

Abstract
Extrahepatic cholangiocarcinoma (ECC), a malignant tumor of biliary origin, has a poor prognosis with limited treatment options. The KRAS oncogene is the most commonly mutated gene in ECC and one of the factors that predicts a poor prognosis and low survival rate. L1 cell adhesion molecule (L1CAM) is expressed in ECC cells and acts as an independent poor prognostic factor in predicting patient survival. In this study we investigate the functional significance of L1CAM in ECC cells with activating KRAS mutation. We selected an ECC cell line, EGI-1, with activating KRAS mutation, and then confirmed its expression of L1CAM by RT-PCR, western blot analysis, and flow cytometry. The suppression of L1CAM expression (using a specific lentivirus-delivered shRNA) significantly decreased the migratory and invasive properties of EGI-1 cells, without altering their proliferation or survival. Analyses of signaling effectors in L1CAM-depleted and control EGI-1 cells indicated that L1CAM suppression decreased the levels of both phosphorylated MKK4 and total MKK4, together with c-Jun N-terminal kinase (JNK) phosphorylation. Further, exposure to a JNK inhibitor (SP600125) decreased migration and invasion of EGI-1 cells. These results suggest that L1CAM promotes cellular migration and invasion via the induction of MKK4 expression, leading to JNK activation. Our study is the first to demonstrate a functional role for L1CAM in ECC carrying the activating KRAS mutation. Given that KRAS is the most commonly mutated oncogene in ECC, L1CAM may serve as an attractive therapeutic target for ECC cells with activating KRAS mutation.
Keywords: extrahepatic cholangiocarcinoma, invasion, KRAS mutation, L1CAM, migration
INTRODUCTION
Cholangiocarcinoma is a malignant tumor that originates from the bile duct epithelium (Roberts et al., 1997). Based on its anatomical location in the biliary tree, cholangiocarcinoma is conventionally classified by the World Health Organization as an intrahepatic (ICC) or extrahepatic cholangiocarcinoma (ECC) (Bosman et al., 2010; Patel, 2011). ICC and ECC are biologically distinct, and therefore manifest substantial differences in terms of incidence, mortality, and risk factors (Cardinale et al., 2010). Cholangiocarcinoma has a poor prognosis because it is notoriously difficult to diagnose due to its late clinical presentation, and is refractory to conventional chemotherapy and radiation therapy (Blechacz and Gores, 2008; Blechacz et al., 2011; Khan et al., 2012). Gemcitabine and cisplatin has become the standard regimen for patients with advanced or metastatic cholangiocarcinoma (Ramirez-Merino et al., 2013; Valle et al., 2010). However, response to the combination chemotherapy in cholangiocarcinoma patients is typically limited, and the 5-year survival remains low (Rizvi et al., 2014). Molecular targeting by agents inhibiting growth factor receptor or vescular endothelial growth factor have been effective in several types of solid tumors (Cunningham et al., 2004; Giusti et al., 2009; Jia and Cai, 2016; Slamon et al., 2001; Smith, 2006). Targeted therapies have also been attempted for cholangiocarcinoma, but to date the results have shown no obvious improvement in clinical outcomes (Bengala et al., 2010; Lee et al., 2012; Lubner et al., 2010; Philip et al., 2006). Thus, new effective therapeutic targets for cholangiocarcinoma are urgently needed.
The L1 cell adhesion molecule (L1CAM) is a 200–220 kDa transmembrane glycoprotein comprising six Ig-like domains, five fibronectin-type III repeats, a transmembrane domain, and a short cytoplasmic tail (Brummendorf and Rathjen, 1993). L1CAM was originally identified as a neural cell adhesion molecule that plays an essential role in the development of the nervous system (Grumet and Edelman, 1988). Subsequently, L1CAM has been found to be aberrantly expressed in a variety of malignant tumors, including ovarian cancer, melanoma, breast cancer, gastric cancer, colorectal cancer, non-small cell lung cancer, pancreatic cancer, neuroblastoma, and cholangiocarcinoma, and its expression correlates with a poor prognosis and metastasis (Altevogt et al., 2016; Chen et al., 2013; Jung et al., 2011; Li et al., 2009; Min et al., 2010; Samatov et al., 2016; Weidle et al., 2009). Studies on the cellular functions of L1CAM have demonstrated its promotion of cellular proliferation, migration, invasion, and chemoresistance (Kiefel et al., 2012; Raveh et al., 2009). Recently, monoclonal antibodies (mAb) against L1CAM were shown to inhibit the growth and dissemination of tumors in ovarian carcinoma or ICC xenograft mouse models (Arlt et al., 2006; Cho et al., 2016; Wolterink et al., 2010). This suggests that L1CAM could serve as a promising new anticancer drug target.
RAS is one of the most commonly mutated oncogenes in human cancer (Bos, 1989; De Luca et al., 2012). Mutations in codons 12, 13, 61, or 146 of one of the three RAS genes (HRAS, KRAS, and NRAS) are sufficient to convert these into oncogenes. Mutational profiling of ECC identified that KRAS was the most commonly mutated gene (Churi et al., 2014; Putra et al., 2015; Simbolo et al., 2014; Voss et al., 2013). RAS encodes a family of membrane-bound 21-kDa guanosine triphosphate (GTP)-binding proteins that function as switches in a wide variety of signaling pathways. Critically, these pathways regulate transcription, cell growth, proliferation, and migration (Downward, 1998; Shields et al., 2000; Vojtek and Der, 1998). Activated RAS cooperates with multiple downstream effectors, including three distinct mitogen-activated protein kinase (MAPK) cascades (ERK, JNK, and p38), and the phosphoinositide 3-kinase (PI3K)-AKT signaling pathway, all of which are important in establishing and maintaining the transformed state (Downward, 1998; Shields et al., 2000; Vojtek and Der, 1998).
We previously identified that L1CAM is expressed in ECC tumors and correlates with a poor prognosis and metastasis (Li et al., 2009). High L1CAM expression is an independent poor prognostic factor in predicting the overall survival of patients with ECC (Li et al., 2009). However, the functions of L1CAM in ECC cells have yet to be reported. Nor has the functional significance of L1CAM in ECC cells carrying the KRAS oncogene been elucidated. In this study, we modulated L1CAM expression in an ECC (EGI-1) cell line with activating KRAS mutation (Xu et al., 2010), and investigated its functional significance.
