

Abstract
Despite the importance of the receptor activator of nuclear factor (NF)-kappaB ligand (RANKL)-RANK signaling mechanisms on osteoclast differentiation, little has been studied on how RANK expression is regulated or what regulates its expression during osteoclastogenesis. We show here that insulin signaling increases RANK expression, thus enhancing osteoclast differentiation by RANKL. Insulin stimulation induced RANK gene expression in time- and dose-dependent manners and insulin receptor shRNA completely abolished RANK expression induced by insulin in bone marrow-derived monocyte/macrophage cells (BMMs). Moreover, the addition of insulin in the presence of RANKL promoted RANK expression. The ability of insulin to regulate RANK expression depends on extracellular signal-regulated kinase 1/2 (ERK1/2) since only PD98059, an ERK1/2 inhibitor, specifically inhibited its expression by insulin. However, the RANK expression by RANKL was blocked by all three mitogen-activated protein (MAP) kinases inhibitors. The activation of RANK increased differentiation of BMMs into tartrate-resistant acid phosphatase-positive (TRAP+) osteoclasts as well as the expression of dendritic cell-specific transmembrane protein (DC-STAMP) and d2 isoform of vacuolar (H+) ATPase (v-ATPase) Vo domain (Atp6v0d2), genes critical for osteoclastic cell-cell fusion. Collectively, these results suggest that insulin induces RANK expression via ERK1/2, which contributes to the enhancement of osteoclast differentiation.
Keywords: ERK1/2, insulin, osteoclastogenesis, RANK
INTRODUCTION
Bone homeostasis is maintained by balanced activity between two bone-specific cell types, osteoblasts and osteoclasts (Harada and Rodan, 2003; Teitelbaum, 2000). Osteoclasts are derived from hematopoietic progenitors of the monocyte/macrophage lineage and two cytokines, RANKL and macrophage colony-stimulating factor (M-CSF) are essential for osteoclast differentiation (Hsu et al., 1999; Lacey et al., 1998; Suda et al., 1999; Yasuda et al., 1998). Imbalance caused by an elevation of osteoclast numbers or activity results in low bone mass such as is seen in various bone-related diseases including postmenopausal osteoporosis (Manolagas, 2000; Rodan and Martin, 2000).
Binding of RANKL to its receptor on osteoclast precursors, RANK, initiates intracellular signal transduction and activates distinct signaling cascades mediated by c-Jun N-terminal Kinase 1/2 (JNK1/2), p38 and ERK1/2 MAP kinases (Boyle et al., 2003), thus finally induces the expression or activation of osteoclast-specific transcription factors including c-Fos, tartrate-resistant acid phosphatase (TRAP), microphthalmia transcription factor (Mitf) and nuclear factor of activated T-cells cytoplasmic 1 (NFATc1) for osteoclast (Fleischmann et al., 2000; Kim et al., 2008; Lee, 2010; Luchin et al., 2001; Matsuo et al., 2000; Takayanagi et al., 2002). Mononuclear osteoclast precursors undergo cell-cell fusion for the maturation and bone-resorbing function of multinucleated osteoclasts by RANKL. DC-STAMP or Atp6v0d2 are key regulators for osteoclast maturation since deficiency of these genes leads to osteopetrosis in mice due to defects in the cell-cell fusion process and in bone resorption (Lee et al., 2006; Yagi et al., 2005).
Recently, several studies have demonstrated that insulin receptor signaling in osteoblasts and osteoclasts regulates bone metabolism (Ferron et al., 2010; Fulzele et al., 2010; Oh et al., 2015a; Yang et al., 2010). Mice lacking insulin receptors in osteoblasts developed postnatal osteopenia and an impairment of osteoblast proliferation and differentiation (Ferron et al., 2010; Fulzele et al., 2007; Ogata et al., 2000). Treatment with insulin enhanced osteoclast differentiation and maturation by RANKL through a subset of osteoclast marker genes (Oh et al., 2015a). Moreover, patients with metabolic diseases such as diabetes mellitus exhibit altered bone metabolism (Erbagci et al., 2002; Isaia et al., 1999; Kemink et al., 2000; Vestergaard, 2007). However, the correlation between RANK expression and insulin has been little defined.
Here, we present for the first time that insulin regulates RANK expression through ERK1/2-dependent signaling pathways, which contribute to the acceleration of osteoclast differentiation by RANKL.
