PE. Systemic lupus erythematosus: an autoimmune disease of B cell hyperactivity. Nat Immunol. 2001;2:764–766. doi: 10.1038/ni0901-764. [DOI] [PubMed] [Google Scholar]
- Cassese G, Lindenau S, de Boer B, Arce S, Hauser A, Riemekasten G, Berek C, Hiepe F, Krenn V, Radbruch A, et al. Inflamed kidneys of NZB/W mice are a major site for the homeostasis of plasma cells. Eur J Immunol. 2001;31:2726–2732. doi: 10.1002/1521-4141(200109)31:9<2726::AID-IMMU2726>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- Takemura S, Braun A, Crowson C, Kurtin PJ, Cofield RH, O'Fallon WM, Goronzy JJ, Weyand CM. Lymphoid neogenesis in rheumatoid synovitis. J Immunol. 2001;167:1072–1080. doi: 10.4049/jimmunol.167.2.1072. [DOI] [PubMed] [Google Scholar]
- Moore PA, Belvedere O, Orr A, Pieri K, LaFleur DW, Feng P, Soppet D, Charters M, Gentz R, Parmelee D, et al. BLyS: member of the tumor necrosis factor family and B lymphocyte stimulator. Science. 1999;285:260–263. doi: 10.1126/science.285.5425.260. [DOI] [PubMed] [Google Scholar]
- Nardelli B, Belvedere O, Roschke V, Moore PA, Olsen HS, Migone TS, Sosnovtseva S, Carrell JA, Feng P, Giri JG, et al. Synthesis and release of B-lymphocyte stimulator from myeloid cells. Blood. 2001;97:198–204. doi: 10.1182/blood.V97.1.198. [DOI] [PubMed] [Google Scholar]
- Scapini P, Nardelli B, Nadali G, Calzetti F, Pizzolo G, Montecucco C, Cassatella MA. G-CSF-stimulated neutrophils are a prominent source of functional BLyS. J Exp Med. 2003;197:297–302. doi: 10.1084/jem.20021343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorelik L, Gilbride K, Dobles M, Kalled SL, Zandman D, Scott ML. Normal B cell homeostasis requires B cell activation factor production by radiation-resistant cells. J Exp Med. 2003;198:937–945. doi: 10.1084/jem.20030789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huard B, Arlettaz L, Ambrose C, Kindler V, Mauri D, Roosnek E, Tschopp J, Schneider P, French LE. BAFF production by antigen-presenting cells provides T cell co-stimulation. Int Immunol. 2004;16:467–475. doi: 10.1093/intimm/dxh043. [DOI] [PubMed] [Google Scholar]
- Gross JA, Johnston J, Mudri S, Enselman R, Dillon SR, Madden K, Xu W, Parrish-Novak J, Foster D, Lofton-Day C, et al. TACI and BCMA are receptors for a TNF homologue implicated in B-cell autoimmune disease. Nature. 2000;404:995–999. doi: 10.1038/35010115. [DOI] [PubMed] [Google Scholar]
- Mackay F, Woodcock SA, Lawton P, Ambrose C, Baetscher M, Schneider P, Tschopp J, Browning JL. Mice transgenic for BAFF develop lymphocytic disorders along with autoimmune manifestations. J Exp Med. 1999;190:1697–1710. doi: 10.1084/jem.190.11.1697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cancro MP. Peripheral B-cell maturation: the intersection of selection and homeostasis. Immunol Rev. 2004;197:89–101. doi: 10.1111/j.0105-2896.2004.0099.x. [DOI] [PubMed] [Google Scholar]
- Avery DT, Kalled SL, Ellyard JI, Ambrose C, Bixler SA, Thien M, Brink R, Mackay F, Hodgkin PD, Tangye SG. BAFF selectively enhances the survival of plasmablasts generated from human memory B cells. J Clin Invest. 2003;112:286–297. doi: 10.1172/JCI200318025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Connor BP, Raman VS, Erickson LD, Cook WJ, Weaver LK, Ahonen C, Lin LL, Mantchev GT, Bram RJ, Noelle RJ. BCMA is essential for the survival of long-lived bone marrow plasma cells. J Exp Med. 2004;199:91–98. doi: 10.1084/jem.20031330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seshasayee D, Valdez P, Yan M, Dixit VM, Tumas D, Grewal IS. Loss of TACI causes fatal lymphoproliferation and autoimmunity, establishing TACI as an inhibitory BLyS receptor. Immunity. 2003;18:279–288. doi: 10.1016/S1074-7613(03)00025-6. [DOI] [PubMed] [Google Scholar]