MATERIALS AND METHODS
Antibodies and reagents
Hexadimethrine bromide and puromycin dihydrochloride were purchased from Sigma Aldrich (USA). Antibodies raised against glyceraldehyde 3-phosphate dehydrogenase (GAPDH), β-actin, Ras, ERK p44/p42, phospho-ERK p44/p42, JNK, phospho-JNK, mitogen-activated protein kinase kinase 4 (MKK4), or phospho-MKK4 were purchased from Cell Signaling Technology (USA). The ERK (U0126) and JNK inhibitors (SP600125) were purchased from Sigma Aldrich and AbMole BioScience (USA), respectively.
Cell lines and cell culture
The human ECC cell lines, EGI-1 and TFK-1 (Saijyo et al., 1995), were purchased from the DSMZ (German Collection of Microorganisms and Cell Cultures, Human and Animal Cell Lines, Germany). EGI-1 cells were cultured in DMEM (Welgene, Korea) containing 10% fetal bovine serum (FBS, Atlas, USA) and MEM amino acids (both essential and non-essential, Gibco Life Technologies, USA). Lenti-X 293T cells (Clontech Laboratories, Inc., USA) were maintained in DMEM supplemented with 10% FBS; TFK-1 cells were cultured in RPMI-1640 media (Welgene), supplemented with 10% FBS. Cells were incubated at 37°C in a humidified atmosphere of 5% CO2.
Flow cytometry
For analysis of L1CAM expression at the cell surface, cells were incubated with the human anti-L1CAM mAb, Ab417 (Cho et al., 2016), for 1 h at 4°C. After washing twice with phosphate buffered saline (PBS), cells were incubated with an anti-human IgG-FITC conjugate (Sigma) for 1 h at 4°C. After washing, propidium iodide (PI) was added to cells (Sigma), with PI-negative cells subsequently analyzed for bound antibody using a FACSCalibur flow cytometer (Becton-Dickinson, USA). Data acquisition and analyses were with the CellQuest software (Becton-Dickinson).
Western blot analysis
Cells were lysed using lysis buffer 6 (R&D systems, USA) containing protease (Roche, Germany) and phosphatase inhibitor cocktails (Roche). Protein concentration was calculated using the Pierce BCA Protein Assay Kit (Thermo Scientific, USA). Protein samples were resolved by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and then transferred onto a nitrocellulose membrane (GE Healthcare, Germany). L1CAM expression was detected using an anti-L1CAM mAb, A10-A3 (Min et al., 2010; Wei et al., 2011), which recognizes the ectodomain of L1CAM. Primary antibodies to GAPDH, β-actin, Ras, ERK p44/p42, phospho-ERK p44/p42, JNK, phospho-JNK, MKK4 or phospho-MKK4, were diluted 1:5,000 to 1:10,000, with incubations of 1 h at room temperature, or overnight at 4°C.
Analysis of KRAS mutation
Genomic DNA from EGI-1 and TFK-1 cells was isolated using the AccuPrep genomic DNA extraction kit (Bioneer, Korea) according to the manufacturer’s instructions. Extracted DNA was then subject to polymerase chain reaction (PCR) amplification of exon 2 of the KRAS gene (containing codons 12 and 13) using the following PCR primers: KRAS-U (5′-TAA GGC CTG CTG AAA ATG AC-3′) and KRAS-D (5′-AAA CAA GAT TTA CCT CTA TTG TTG GA-3′) as described previously (Xu et al., 2010). Amplification followed an initial denaturation at 94°C for 2 min, with 35 cycles of denaturation at 94°C, annealing at 55°C, and extension at 72°C (each for 1 min), with a final 8 min extension. DNA sequencing was done by Macrogen, Inc. (Korea).
Activated Ras pull down assay
Ras activation was determined using a pull-down assay (Ras activation assay, Cell signaling technology) according to the manufacturer’s instructions. Cell lysates were prepared using lysis buffer (R&D systems) with 500 μg of cell lysate used in each assay. Samples were incubated at 4°C for 1 h with GST-Raf-RBD (Ras binding domain) beads, which specifically recognize the GTP-bound (active) form of Ras. After removing unbound proteins by centrifugation and washing, glutathione resin-bound GTPase was eluted in SDS buffer. Western blot analysis of the eluate was used to quantitate protein levels.
L1CAM knockdown by lentivirus-mediated shRNA
Knockdown of L1CAM in EGI-1 cells was achieved using lentivirus-mediated delivery of a short hairpin RNA (shRNA) specific for the mRNA of L1CAM, using the Lenti-X lentiviral expression system (Clontech). The L1CAM shRNA (5′-CCG GTC CGC AGG TAT GCA CGC GTG AAT TCT CGA CCT CGA GAC AAA TGG CAG TAT TCA TCC ACA ATT TT - 3′) was used to down-regulate L1CAM expression; a control shRNA (5′-CCG G GC CAA TGC CTA CAT CTA CGT TCT CGA GAA CGT AGA TGT AGG CAT TGC TTT TT - 3′) was also generated. Lentiviral and packaging vectors were then cotransfected into Lenti-X 293T cells with packaging reagents, according to the manufacturer’s instructions. After 48 h, virus was harvested from the supernatant and added to the EGI-1 cells plated in six-well plates, which were then incubated at 37°C for 18 h, with the inclusion of 8 μg/ml hexadimethrine bromide (Sigma). The culture medium was removed the next day, and replaced with fresh medium containing 1.2 μg/ml puromycin for selection. Puromycin resistant clones were isolated after a 10 days selection period.
Reverse transcriptase - polymerase chain reaction (RT-PCR)
Total RNA was extracted from EGI-1 cells using the RNeasy mini kit (Qiagen, USA). The cDNA template was synthesized using the GoScript reverse transcription (RT) system kit (Promega, USA), according to the manufacturer’s instructions. PCR was performed by mixing cDNA with the AccuPower PCR PreMix (Bioneer, Korea) and gene specific primers (Supplementary Fig. 1). PCR products were subsequently resolved by electrophoresis using a 1.2% agarose gel.