MATERIALS AND METHODS
Isolation of bone marrow precursors and in vitro osteoclastogenesis
Isolation of bone marrow precursors was performed as described in previous research (Kim and Lee, 2014). In brief, bone marrow cells were flushed out from the femur of 4–6-week-old C57BL/6 mice with a sterile 21-gauge syringe and incubated in alpha-MEM media containing 10% FBS and 10 ng/ml of M-CSF (R&D Systems). After 24 h, non-adherent cells were harvested and cultured in the presence of M-CSF (20 ng/ml) for 3 days. After washing out the non-adherent cells, adherent cells were used as BMMs. Culture media was changed every two days. For in vitro osteoclastogenesis experiments, isolated BMMs were further cultured in the presence of 200 ng/ml of RANKL (provided by Dr. S.Y. Lee) and 30 ng/ml of M-CSF. After 5 days, the cells were fixed and stained for tartrate-resistant acid phosphatase (TRAP) using the TRAP staining kit (Sigma). Pink-colored TRAP-positive multinucleated (>3 nuclei) cells (MNCs) were counted as osteoclast-like cells. The cells were observed using a Zeiss Axiovert 200 microscope and images were obtained with an AxioCam HR (Carl Zeiss) equipped with Axio Vision 3.1 software (Carl Zeiss).
Western blot analyses
BMMs stimulated with 10 nM of insulin were lysed in lysis buffer (20 mM Tris-HCl, pH7.5, 150 mM NaCl, 1 mM Na2EDTA, 1 mM EGTA, 1% Triton, 2.5 mM sodium pyrophosphate, 1 mM beta-glycerophosphate, 1 mM Na3VO4, 1 μg/ml leupeptin, 1 mM phenylmethylsulfonylfluoride) and supernatants were prepared by centrifugation, electrophoresed on a 10% SDS-polyacrylamide gel and blotted onto a polyvinylidene difluoride membrane. Immunoblotting was performed with polyclonal antibodies specific to insulin receptor, β-actin (as a loading control) (Cell Signaling Technology, USA), and RANK (Santa Cruz Biotechnology Inc.), followed by HRP-conjugated secondary antibodies and enhanced using an ECL detection kit (Amersham Biosciences) (Lee and Lee, 2014).
RNA isolation and real-time PCR
According to the manufacturer’s protocol, total RNA was isolated using TRIZOL and reverse transcribed using SuperscriptIII reverse transcriptase (Invitrogen). PCRs were performed with the Brilliant UltraFast SYBR Green QPCR Master Mix (Agilent Technologies) and primers of specific genes and hprt (for endogenous control) from QIAGEN in triplicates on an Mx3000P instrument (Agilent Technologies). The thermal cycling conditions were as follows: 3 min at 95°C, followed by 40 cycles of 95°C for 10 s, 60°C for 20 s, and 1 cycle of 95°C for 1 min, 55°C for 30 s, and 95°C for 30 s. All quantitation were normalized to an hprt (Oh et al., 2015b).
Lentiviral-mediated gene transduction
Lentiviral-mediated gene transduction was performed as described in previous research (Oh et al., 2015a). In brief, the lentiviral packaging was done in accordance with the Lentiviral packaging system (OriGene). HEK293T cells were transfected with premixed packaging plasmids and pGFP-C-InsR shRNA lentiviral vector using transfection reagent (MegaTran). The supernatants collected 48 h after transfection were used as the viral stocks. For lentiviral infection, the BMMs were incubated with the lentivirus stock and polybrene (10 μg/ml) for 6 h. Two days after exposure to virus, the infected cells were replaced with a complete medium containing puromycin (2 μg/ml) to select for insulin receptor shRNA expressing cells, and the total cell lysates were subjected to a western blot analysis.
Transfection and luciferase reporter assay
The RANK promoter-luciferase plasmid was purchased from Switchgear Genomics. Plasmid DNA was mixed with FuGENE6 and transfected into the RAW 264.7 cells. After 12 h of transfection, cells were treated with RANKL and/or insulin for 24 h then assayed for luciferase activity. Luciferase activity was measured with the Dual Luciferase Reporter Assay System (Promega) according to the manufacturer’s instructions.
RAW 264.7 cells with stable expression of the RANK
RAW 264.7 cells were transduced with pMX-puro-FLAG-RANK and then selected in DMEM containing 10% FBS and 4 μg/ml of puromycin. Puromycin-resistant clones (RAW-RANK cells) were examined for osteoclast formation or subjected to real-time PCR by incubating with or without an anti-FLAG antibody (2 μg/ml) (Choi et al., 2013).
Statistical analysis
Results are presented as means ± standard deviations (SD) from at least 3 independent experiments and statistical analyses were determined using Student’s t test, if not, indicated. P < 0.05 was considered to be statistically significant.