- Varfolomeev E, Kischkel F, Martin F, Seshasayee D, Wang H, Lawrence D, Olsson C, Tom L, Erickson S, French D, et al. APRIL-deficient mice have normal immune system development. Mol Cell Biol. 2004;24:997–1006. doi: 10.1128/MCB.24.3.997-1006.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Litinskiy MB, Nardelli B, Hilbert DM, He B, Schaffer A, Casali P, Cerutti A. DCs induce CD40-independent immunoglobulin class switching through BLyS and APRIL. Nat Immunol. 2002;3:822–829. doi: 10.1038/ni829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roschke V, Sosnovtseva S, Ward CD, Hong JS, Smith R, Albert V, Stohl W, Baker KP, Ullrich S, Nardelli B, et al. BLyS and APRIL form biologically active heterotrimers that are expressed in patients with systemic immune-based rheumatic diseases. J Immunol. 2002;169:4314–4321. doi: 10.4049/jimmunol.169.8.4314. [DOI] [PubMed] [Google Scholar]
- Kolfschoten GM, Pradet-Balade B, Hahne M, Medema JP. TWEPRIL; a fusion protein of TWEAK and APRIL. Biochem Pharmacol. 2003;66:1427–1432. doi: 10.1016/S0006-2952(03)00493-3. [DOI] [PubMed] [Google Scholar]
- Gavin AL, Ait-Azzouzene D, Ware CF, Nemazee D. DeltaBAFF, an alternate splice isoform that regulates receptor binding and biopresentation of the B cell survival cytokine, BAFF. J Biol Chem. 2003;278:38220–38228. doi: 10.1074/jbc.M306852200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Do RK, Hatada E, Lee H, Tourigny MR, Hilbert D, Chen-Kiang S. Attenuation of apoptosis underlies B lymphocyte stimulator enhancement of humoral immune response. J Exp Med. 2000;192:953–964. doi: 10.1084/jem.192.7.953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorelik L, Cutler AH, Thill G, Miklasz SD, Shea DE, Ambrose C, Bixler SA, Su L, Scott ML, Kalled SL. Cutting edge: BAFF regulates CD21/35 and CD23 expression independent of its B cell survival function. J Immunol. 2004;172:762–766. doi: 10.4049/jimmunol.172.2.762. [DOI] [PubMed] [Google Scholar]
- Hase H, Kanno Y, Kojima M, Hasegawa K, Sakurai D, Kojima H, Tsuchiya N, Tokunaga K, Masawa N, Azuma M, et al. BAFF/BLyS can potentiate B-cell selection with the B-cell coreceptor complex. Blood. 2004;103:2257–2265. doi: 10.1182/blood-2003-08-2694. [DOI] [PubMed] [Google Scholar]
- Lesley R, Xu Y, Kalled SL, Hess DM, Schwab SR, Shu HB, Cyster JG. Reduced competitiveness of autoantigen-engaged B cells due to increased dependence on BAFF. Immunity. 2004;20:441–453. doi: 10.1016/S1074-7613(04)00079-2. [DOI] [PubMed] [Google Scholar]
- Thien M, Phan TG, Gardam S, Amesbury M, Basten A, Mackay F, Brink R. Excess BAFF rescues self-reactive B cells from peripheral deletion and allows them to enter forbidden follicular and marginal zone niches. Immunity. 2004;20:785–798. doi: 10.1016/j.immuni.2004.05.010. [DOI] [PubMed] [Google Scholar]
- Rahman ZS, Rao SP, Kalled SL, Manser T. Normal induction but attenuated progression of germinal center responses in BAFF and BAFF-R signaling-deficient mice. J Exp Med. 2003;198:1157–1169. doi: 10.1084/jem.20030495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramanujam M, Wang X, Huang W, Schiffer L, Grimaldi C, Akkerman A, Diamond B, Madaio M, Davidson A. Mechanism of action of TACI-Ig in murine SLE. J Immunol. 2004. [DOI] [PubMed]
- Schiemann B, Gommerman JL, Vora K, Cachero TG, Shulga-Morskaya S, Dobles M, Frew E, Scott ML. An essential role for BAFF in the normal development of B cells through a BCMA-independent pathway. Science. 2001;293:2111–2114. doi: 10.1126/science.1061964. [DOI] [PubMed] [Google Scholar]
- Gross JA, Dillon SR, Mudri S, Johnston J, Littau A, Roque R, Rixon M, Schou O, Foley KP, Haugen H, et al. TACI-Ig neutralizes molecules critical for B cell development and autoimmune disease. impaired B cell maturation in mice lacking BLyS. Immunity. 2001;15:289–302. doi: 10.1016/S1074-7613(01)00183-2. [DOI] [PubMed] [Google Scholar]
- Wang H, Marsters SA, Baker T, Chan B, Lee WP, Fu L, Tumas D, Yan M, Dixit VM, Ashkenazi A, et al. TACI–ligand interactions are required for T cell activation and collagen-induced arthritis in mice. Nat Immunol. 2001;2:632–637. doi: 10.1038/89782. [DOI] [PubMed] [Google Scholar]