Cell proliferation and survival assays
Cell viability was assayed using a Vi-CELL counter (Beckman Coulter, USA). For the cell proliferation assay, cells (2 × 105) were seeded in 6-well, cell culture plates (SPL, Korea), in duplicate. For the survival assay, gemcitabine (Sigma) or cis-diamineplatinum( II) dichloride (cisplatin, Sigma) was added to the cells. Three days later, after washing with PBS (Welgene), cells were detached using 0.05% trypsin-EDTA (Gibco) for 10 min, with viable cells counted following appropriate dilution.
Wound healing assay
Monolayers of EGI-1 cells were cultured to a density of 90–100% and then scratched using a pipette tip. After washing cells, fresh medium supplemented with either 10% or 1% FBS was added. Cell migration was observed with an optical microscope (X 200) with images captured using a Nikon camera. The distance between the leading edges of the wound was calculated using the micrographs. For inhibitor studies, 10 μM of either the JNK (SP600125) or ERK inhibitor (U0126) were added prior to scratching the monolayers.
Cell invasion assay
Cells were cultured in media supplemented with 10% FBS and then starved in media supplemented with 1% FBS for 24 h. The basement membrane growth factor-reduced matrigel (BD Biosciences, 30 μg) in coating buffer (0.01M Tris pH 8.0, 0.7% NaCl) was added into the transwell insert (8.0 μm pore size, Corning) and incubated for 2 h at 37°C. The lower chambers were filled with media containing 5% FBS, with 105 cells added to each upper chamber. After 24 h, the inner surface of the insert was cleaned with a cotton swab, with migratory cells then fixed and stained with 0.1% crystal violet (Sigma) in 2% ethanol (Merck Millipore, Germany), with lysis in 30% acetic acid prior to reading the optical density (OD) at 590 nm on a microtiter plate reader (Versa max, Molecular Devices, USA). The invasion percentage was calculated from OD values obtained from triplicate experiments.
Statistics
Data significance was assessed using the two-tailed unpaired t-test. Data shown indicate the mean ± SD. Statistical significance is indicated by asterisks: *p < 0.05, **p < 0.01, and ***p < 0.001.
RESULTS
Analyses of L1CAM expression and KRAS mutation in ECC cells
We examined L1CAM expression by flow cytometry and western blot analysis in two ECC cell lines, EGI-1 and TFK-1, which were known to carry the KRAS oncogene and wild-type KRAS, respectively. TFK-1 cells were used as a control. EGI-1 cells expressed a much higher level of L1CAM vs. TFK-1, with their respective L1CAM expression levels unaffected by reduced serum concentration in the growth medium (Figs. 1A and 1B). DNA sequencing of PCR-amplified KRAS allowed us to confirm that EGI-1 cells carry the G12D mutation, vs. wild-type KRAS in TFK-1 cells (Fig. 1C). Accordingly, much higher levels of Ras-GTP (active RAS) and its downstream signaling effector, p-ERK, were identified in EGI-1 cells compared to TFK-1 cells (Fig. 1D). Additionally, levels of Ras-GTP and p-ERK in EGI-1 cells were unaffected by reduced serum concentration (1% FBS), whereas they decreased in TFK-1 cells, supporting the notion that RAS-ERK signaling is constitutively active in EGI-1 cells.
Fig. 1.
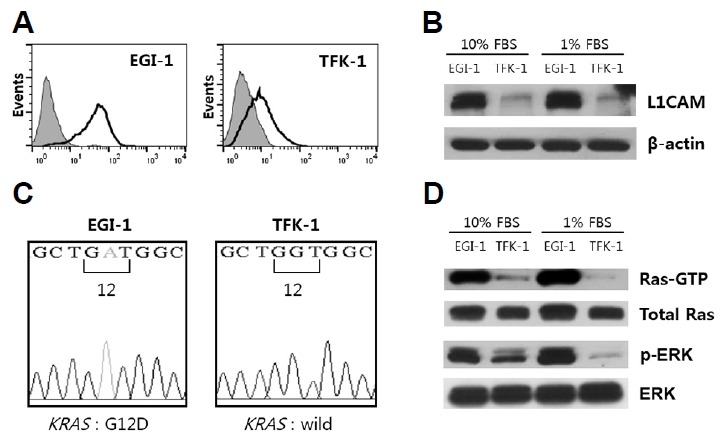
Analyses of L1CAM expression and KRAS mutation in EGI-1 and TFK-1 cells.
(A) Cell surface expression of L1CAM was confirmed by flow cytometry with an anti-L1CAM mAb (black line) vs. a negative control (grey). (B) Western blot analyses of L1CAM expression in cells cultured in medium supplemented with 10% FBS vs. 1% FBS; β-actin was used as a loading control. (C) Electropherogram showing the DNA sequence of exon 2 of the KRAS gene at codon 12. The EGI-1 cell line carries the G12D mutation while the TFK-1 cell line contains the wild type KRAS gene. (D) Western blot analyses of RAS and ERK activation in cells cultured in media supplemented with either 10% or 1% FBS. Results are representative of three independent experiments.
Functional analysis of L1CAM in EGI-1 cells
To investigate the functional significance of L1CAM in EGI-1 cells, L1CAM expression was down-regulated using lentiviral delivery of a short hairpin RNA (shRNA), with a control shRNA transduced as a negative control. Following puromycin selection, EGI-1 cells that were transduced with either shRNA construct were established. L1CAM shRNA effectively down-regulated both L1CAM mRNA levels (Fig. 2A) and total L1CAM protein levels (Fig. 2B) compared with control shRNA. Levels of cell surface L1CAM decreased in the L1CAM shRNA cells (Fig. 2C). In contrast, TFK-1 cells transduced with the L1CAM shRNA could not be established because they failed to grow, implying that L1CAM plays an essential role in the growth of TFK-1 cells.
Fig. 2.