RESULTS
Insulin induces the expression of RANK in BMMs
A previous study in which the treatment of insulin enhanced osteoclast differentiation and maturation by RANKL (Oh et al., 2015a) led us to explore the regulatory mechanism of insulin on osteoclastogenesis, especially whether insulin regulates the expression of RANK, a receptor for RANKL, a key cytokine for osteoclast differentiation. We first examined whether RANKL induces RANK expression. As expected, the stimulation with RANKL notably induced the expression of RANK (Fig. 1A). Interestingly, insulin alone also increased RANK expression in time- and dose-dependent manners (Figs. 1B–1D).
Fig. 1.
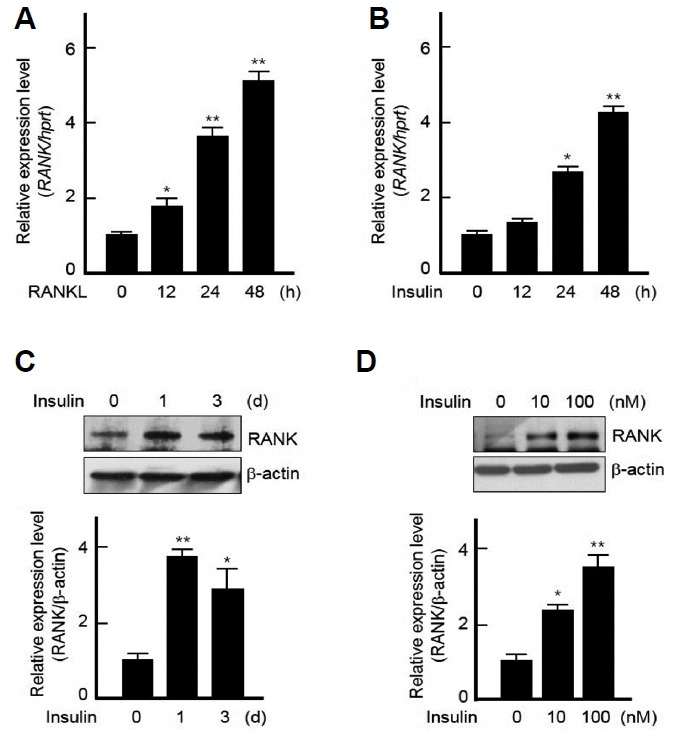
Insulin regulates RANK expression in BMMs.
(A, B) BMMs were treated with RANKL (200 ng/ml) or insulin (10 nM) for indicated times and then isolated total RNAs were subjected to real-time PCR using RANK-specific primers. All quantitation were normalized to hprt. (C, D) Isolated BMMs were treated with 10 nM of insulin for indicated times (C) or with indicated doses of insulin for 3 days. (D) and then the cell lysates were analyzed by Western blotting using specific antibodies against RANK and β-actin (upper panels). Protein bands were quantified by densitometry, and levels of RANK were normalized to levels of β-actin (lower panels). Results are representative of at least three independent experiments. *p < 0.05 and **p < 0.005 vs. non-treated cells.
To investigate this increase by insulin through an insulin receptor, insulin receptor shRNA was used. Insulin receptor knock-down with insulin receptor shRNA remarkably abolished not only the basal expression but also the induction of RANK by insulin in BMMs (Fig. 2). These data suggest that insulin regulates the expression of RANK through insulin receptor signaling in BMMs.
Fig. 2.
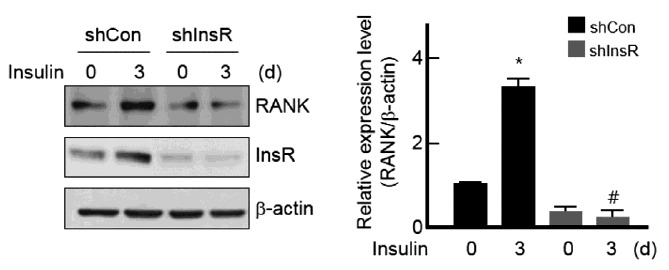
Suppression of RANK expression by knock-down of insulin receptor.
BMMs were infected with lentiviruses expressing control shRNA (shCon) or insulin receptor-specific shRNA (shInsR) and then stimulated with or without 10 nM of insulin for 3 days. Western blot analysis was performed to detect the expression of RANK, insulin receptor (InsR) and β-actin (left). Protein bands were quantified by densitometry, and levels of RANK were normalized to levels of β-actin (right). Results are representative of at least three independent experiments. *p < 0.05 vs. non-treated cells and #p < 0.05 vs. insulin-treated cells.