- Stein JV, Lopez-Fraga M, Elustondo FA, Carvalho-Pinto CE, Rodriguez D, Gomez-Caro R, De Jong J, Martinez AC, Medema JP, Hahne M. APRIL modulates B and T cell immunity. J Clin Invest. 2002;109:1587–1598. doi: 10.1172/JCI200215034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheema GS, Roschke V, Hilbert DM, Stohl W. Elevated serum B lymphocyte stimulator levels in patients with systemic immune-based rheumatic diseases. Arthritis Rheum. 2001;44:1313–1319. doi: 10.1002/1529-0131(200106)44:6<1313::AID-ART223>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- Petri M, Stohl W, Chatham W, McCune J, Butler T, Ryel J, Zhong J, Recta J, Freimuth W. BLyS plasma concentrations correlate with disease activity and levels of anti-dsDNA autoantibodies and immunoglobulins (IgG) in a SLE patient observational study [abstract] Arthritis Rheum. 2003;48:S655. [Google Scholar]
- Tan SM, Xu D, Roschke V, Perry JW, Arkfeld DG, Ehresmann GR, Migone TS, Hilbert DM, Stohl W. Local production of B lymphocyte stimulator protein and APRIL in arthritic joints of patients with inflammatory arthritis. Arthritis Rheum. 2003;48:982–992. doi: 10.1002/art.10860. [DOI] [PubMed] [Google Scholar]
- Groom J, Kalled SL, Cutler AH, Olson C, Woodcock SA, Schneider P, Tschopp J, Cachero TG, Batten M, Wheway J, et al. Association of BAFF/BLyS overexpression and altered B cell differentiation with Sjogren's syndrome. J Clin Invest. 2002;109:59–68. doi: 10.1172/JCI200214121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magliozzi R, Columba-Cabezas S, Serafini B, Aloisi F. Intracerebral expression of CXCL13 and BAFF is accompanied by formation of lymphoid follicle-like structures in the meninges of mice with relapsing experimental autoimmune encephalomyelitis. J Neuroimmunol. 2004;148:11–23. doi: 10.1016/j.jneuroim.2003.10.056. [DOI] [PubMed] [Google Scholar]
- Martin F, Chan AC. Pathogenic roles of B cells in human auto-immunity; insights from the clinic. Immunity. 2004;20:517–527. doi: 10.1016/S1074-7613(04)00112-8. [DOI] [PubMed] [Google Scholar]
- Kayagaki N, Yan M, Seshasayee D, Wang H, Lee W, French DM, Grewal IS, Cochran AG, Gordon NC, Yin J, et al. BAFF/BLyS receptor 3 binds the B cell survival factor BAFF ligand through a discrete surface loop and promotes processing of NF-κB2. Immunity. 2002;17:515–524. doi: 10.1016/S1074-7613(02)00425-9. [DOI] [PubMed] [Google Scholar]
- Baker KP, Edwards BM, Main SH, Choi GH, Wager RE, Halpern WG, Lappin PB, Riccobene T, Abramian D, Sekut L, et al. Generation and characterization of LymphoStat-B, a human monoclonal antibody that antagonizes the bioactivities of B lymphocyte stimulator. Arthritis Rheum. 2003;48:3253–3265. doi: 10.1002/art.11299. [DOI] [PubMed] [Google Scholar]
- Pelletier M, Thompson JS, Qian F, Bixler SA, Gong D, Cachero T, Gilbride K, Day E, Zafari M, Benjamin C, et al. Comparison of soluble decoy IgG fusion proteins of BAFF-R and BCMA as antagonists for BAFF. J Biol Chem. 2003;278:33127–33133. doi: 10.1074/jbc.M305754200. [DOI] [PubMed] [Google Scholar]
- Patel DR, Wallweber HJ, Yin J, Shriver SK, Marsters SA, Gordon NC, Starovasnik MA, Kelley RF. Engineering an APRIL-specific B-cell maturation antigen (BCMA) J Biol Chem. 2004. [DOI] [PubMed]
- Furie R, Stohl W, Ginzler E, Becker M, Mishra N, Chatham W, Merrill JT, Weinstein A, McCune WJ, Zhong J, et al. Pharmacokinetic and pharmacodynamic results of a Phase I single and double dose-escalation study of LymphoStat-B (human monoclonal antibody to BLyS) in SLE patients [abstract] Arthritis Rheum. 2003;48:S377. [Google Scholar]
- Martin F, Kearney JF. B-cell subsets and the mature preimmune repertoire. Marginal zone and B1 B cells as part of a 'natural immune memory'. Immunol Rev. 2000;175:70–79. [PubMed] [Google Scholar]
- Mackay F, Schneider P, Rennert P, Browning J. BAFF and APRIL: a tutorial on B cell survival. Annu Rev Immunol. 2003;21:231–264. doi: 10.1146/annurev.immunol.21.120601.141152. [DOI] [PubMed] [Google Scholar]
- Balazs M, Martin F, Zhou T, Kearney J. Blood dendritic cells interact with splenic marginal zone B cells to initiate T-independent immune responses. Immunity. 2002;17:341–352. doi: 10.1016/S1074-7613(02)00389-8. [DOI] [PubMed] [Google Scholar]
- Miller DJ, Hanson KD, Carman JA, Hayes CE. A single autosomal gene defect severely limits IgG but not IgM responses in B lymphocyte-deficient A/WySnJ mice. Eur J Immunol. 1992;22:373–379. doi: 10.1002/eji.1830220213. [DOI] [PubMed] [Google Scholar]