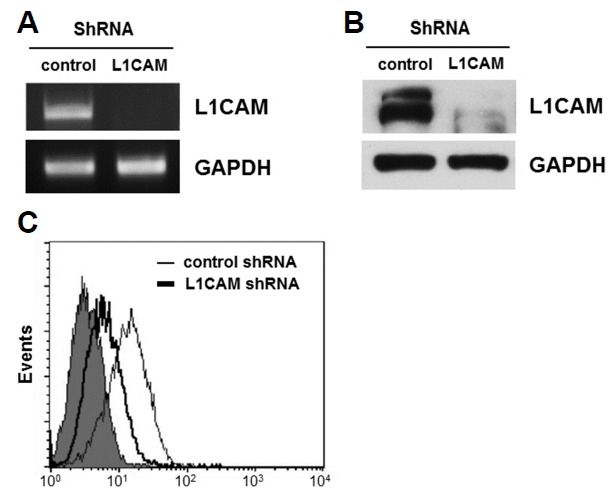
Establishment of L1CAM-depleted cells.
L1CAM shRNA, or a control shRNA, were transduced using lentiviral delivery into EGI-1 cells; a stable L1CAM-knockdown cell line was subsequently established. The expression level of L1CAM was determined by RT-PCR (A), Western blot analysis (B) and flow cytometry (C). Results are representative of three independent experiments.
To examine the effects of L1CAM knockdown in EGI-1 cells, the migration, invasion, proliferation, and survival of L1CAM-depleted cells were compared with those of controls. The L1CAM-depleted cells exhibited significantly reduced migration and invasion compared with control cells (Figs. 3A and 3B). This difference in migration was more pronounced in media supplemented with 1% FBS vs. 10% FBS (Supplementary Fig. 2). However, L1CAM knockdown in EGI-1 cells neither affected cell proliferation nor survival following exposure to chemotherapeutic drugs such as gemcitabine or cisplatin (Figs. 3C and 3D).
Fig. 3.
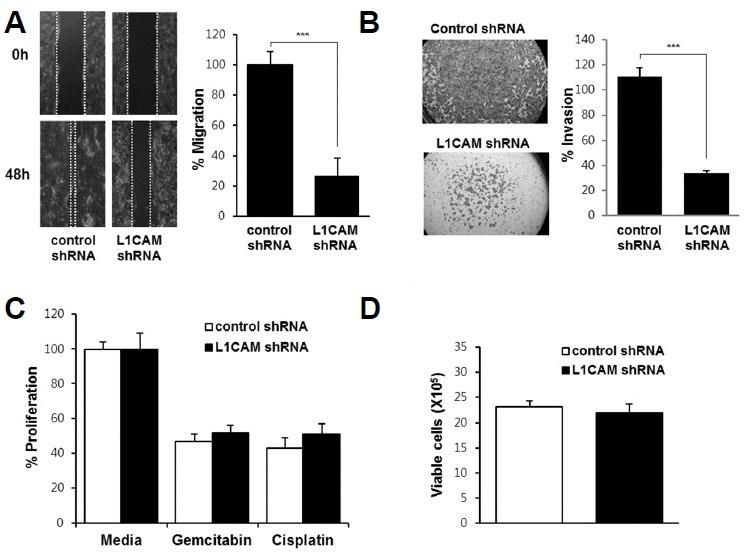
Effects of L1CAM knockdown on migration (A), invasion (B), survival (C), and proliferation (D) of EGI-1 cells.
(A) A wound healing assay was performed, with photographs taken immediately after scratching the monolayer (i.e. wound induction) and 48 h later (lower panel). Cell migration was quantified by measuring the distance between the leading edges on either side of the scratch site, plotted as a percentage relative to the zero time point (right panel). (B) A cell invasion assay was performed, with invasive cells quantified by measuring their absorbance at 590 nm. (C) L1CAM-depleted and control cells were treated with 0.5 μg/ml gemcitabin or cisplatin for 72 h and then subjected to the WST-1 cell proliferation assay. (D) L1CAM-depleted and control cells were cultured for 72 h and then cell numbers counted. Three independent experiments were performed in duplicate. Data are expressed as the mean ± SD (***p < 0.001).
L1CAM knockdown decreases JNK signaling in EGI-1 cells
To investigate whether L1CAM knockdown in EGI-1 cells influences Ras activation, or it’s downstream signaling pathway effectors (Raf–ERK, MEKK–JNK, or PI3K–AKT), we compared protein levels of Ras-GTP, P-ERK, P-JNK, and P-AKT between L1CAM-depleted and control cells. As shown in Fig. 4A, levels of Ras-GTP, P-ERK, and P-AKT were comparable for L1CAM-depleted and control cells, whereas the level of P-JNK was substantially lower in the L1CAM-depleted cells vs. controls.
Fig. 4.
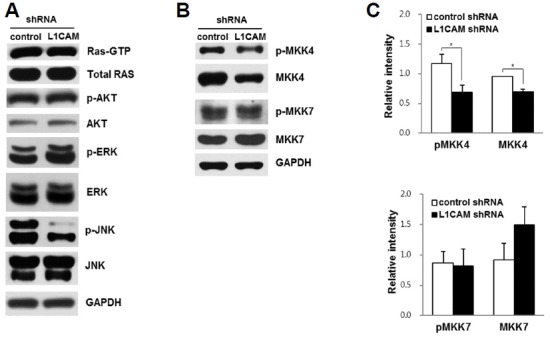
L1CAM knockdown in EGI-1 cells affects JNK activation.
(A) Western blot analyses of cell signaling effectors in L1CAM-depleted and control cells. (B, C) Levels of phospho- and total MKK4 and MKK7 were analyzed in L1CAM-depleted and control cells by Western blot analysis. Results are representative of three independent experiments. Data are expressed as the mean ± SD (* p < 0.05).
JNK is an evolutionary conserved MAPK and plays an important role in converting extracellular stimuli into a wide range of cellular responses (Weston and Davis, 2007). Specific stimuli trigger the activation of MAPKK kinases, which phosphorylate and activate the kinases, MKK4 and MKK7, which, in turn, phosphorylate (and activate) JNK (Davis, 2000; Weston and Davis, 2002). To understand the mechanism of JNK activation by L1CAM, levels of the MKK4 and MKK7 phospho-forms were compared in L1CAM-depleted vs. control cells. L1CAM knockdown decreased the levels of both phosphorylated MKK4 and total MKK4 protein, but not MKK7 phosphorylation (Figs. 4B and 4C).