Insulin signaling enhances RANK expression by RANKL
We next explored whether insulin enhances the expression of RANK by RANKL using RAW264.7 cells transiently transfected with plasmids containing a promoter region of RANK gene in the reporter plasmid. As shown in Fig. 3A, luciferase activity of this construct was strongly up-regulated by both RANKL and insulin. Especially, the addition of insulin in the presence of RANKL potentiated the RANK expression compared to when treated with RANKL or insulin alone (Fig. 3A). The same was true when it was measured at protein levels (Fig. 3B). These results demonstrate that insulin signaling potentiates RANK expression by RANKL during osteoclast differentiation.
Fig. 3.
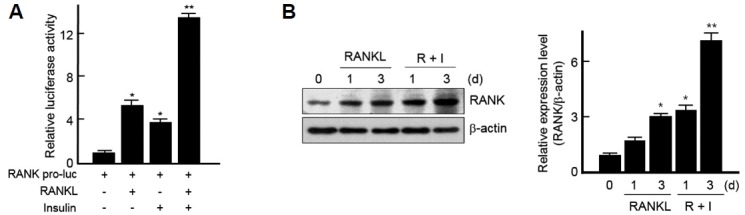
Insulin enhances RANK expression by RANKL.
(A) RANK promoter-luciferase reporter plasmids (RANK pro-luc) were transiently transfected into the RAW 264.7 cells. After 12 h of transfection, cells were treated with RANKL and/or insulin for 24 h then assayed for luciferase activity. The relative luciferase activity was normalized to the control activity. *p < 0.05 and **p < 0.005 vs. cells transfected with RANK promoter-luciferase reporter plasmids (RANK pro-luc) alone. (B) Isolated BMMs were treated with RANKL alone or RANKL and 10 nM of insulin for indicated times and then subjected to Western blotting using specific antibodies against RANK (left). Protein bands were quantified by densitometry, and levels of RANK were normalized to levels of β-actin. The fold increase in the stimulated cells compared with untreated cells is shown (right). Results are representative of at least three independent experiments. *p < 0.05 and **p < 0.005 vs. non-treated cells.
Insulin regulates RANK expression through ERK1/2 MAP kinase signaling
To gain insight into the regulatory pathway of RANK gene expression, MAP kinases inhibitors were pretreated. RANK expression by RANKL was down-regulated by all three MAP kinases inhibitors: SB203580, PD98059 and SP600125, inhibitors of p38, ERK1/2 and JNK1/2 MAP kinase, respectively. Interestingly, pretreatment with SP600125 completely abolished the expression of RANK induced by RANKL (Fig. 4A). However, the expression of RANK by insulin was diminished only by PD98059, an inhibitor of ERK1/2 (Fig. 4B). This data indicates that the induction of RANK by insulin stimulation occurs via ERK1/2 MAP kinase rather than p38 and JNK1/2.
Fig. 4.
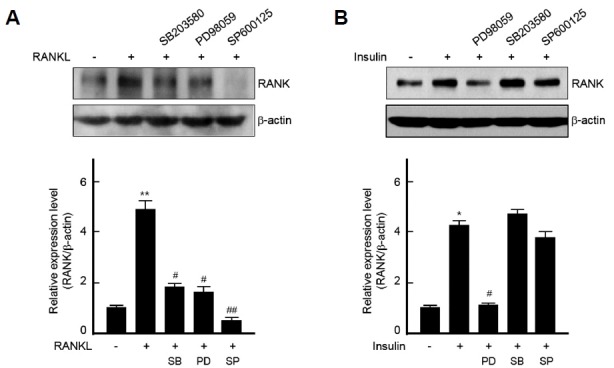
The regulation of RANK expression by MAP kinases signaling in BMMs.
(A, B) BMMs were pretreated with SB203580 (20 μM), PD98059 (20 μM) or SP600125 (20 μM) for 1 h and then incubated with RANKL (200 ng/ml) for 5 days (A) or with insulin (100 nM) for 3 days (B). Total cell lysates were analyzed by Western blot analysis (upper panels). Protein bands were quantified by densitometry, and levels of RANK were normalized to levels of β-actin (lower panels). Results are representative of at least three independent experiments. *p < 0.05 and **p < 0.005 vs. non-treated cells and #p < 0.05 and ##p < 0.005 vs. RANKL- or insulin-treated cells.
The activation of RANK up-regulates osteoclast differentiation
To observe the direct evidence that RANK is involved in osteoclast differentiation, we used RAW 264.7 cell lines stably expressing FLAG-tagged RANK (denoted as RAW-RANK) (Choi et al., 2013). The addition of anti-FLAG antibody to activate downstream signaling of RANK significantly increased the number of TRAP-positive osteoclasts in RAW-RANK cells. In the absence of anti-FLAG antibody, RAW-RANK cells failed to differentiate into TRAP-positive osteoclasts (Fig. 5A).
Fig. 5.

RANK up-regulates osteoclast differentiation.
(A) RAW 264.7 cells stably expressing FLAG-tagged RANK (RAW-RANK) were incubated with or without an anti-FLAG antibody (α-FLAG, 2 μg/ml) for 5 days and then TRAP staining was performed (original magnification, X100) (left) and the number of TRAP+ multinucleated cells (MNCs) were counted (right). Results are representative of at least three independent experiments. **p < 0.005 vs. non-treated cells. (B) RAW-RANK cells were treated with α-FLAG (2 μg/ml) for 48 h and then subjected to real-time PCR using Atp6v0d2- or DC-STAMP-specific primers. All quantitation were normalized to hprt and results are representative of at least three independent experiments. **p < 0.005 vs. non-treated cells.
Since we have reported insulin enhanced osteoclast differentiation by inducing the expression of osteoclast marker genes including Atp6v0d2 and DC-STAMP, key genes regulating osteoclast fusion (Oh et al., 2015a), it was investigated whether the activation of RANK regulates the expression of Atp6v0d2 and DC-STAMP as insulin did. The stimulation with an anti-FLAG antibody in RAW-RANK cells caused a remarkable increase of the expression of Atp6v0d2 and DC-STAMP (Fig. 5B). Collectively, these results suggest that insulin stimulation induces transcriptional up-regulation of RANK via ERK1/2 MAP kinase signaling, and, the cells highly expressing RANK by insulin are more susceptible to binding RANKL, thus contributing to the enhancement of RANKL-mediated osteoclastogenesis (Fig. 6).
Fig. 6.
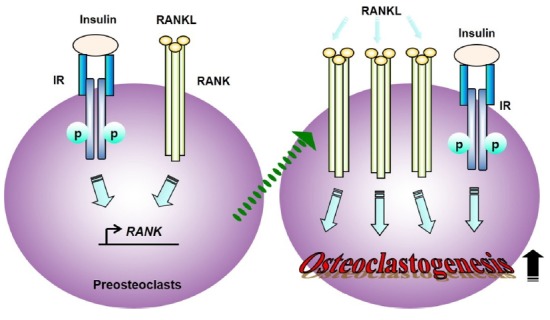
Schematic representation of regulatory mechanism model of osteoclastogenesis by insulin.
The cells expressing increased level of RANK by insulin are more susceptible to binding RANKL, thus regulating expression of osteoclast marker genes and finally enhancing osteoclastogenesis in the presence of RANKL.
DISCUSSION
Mononuclear osteoclast precursors fuse and differentiate to form bone-resorbing multinuclear osteoclasts in the presence of RANKL in the canonical pathway. The study on RANKL-RANK-mediated signaling mechanisms in osteoclast differentiation is necessary to understand bone metabolism. Several cytokines and growth factors have been described as a substitute for RANKL which are able to induce osteoclastogenesis in the non-canonical pathways. These include transforming growth factor beta (TGF-β), interleukin-6 (IL-6) and insulin-like growth factor-I (IGF-I) (Hemingway et al., 2011; Itonaga et al., 2004; Kudo et al., 2003). Here, we show for the first time that stimulation with insulin regulates RANK expression through ERK1/2 MAP kinase signaling, which contributes to the enhancement of osteoclast differentiation by RANKL.
Insulin has been proposed to be an anabolic agent that regulates proliferation and differentiation of osteoblasts and osteoclasts in bone metabolism (Thrailkill et al., 2005). Moreover, our previous study has demonstrated that insulin enhanced osteoclast differentiation and fusion by RANKL (Oh et al., 2015a). Although the importance of RANK in the differentiation of osteoclasts has been clearly demonstrated in rank−/− mice (Dougall et al., 1999; Li et al., 2000), the regulatory mechanisms about RANK expression during osteoclast differentiation have been little studied.
Surprisingly, insulin treatment significantly induced RANK expression in dose- and time-dependent manners in BMMs and the addition of insulin in the presence of RANKL enhanced RANK expression. These results were supported by insulin receptor shRNA since it blocked the expression of RANK by insulin.