JNK signaling is involved in the migration and invasion of EGI-1 cells
To examine whether JNK signaling contributes to the migration and invasion of EGI-1 cells, cells were exposed to a specific JNK inhibitor (SP600125) and then subjected to the cell proliferation, migration, and invasion assays. The JNK inhibitor significantly decreased cell migration and invasion, as well as JNK phosphorylation (Figs. 5A–5C), but not cell proliferation (Fig. 5D). These data indicated that the JNK inhibitor specifically decreases cell migration and invasion.
Fig. 5.
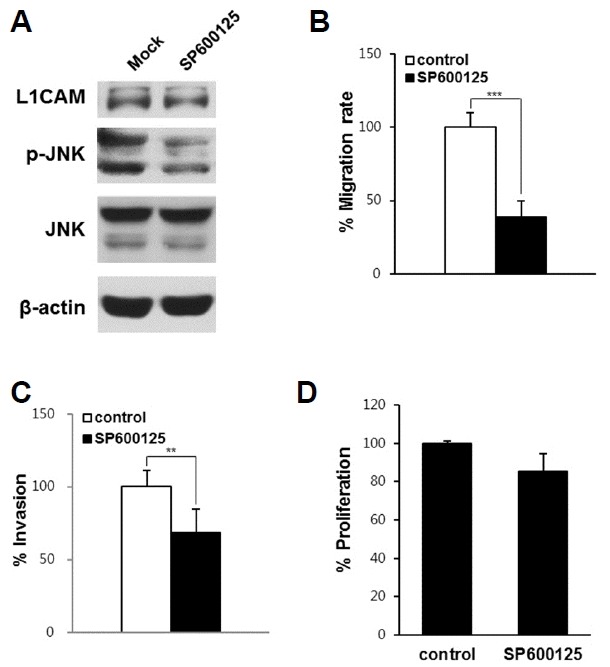
Effects of JNK inhibitor on migration, invasion, and the proliferation of EGI-1 cells.
EGI-1 cells were exposed to 10 μM SP600125 or PBS, and then subjected to western blot analysis (A), the wound healing assay (B), an invasion assay (C), and a cell proliferation assay (D). Three independent experiments were performed in duplicate. Data are expressed as the mean ± SD (**p < 0.01; *** p < 0.001).
DISCUSSION
L1CAM is aberrantly expressed in a variety of malignant tumors, where it has been shown to play crucial roles in promoting tumor progression. In a clinicopathologic study, we previously identified that L1CAM expression in ECC acts as an independent poor prognostic factor. However, the functional mechanisms of L1CAM in ECC cells have yet to be elucidated. Given that KRAS is the most commonly mutated gene in ECC, with this mutation correlating with a poor prognosis and low survival rate, we opted to assess the functional significance of L1CAM in ECC cells on a background of elevated RAS signaling. To achieve this, we analyzed the L1CAM-positive ECC cell line (EGI-1) containing the KRAS activating mutation. L1CAM knockdown in EGI-1 cells significantly decreased cell migration and invasion, but not cell proliferation or survival, indicating that L1CAM promotes the migration and invasion of EGI-1 cells. In contrast, the TFK-1 cells transduced with the L1CAM shRNA failed to grow, suggesting that L1CAM plays an essential role in the growth of TFK-1 cells. The differential effects of L1CAM knockdown in these two cell lines may represent that L1CAM signaling pathway in the TFK-1 cells is different from that in EGI-1 cells. In fact, we previously observed that L1CAM overexpression in gallbladder carcinoma SNU308 or ICC cells with wild type KRAS increased cell proliferation, migration and invasion through activation of FAK and AKT signaling (Jung et al., 2011; Min et al., 2010). To elucidate the functional role of L1CAM in the ECC cells with wild type KRAS, the effect of L1CAM overexpression in TFK-1 cells needs to be examined. Taken together, to our knowledge, our study is the first to show that L1CAM promotes the migration and invasion of ECC cells carrying KRAS oncogene.
JNK’s role in carcinogenesis is context-dependent. JNK can suppress tumorigenesis by negatively regulating the cell cycle and by inducing apoptosis (Dhanasekaran and Reddy, 2008; Kim et al., 2013; Taylor et al., 2013; Wagner and Nebreda, 2009). Conversely, JNK’s oncogenic activity is achieved through its promotion of inflammation, proliferation, invasion, and angiogenesis (Mingo-Sion et al., 2004; Vivanco et al., 2007; Wagner and Nebreda, 2009). Regarding the role of JNK in the tumor progression of cholangiocarcinoma, two recent studies showed that JNK promotes proliferation and invasion by cholangiocarcinoma cells via induction of the mTOR-regulated, glucose-regulated protein 78 (GRP78) (Feng et al., 2014), whereas a JNK inhibitor (SP600125) enhanced TGF-β1-induced apoptosis of RBE (ICC) cells via a SMAD-dependent caspase activation (Lin et al., 2013). However, the mechanism by which JNK is activated in cholangiocarcinoma cells had yet to be elucidated. In the present study, we found that L1CAM induced MKK4 and JNK activation, which, in turn, promotes the migration and invasion of EGI-1 cells. These data suggest that L1CAM promotes the migration and invasion of EGI-1 cells via the induction of JNK activation. To our knowledge, our study is the first to show the association between L1CAM and JNK signaling in cancer cells.
The prognostic relevance of L1CAM has been demonstrated in several different cancers, with L1CAM shown to be a promising target for anti-cancer therapy (Altevogt et al., 2016; Samatov et al., 2016). However, its functional role in cells with activating KRAS mutation has not yet been clearly shown. In this study, we found that L1CAM contributes to the migration and invasion of ECC cells with a KRAS activating mutation. Given that KRAS is the most commonly mutated oncogene in ECC cells, L1CAM may serve as an attractive therapeutic target for ECC carrying KRAS oncogene.