RANKL treatment activates all three MAP kinases and these all were involved in the RANK expression by RANKL. Interestingly, an inhibitor of JNK1/2, SP600125, completely abolished the expression of RANK induced by RANKL, which suggests that RANK expression by RANKL is more dependent on JNK1/2 signaling compared to other MAP kinases. Conversely, the induction of RANK by insulin was blocked only by PD98059, an inhibitor of ERK1/2. This can be explained by that insulin stimulation specifically activated ERK1/2 MAP kinase among three MAP kinases during osteoclast differentiation (Oh et al., 2015a). This result indicates that the activation of ERK1/2 by insulin stimulation contributes the induction of RANK.
It is well known that Atp6v0d2 and DC-STAMP are important molecules that regulate osteoclastic cell fusion (Lee et al., 2006; Yagi et al., 2005). The activation of RANK significantly increased osteoclast differentiation, as well as the expression of DC-STAMP and Atp6v0d2, by stimulation with an anti-FLAG antibody, which is consistent with the results that insulin stimulation increased the expression of DC-STAMP and Atp6v0d2 (Oh et al., 2015a). Collectively, these results imply that RANK induced by insulin contributes to the expression of DC-STAMP and Atp6v0d2, thus triggering differentiation and maturation of osteoclasts.
Although RANK was overexpressed in the cells, it was not enough to induce osteoclast differentiation since RAW-RANK cells failed to differentiate into osteoclasts in the absence of stimulation with an anti-FLAG antibody. This may give hints about the results that even though insulin increased the RANK expression, insulin alone failed to induce osteoclast differentiation (Oh et al., 2015a).
Although questions about how the expression of RANK is regulated in detail remain to be elucidated, taken together, our study advances our understanding of pathological bone diseases and provides a basis for rational development of therapeutics for bone-related metabolic diseases.
ACKNOWLEDGMENTS
We thank Dr.S.Y.Lee for providing the RANKL. This work was supported by Basic Science Research Program through the National Research Foundation of Korea funded by the Ministry of Education, Science and Technology (NRF-2016 R1D1A1B01012205) and the Soonchunhyang University Research Fund.
REFERENCES
- Boyle W.J., Simonet W.S., Lacey D.L. Osteoclast differentiation and activation. Nature. 2003;423:337–342. doi: 10.1038/nature01658. [DOI] [PubMed] [Google Scholar]
- Choi H.K., Kang H.R., Jung E., Kim T.E., Lin J.J., Lee S.Y. Early estrogen-induced gene 1, a novel RANK signaling component, is essential for osteoclastogenesis. Cell Res. 2013;23:524–536. doi: 10.1038/cr.2013.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dougall W.C., Glaccum M., Charrier K., Rohrbach K., Brasel K., De Smedt T., Daro E., Smith J., Tometsko M.E., Maliszewski C.R., et al. RANK is essential for osteoclast and lymph node development. Genes Dev. 1999;13:2412–2424. doi: 10.1101/gad.13.18.2412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erbagci A.B., Araz M., Erbagci A., Tarakcioglu M., Namiduru E.S. Serum prolidase activity as a marker of osteoporosis in type 2 diabetes mellitus. Clin Biochem. 2002;35:263–268. doi: 10.1016/s0009-9120(02)00305-3. [DOI] [PubMed] [Google Scholar]
- Ferron M., Wei J., Yoshizawa T., Del Fattore A., DePinho R.A., Teti A., Ducy P., Karsenty G. Insulin signaling in osteoblasts integrates bone remodeling and energy metabolism. Cell. 2010;142:296–308. doi: 10.1016/j.cell.2010.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleischmann A., Hafezi F., Elliott C., Reme C.E., Ruther U., Wagner E.F. Fra-1 replaces c-Fos-dependent functions in mice. Genes Dev. 2000;14:2695–2700. doi: 10.1101/gad.187900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fulzele K., DiGirolamo D.J., Liu Z., Xu J., Messina J.L., Clemens T.L. Disruption of the insulin-like growth factor type 1 receptor in osteoblasts enhances insulin signaling and action. J Biol Chem. 2007;282:25649–25658. doi: 10.1074/jbc.M700651200. [DOI] [PubMed] [Google Scholar]
- Fulzele K., Riddle R.C., DiGirolamo D.J., Cao X., Wan C., Chen D., Faugere M.C., Aja S., Hussain M.A., Bruning J.C., et al. Insulin receptor signaling in osteoblasts regulates postnatal bone acquisition and body composition. Cell. 2010;142:309–319. doi: 10.1016/j.cell.2010.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harada S., Rodan G.A. Control of osteoblast function and regulation of bone mass. Nature. 2003;423:349–355. doi: 10.1038/nature01660. [DOI] [PubMed] [Google Scholar]
- Hemingway F., Taylor R., Knowles H.J., Athanasou N.A. RANKL-independent human osteoclast formation with APRIL, BAFF, NGF, IGF I and IGF II. Bone. 2011;48:938–944. doi: 10.1016/j.bone.2010.12.023. [DOI] [PubMed] [Google Scholar]
- Hsu H., Lacey D.L., Dunstan C.R., Solovyev I., Colombero A., Timms E., Tan H.L., Elliott G., Kelley M.J., Sarosi I., et al. Tumor necrosis factor receptor family member RANK mediates osteoclast differentiation and activation induced by osteoprotegerin ligand. Proc Natl Acad Sci USA. 1999;96:3540–3545. doi: 10.1073/pnas.96.7.3540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itonaga I., Sabokbar A., Sun S.G., et al. Transforming growth factor-beta induces osteoclast formation in the absence of RANKL. Bone. 2004;34:57–64. doi: 10.1016/j.bone.2003.08.008. [DOI] [PubMed] [Google Scholar]
- Isaia G.C., Ardissone P., Di Stefano M., Ferrari D., Martina V., Porta M., Tagliabue M., Molinatti G.M. Bone metabolism in type 2 diabetes mellitus. Acta Diabetol. 1999;36:35–38. doi: 10.1007/s005920050142. [DOI] [PubMed] [Google Scholar]
- Kemink S.A., Hermus A.R., Swinkels L.M., Lutterman J.A., Smals A.G. Osteopenia in insulin-dependent diabetes mellitus; prevalence and aspects of pathophysiology. J Endocrinol Invest. 2000;23:295–303. doi: 10.1007/BF03343726. [DOI] [PubMed] [Google Scholar]
- Kim H.S., Lee N.K. Gene expression profiling in osteoclast precursors by insulin using microarray analysis. Mol Cells. 2014;37:827–832. doi: 10.14348/molcells.2014.0223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim K., Lee S.H., Ha Kim J., Choi Y., Kim N. NFATc1 induces osteoclast fusion via up-regulation of Atp6v0d2 and the dendritic cell-specific transmembrane protein (DC-STAMP) Mol Endocrinol. 2008;22:176–185. doi: 10.1210/me.2007-0237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kudo O., Sabokbar A., Pocock A., Itonaga I., Fujikawa Y., Athanasou N.A. Interleukin-6 and interleukin-11 support human osteoclast formation by a RANKL-independent mechanism. Bone. 2003;32:1–7. doi: 10.1016/s8756-3282(02)00915-8. [DOI] [PubMed] [Google Scholar]
- Lacey D.L., Timms E., Tan H.L., Kelley M.J., Dunstan C.R., Burgess T., Elliott R., Colombero A., Elliott G., Scully S., et al. Osteoprotegerin ligand is a cytokine that regulates osteoclast differentiation and activation. Cell. 1998;93:165–176. doi: 10.1016/s0092-8674(00)81569-x. [DOI] [PubMed] [Google Scholar]
- Lee N.K. Molecular Understanding of Osteoclast Differentiation and Physiology. EnM. 2010;25:264–269. [Google Scholar]
- Lee J.Y., Lee N.K. Up-regulation of cyclinD1 and Bcl2A1 by insulin is involved in osteoclast proliferation. Life sci. 2014;114:57–61. doi: 10.1016/j.lfs.2014.07.006. [DOI] [PubMed] [Google Scholar]
- Lee S.H., Rho J., Jeong D., Sul J.Y., Kim T., Kim N., Kang J.S., Miyamoto T., Suda T., Lee S.K., et al. v-ATPase V0 subunit d2-deficient mice exhibit impaired osteoclast fusion and increased bone formation. Nat Med. 2006;12:1403–1409. doi: 10.1038/nm1514. [DOI] [PubMed] [Google Scholar]
- Li J., Sarosi I., Yan X.Q., Morony S., Capparelli C., Tan H.L., McCabe S., Elliott R., Scully S., Van G., et al. RANK is the intrinsic hematopoietic cell surface receptor that controls osteoclastogenesis and regulation of bone mass and calcium metabolism. Proc Natl Acad Sci USA. 2000;97:1566–1571. doi: 10.1073/pnas.97.4.1566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luchin A., Suchting S., Merson T., Rosol T.J., Hume D.A., Cassady A.I., Ostrowski M.C. Genetic and physical interactions between Microphthalmia transcription factor and PU.1 are necessary for osteoclast gene expression and differentiation. J Biol Chem. 2001;276:36703–36710. doi: 10.1074/jbc.M106418200. [DOI] [PubMed] [Google Scholar]