Supplementary data
ACKNOWLEDGMENTS
This study was supported by a grant (NRF-2015R1D1A1A01 057197) from the Basic Science Research Program, through the National Research Foundation of Korea, funded by the Ministry of Education, and by a grant (KDDF-201212-12) from the Korea Drug Development Fund (KDDF), funded by the Ministry of Science, ICT, and Future Planning, the Ministry of Trade, Industry & Energy, and the Ministry of Health & Welfare.
Footnotes
Note: Supplementary information is available on the Molecules and Cells website (www.molcells.org).
REFERENCES
- Altevogt P., Doberstein K., Fogel M. L1CAM in human cancer. Int J Cancer. 2016;138:1565–1576. doi: 10.1002/ijc.29658. [DOI] [PubMed] [Google Scholar]
- Arlt M.J., Novak-Hofer I., Gast D., Gschwend V., Moldenhauer G., Grunberg J., Honer M., Schubiger P.A., Altevogt P., Kruger A. Efficient inhibition of intra-peritoneal tumor growth and dissemination of human ovarian carcinoma cells in nude mice by anti-L1-cell adhesion molecule monoclonal antibody treatment. Cancer Res. 2006;66:936–943. doi: 10.1158/0008-5472.CAN-05-1818. [DOI] [PubMed] [Google Scholar]
- Bengala C., Bertolini F., Malavasi N., Boni C., Aitini E., Dealis C., Zironi S., Depenni R., Fontana A., Del Giovane C., et al. Sorafenib in patients with advanced biliary tract carcinoma: a phase II trial. Br J Cancer. 2010;102:68–72. doi: 10.1038/sj.bjc.6605458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blechacz B., Gores G.J. Cholangiocarcinoma: advances in pathogenesis, diagnosis, and treatment. Hepatology. 2008;48:308–321. doi: 10.1002/hep.22310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blechacz B., Komuta M., Roskams T., Gores G.J. Clinical diagnosis and staging of cholangiocarcinoma. Nat Rev Gastroenterol Hepatol. 2011;8:512–522. doi: 10.1038/nrgastro.2011.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bos J.L. ras oncogenes in human cancer: a review. Cancer Res. 1989;49:4682–4689. [PubMed] [Google Scholar]
- Bosman F.T., Carneiro F., Hruban R.H., Theise N.D. WHO classification of tumours of the digestive system. World Health Organization; 2010. [Google Scholar]
- Brummendorf T., Rathjen F.G. Axonal glycoproteins with immunoglobulin- and fibronectin type III-related domains in vertebrates: structural features, binding activities, and signal transduction. J Neurochem. 1993;61:1207–1219. doi: 10.1111/j.1471-4159.1993.tb13611.x. [DOI] [PubMed] [Google Scholar]
- Cardinale V., Semeraro R., Torrice A., Gatto M., Napoli C., Bragazzi M.C., Gentile R., Alvaro D. Intra-hepatic and extra-hepatic cholangiocarcinoma: New insight into epidemiology and risk factors. World J Gastrointestinal Oncol. 2010;2:407–416. doi: 10.4251/wjgo.v2.i11.407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen D.L., Zeng Z.L., Yang J., Ren C., Wang D.S., Wu W.J., Xu R.H. L1cam promotes tumor progression and metastasis and is an independent unfavorable prognostic factor in gastric cancer. J Hematol Oncol. 2013;6:43. doi: 10.1186/1756-8722-6-43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho S., Park I., Kim H., Jeong M.S., Lim M., Lee E.S., Kim J.H., Kim S., Hong H.J. Generation, characterization and preclinical studies of a human anti-L1CAM monoclonal antibody that cross-reacts with rodent L1CAM. MAbs. 2016;8:414–425. doi: 10.1080/19420862.2015.1125067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Churi C.R., Shroff R., Wang Y., Rashid A., Kang H.C., Weatherly J., Zuo M., Zinner R., Hong D., Meric-Bernstam F., et al. Mutation profiling in cholangiocarcinoma: prognostic and therapeutic implications. PloS One. 2014;9:e115383. doi: 10.1371/journal.pone.0115383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunningham D., Humblet Y., Siena S., Khayat D., Bleiberg H., Santoro A., Bets D., Mueser M., Harstrick A., Verslype C., et al. Cetuximab monotherapy and cetuximab plus irinotecan in irinotecan-refractory metastatic colorectal cancer. N Eng J Med. 2004;351:337–345. doi: 10.1056/NEJMoa033025. [DOI] [PubMed] [Google Scholar]
- Davis R.J. Signal transduction by the JNK group of MAP kinases. Cell. 2000;103:239–252. doi: 10.1016/s0092-8674(00)00116-1. [DOI] [PubMed] [Google Scholar]
- De Luca A., Maiello M.R., D’Alessio A., Pergameno M., Normanno N. The RAS/RAF/MEK/ERK and the PI3K/AKT signalling pathways: role in cancer pathogenesis and implications for therapeutic approaches. Exp Opin Therapeutic Targets. 2012;16(Suppl 2):S17–27. doi: 10.1517/14728222.2011.639361. [DOI] [PubMed] [Google Scholar]
- Dhanasekaran D.N., Reddy E.P. JNK signaling in apoptosis. Oncogene. 2008;27:6245–6251. doi: 10.1038/onc.2008.301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Downward J. Signal transduction. New exchange, new target Nature. 1998;396:416–417. doi: 10.1038/24743. [DOI] [PubMed] [Google Scholar]