- Manolagas S.C. Birth and death of bone cells: basic regulatory mechanisms and implications for the pathogenesis and treatment of osteoporosis. Endocr Rev. 2000;21:115–137. doi: 10.1210/edrv.21.2.0395. [DOI] [PubMed] [Google Scholar]
- Matsuo K., Owens J.M., Tonko M., Elliott C., Chambers T.J., Wagner E.F. Fosl1 is a transcriptional target of c-Fos during osteoclast differentiation. Nat Genet. 2000;24:184–187. doi: 10.1038/72855. [DOI] [PubMed] [Google Scholar]
- Ogata N., Chikazu D., Kubota N., Terauchi Y., Tobe K., Azuma Y., Ohta T., Kadowaki T., Nakamura K., Kawaguchi H. Insulin receptor substrate-1 in osteoblast is indispensable for maintaining bone turnover. J Clin Invest. 2000;105:935–943. doi: 10.1172/JCI9017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh J.H., Lee J.Y., Joung S.H., Oh Y.T., Kim H.S., Lee N.K. Insulin enhances RANKL-induced osteoclastogenesis via ERK1/2 activation and induction of NFATc1 and Atp6v0d2. Cell Signal. 2015a;27:2325–2331. doi: 10.1016/j.cellsig.2015.09.002. [DOI] [PubMed] [Google Scholar]
- Oh J.H., Lee J.Y., Park J.H., No J.H., Lee N.K. Obatoclax regulates the proliferation and fusion of osteoclast precursors through the inhibition of ERK activation by RANKL. Mol Cells. 2015b;38:279–284. doi: 10.14348/molcells.2015.2340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodan G.A., Martin T.J. Therapeutic approaches to bone diseases. Science. 2000;289:1508–1514. doi: 10.1126/science.289.5484.1508. [DOI] [PubMed] [Google Scholar]
- Suda T., Takahashi N., Udagawa N., Jimi E., Gillespie M.T., Martin T.J. Modulation of osteoclast differentiation and function by the new members of the tumor necrosis factor receptor and ligand families. Endocr Rev. 1999;20:345–357. doi: 10.1210/edrv.20.3.0367. [DOI] [PubMed] [Google Scholar]
- Takayanagi H., Kim S., Koga T., Nishina H., Isshiki M., Yoshida H., Saiura A., Isobe M., Yokochi T., Inoue J., et al. Induction and activation of the transcription factor NFATc1 (NFAT2) integrate RANKL signaling in terminal differentiation of osteoclasts. Dev Cell. 2002;3:889–901. doi: 10.1016/s1534-5807(02)00369-6. [DOI] [PubMed] [Google Scholar]
- Teitelbaum S.L. Bone resorption by osteoclasts. Science. 2000;289:1504–1508. doi: 10.1126/science.289.5484.1504. [DOI] [PubMed] [Google Scholar]
- Thrailkill K.M., Lumpkin C.K., Jr, Bunn R.C., Kemp S.F., Fowlkes J.L. Is insulin an anabolic agent in bone? Dissecting the diabetic bone for clues. Am J Physiol Endocrinol Metab. 2005;289:E735–745. doi: 10.1152/ajpendo.00159.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vestergaard P. Discrepancies in bone mineral density and fracture risk in patients with type 1 and type 2 diabetes-a meta-analysis. Osteoporos Int. 2007;18:427–444. doi: 10.1007/s00198-006-0253-4. [DOI] [PubMed] [Google Scholar]
- Yagi M., Miyamoto T., Sawatani Y., Iwamoto K., Hosogane N., Fujita N., Morita K., Ninomiya K., Suzuki T., Miyamoto K., et al. DC-STAMP is essential for cell-cell fusion in osteoclasts and foreign body giant cells. J Exp Med. 2005;202:345–351. doi: 10.1084/jem.20050645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J., Zhang X., Wang W., Liu J. Insulin stimulates osteoblast proliferation and differentiation through ERK and PI3K in MG-63 cells. Cell Biochem Funct. 2010;28:334–341. doi: 10.1002/cbf.1668. [DOI] [PubMed] [Google Scholar]
- Yasuda H., Shima N., Nakagawa N., Yamaguchi K., Kinosaki M., Mochizuki S., Tomoyasu A., Yano K., Goto M., Murakami A., et al. Osteoclast differentiation factor is a ligand for osteoprotegerin/osteoclastogenesis-inhibitory factor and is identical to TRANCE/RANKL. Proc Natl Acad Sci USA. 1998;95:3597–3602. doi: 10.1073/pnas.95.7.3597. [DOI] [PMC free article] [PubMed] [Google Scholar]