- Feng C., He K., Zhang C., Su S., Li B., Li Y., Duan C.Y., Chen S., Chen R., Liu Y., et al. JNK contributes to the tumorigenic potential of human cholangiocarcinoma cells through the mTOR pathway regulated GRP78 induction. PLoS One. 2014;9:e90388. doi: 10.1371/journal.pone.0090388. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Giusti R.M., Cohen M.H., Keegan P., Pazdur R. FDA review of a panitumumab (Vectibix). clinical trial for first-line treatment of metastatic colorectal cancer. Oncologist. 2009;14:284–290. doi: 10.1634/theoncologist.2008-0254. [DOI] [PubMed] [Google Scholar]
- Grumet M., Edelman G.M. Neuron-glia cell adhesion molecule interacts with neurons and astroglia via different binding mechanisms. J Cell Biol. 1988;106:487–503. doi: 10.1083/jcb.106.2.487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia S., Cai J. Update on biomarkers in development of anti-angiogenic drugs in gastric cancer. Anticancer Res. 2016;36:1111–1118. [PubMed] [Google Scholar]
- Jung J., Son Y.S., Park H., Jeon S.K., Lee J.W., Choi S.Y., Kim J.M., Kwon Y.G., Hong H.J., Min J.K. The cell adhesion molecule L1 promotes gallbladder carcinoma progression in vitro and in vivo. Oncol Rep. 2011;25:945–952. doi: 10.3892/or.2011.1181. [DOI] [PubMed] [Google Scholar]
- Khan S.A., Davidson B.R., Goldin R.D., Heaton N., Karani J., Pereira S.P., Rosenberg W.M., Tait P., Taylor-Robinson S.D., Thillainayagam A.V., et al. Guidelines for the diagnosis and treatment of cholangiocarcinoma: an update. Gut. 2012;61:1657–1669. doi: 10.1136/gutjnl-2011-301748. [DOI] [PubMed] [Google Scholar]
- Kiefel H., Bondong S., Hazin J., Ridinger J., Schirmer U., Riedle S., Altevogt P. L1CAM: a major driver for tumor cell invasion and motility. Cell Adh Migr. 2012;6:374–384. doi: 10.4161/cam.20832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim A.D., Kang K.A., Kim H.S., Kim D.H., Choi Y.H., Lee S.J., Hyun J.W. A ginseng metabolite, compound K, induces autophagy and apoptosis via generation of reactive oxygen species and activation of JNK in human colon cancer cells. Cell Death Dis. 2013;4:e750. doi: 10.1038/cddis.2013.273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J., Park S.H., Chang H.M., Kim J.S., Choi H.J., Lee M.A., Jang J.S., Jeung H.C., Kang J.H., Lee H.W., et al. Gemcitabine and oxaliplatin with or without erlotinib in advanced biliary-tract cancer: a multicentre, open-label, randomised, phase 3 study. Lancet Oncol. 2012;13:181–188. doi: 10.1016/S1470-2045(11)70301-1. [DOI] [PubMed] [Google Scholar]
- Li S., Jo Y.S., Lee J.H., Min J.K., Lee E.S., Park T., Kim J.M., Hong H.J. L1 cell adhesion molecule is a novel independent poor prognostic factor of extrahepatic cholangiocarcinoma. Clin Cancer Res. 2009;15:7345–7351. doi: 10.1158/1078-0432.CCR-09-0959. [DOI] [PubMed] [Google Scholar]
- Lin Y., Zhang B., Liang H., Lu Y., Ai X., Chen X. JNK inhibitor SP600125 enhances TGF-β-induced apoptosis of RBE human cholangiocarcinoma cells in a Smad-dependent manner. Mol Med Rep. 2013;8:1623–1629. doi: 10.3892/mmr.2013.1711. [DOI] [PubMed] [Google Scholar]
- Lubner S.J., Mahoney M.R., Kolesar J.L., Loconte N.K., Kim G.P., Pitot H.C., Philip P.A., Picus J., Yong W.P., Horvath L., et al. Report of a multicenter phase II trial testing a combination of biweekly bevacizumab and daily erlotinib in patients with unresectable biliary cancer: a phase II Consortium study. J Clin Oncol. 2010;28:3491–3497. doi: 10.1200/JCO.2010.28.4075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Min J.K., Kim J.M., Li S., Lee J.W., Yoon H., Ryu C.J., Jeon S.H., Lee J.H., Kim J.Y., Yoon H.K., et al. L1 cell adhesion molecule is a novel therapeutic target in intrahepatic cholangiocarcinoma. Clin Cancer Res. 2010;16:3571–3580. doi: 10.1158/1078-0432.CCR-09-3075. [DOI] [PubMed] [Google Scholar]
- Mingo-Sion A.M., Marietta P.M., Koller E., Wolf D.M., Van Den Berg C.L. Inhibition of JNK reduces G2/M transit independent of p53, leading to endoreduplication, decreased proliferation, and apoptosis in breast cancer cells. Oncogene. 2004;23:596–604. doi: 10.1038/sj.onc.1207147. [DOI] [PubMed] [Google Scholar]
- Patel T. Cholangiocarcinoma--controversies and challenges. Nat Rev Gastroenterol Hepatol. 2011;8:189–200. doi: 10.1038/nrgastro.2011.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Philip P.A., Mahoney M.R., Allmer C., Thomas J., Pitot H.C., Kim G., Donehower R.C., Fitch T., Picus J., Erlichman C. Phase II study of erlotinib in patients with advanced biliary cancer. J Clin Oncol. 2006;24:3069–3074. doi: 10.1200/JCO.2005.05.3579. [DOI] [PubMed] [Google Scholar]
- Putra J., de Abreu F.B., Peterson J.D., Pipas J.M., Mody K., Amos C.I., Tsongalis G.J., Suriawinata A.A. Molecular profiling of intrahepatic and extrahepatic cholangiocarcinoma using next generation sequencing. Exp Mol Pathol. 2015;99:240–244. doi: 10.1016/j.yexmp.2015.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramirez-Merino N., Aix S.P., Cortes-Funes H. Chemotherapy for cholangiocarcinoma: An update. World J Gastrointestinal Oncol. 2013;5:171–176. doi: 10.4251/wjgo.v5.i7.171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raveh S., Gavert N., Ben-Ze’ev A. L1 cell adhesion molecule (L1CAM). in invasive tumors. Cancer Lett. 2009;282:137–145. doi: 10.1016/j.canlet.2008.12.021. [DOI] [PubMed] [Google Scholar]
- Rizvi S., Borad M.J., Patel T., Gores G.J. Cholangiocarcinoma: molecular pathways and therapeutic opportunities. Semin Liver Dis. 2014;34:456–464. doi: 10.1055/s-0034-1394144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts S.K., Ludwig J., Larusso N.F. The pathobiology of biliary epithelia. Gastroenterology. 1997;112:269–279. doi: 10.1016/s0016-5085(97)70244-0. [DOI] [PubMed] [Google Scholar]
- Saijyo S., Kudo T., Suzuki M., Katayose Y., Shinoda M., Muto T., Fukuhara K., Suzuki T., Matsuno S. Establishment of a new extrahepatic bile duct carcinoma cell line, TFK-1. Tohoku J Exp Med. 1995;177:61–71. doi: 10.1620/tjem.177.61. [DOI] [PubMed] [Google Scholar]
- Samatov T.R., Wicklein D., Tonevitsky A.G. L1CAM: Cell adhesion and more. Prog Histochem Cytochem. 2016;51:25–32. doi: 10.1016/j.proghi.2016.05.001. [DOI] [PubMed] [Google Scholar]
- Shields J.M., Pruitt K., McFall A., Shaub A., Der C.J. Understanding Ras: ‘it ain’t over ‘til it’s over’. Trends Cell Biol. 2000;10:147–154. doi: 10.1016/s0962-8924(00)01740-2. [DOI] [PubMed] [Google Scholar]
- Simbolo M., Fassan M., Ruzzenente A., Mafficini A., Wood L.D., Corbo V., Melisi D., Malleo G., Vicentini C., Malpeli G., et al. Multigene mutational profiling of cholangiocarcinomas identifies actionable molecular subgroups. Oncotarget. 2014;5:2839–2852. doi: 10.18632/oncotarget.1943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slamon D.J., Leyland-Jones B., Shak S., Fuchs H., Paton V., Bajamonde A., Fleming T., Eiermann W., Wolter J., Pegram M., et al. Use of chemotherapy plus a monoclonal antibody against HER2 for metastatic breast cancer that overexpresses HER2. N Engl J Med. 2001;344:783–792. doi: 10.1056/NEJM200103153441101. [DOI] [PubMed] [Google Scholar]
- Smith I.E. Trastuzumab for early breast cancer. Lancet. 2006;367:107. doi: 10.1016/S0140-6736(06)67951-8. [DOI] [PubMed] [Google Scholar]
- Taylor C.A., Zheng Q., Liu Z., Thompson J.E. Role of p38 and JNK MAPK signaling pathways and tumor suppressor p53 on induction of apoptosis in response to Ad-eIF5A1 in A549 lung cancer cells. Mol Cancer. 2013;12:35. doi: 10.1186/1476-4598-12-35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valle J., Wasan H., Palmer D.H., Cunningham D., Anthoney A., Maraveyas A., Madhusudan S., Iveson T., Hughes S., Pereira S.P., et al. Cisplatin plus gemcitabine versus gemcitabine for biliary tract cancer. N Eng J Med. 2010;362:1273–1281. doi: 10.1056/NEJMoa0908721. [DOI] [PubMed] [Google Scholar]
- Vivanco I., Palaskas N., Tran C., Finn S.P., Getz G., Kennedy N.J., Jiao J., Rose J., Xie W., Loda M., et al. Identification of the JNK signaling pathway as a functional target of the tumor suppressor PTEN. Cancer Cell. 2007;11:555–569. doi: 10.1016/j.ccr.2007.04.021. [DOI] [PubMed] [Google Scholar]
- Vojtek A.B., Der C.J. Increasing complexity of the Ras signaling pathway. J Biol Chem. 1998;273:19925–19928. doi: 10.1074/jbc.273.32.19925. [DOI] [PubMed] [Google Scholar]
- Voss J.S., Holtegaard L.M., Kerr S.E., Fritcher E.G., Roberts L.R., Gores G.J., Zhang J., Highsmith W.E., Halling K.C., Kipp B.R. Molecular profiling of cholangiocarcinoma shows potential for targeted therapy treatment decisions. Hum Pathol. 2013;44:1216–1222. doi: 10.1016/j.humpath.2012.11.006. [DOI] [PubMed] [Google Scholar]
- Wagner E.F., Nebreda A.R. Signal integration by JNK and p38 MAPK pathways in cancer development. Nat Rev Cancer. 2009;9:537–549. doi: 10.1038/nrc2694. [DOI] [PubMed] [Google Scholar]
- Wei C.H., Lee E.S., Jeon J.Y., Heo Y.S., Kim S.J., Jeon Y.H., Kim K.H., Hong H.J., Ryu S.E. Structural mechanism of the antigen recognition by the L1 cell adhesion molecule antibody A10-A3. FEBS Lett. 2011;585:153–158. doi: 10.1016/j.febslet.2010.11.028. [DOI] [PubMed] [Google Scholar]
- Weidle U.H., Eggle D., Klostermann S. L1-CAM as a target for treatment of cancer with monoclonal antibodies. Anticancer Res. 2009;29:4919–4931. [PubMed] [Google Scholar]
- Weston C.R., Davis R.J. The JNK signal transduction pathway. Curr Opin Genet Dev. 2002;12:14–21. doi: 10.1016/s0959-437x(01)00258-1. [DOI] [PubMed] [Google Scholar]
- Weston C.R., Davis R.J. The JNK signal transduction pathway. Curr Opin Cell Biol. 2007;19:142–149. doi: 10.1016/j.ceb.2007.02.001. [DOI] [PubMed] [Google Scholar]
- Wolterink S., Moldenhauer G., Fogel M., Kiefel H., Pfeifer M., Luttgau S., Gouveia R., Costa J., Endell J., Moebius U., et al. Therapeutic antibodies to human L1CAM: functional characterization and application in a mouse model for ovarian carcinoma. Cancer Res. 2010;70:2504–2515. doi: 10.1158/0008-5472.CAN-09-3730. [DOI] [PubMed] [Google Scholar]
- Xu L., Hausmann M., Dietmaier W., Kellermeier S., Pesch T., Stieber-Gunckel M., Lippert E., Klebl F., Rogler G. Expression of growth factor receptors and targeting of EGFR in cholangiocarcinoma cell lines. BMC Cancer. 2010;10:302. doi: 10.1186/1471-2407-10-302. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.