

Abstract
ADAMTS18 dysregulation plays an important role in many disease processes including cancer. We previously found ADAMTS18 as frequently methylated tumor suppressor gene (TSG) for multiple carcinomas, however, its biological functions and underlying molecular mechanisms in breast carcinogenesis remain unknown. Here, we found that ADAMTS18 was silenced or downregulated in breast cancer cell lines. ADAMTS18 was reduced in primary breast tumor tissues as compared with their adjacent noncancer tissues. ADAMTS18 promoter methylation was detected in 70.8% of tumor tissues by methylation‐specific PCR, but none of the normal tissues. Demethylation treatment restored ADAMTS18 expression in silenced breast cell lines. Ectopic expression of ADAMTS18 in breast tumor cells resulted in inhibition of cell migration and invasion. Nude mouse model further confirmed that ADAMTS18 suppressed breast cancer metastasis in vivo. Further mechanistic studies showed that ADAMTS18 suppressed epithelial‐mesenchymal transition (EMT), further inhibited migration and invasion of breast cancer cells. ADAMT18 deregulated AKT and NF‐κB signaling, through inhibiting phosphorylation levels of AKT and p65. Thus, ADAMTS18 as an antimetastatic tumor suppressor antagonizes AKT and NF‐κB signaling in breast tumorigenesis. Its methylation could be a potential tumor biomarker for breast cancer.
Keywords: ADAMTS18, cancer, metastasis, methylation, tumor suppressor
Introduction
Breast cancer is one of the most commonly diagnosed malignant tumors and the leading cause of cancer death in women 1. Data indicate that breast cancer dissemination and distant metastasis is an early onset and inherent genetic property that does not adhere to the multistep tumorigenesis theory 2. Therefore, timely metastasis‐related biomarker testing helps to reduce breast cancer mortality.
A disintegrin and metalloproteinase with thrombospondin motifs (ADAMTS) family was first identified in 1997 3. Compared with members of the metalloproteinases (MMPs) and a disintegrin and metalloproteinases (ADAMs), ADAMTS are secreted proteases, being synthesized and secreted by a variety of cells including macrophagocyte, vascular endothelial cell, fibroblasts, and smooth muscle cells and are usually integrated with the extracellular matrix 4. Currently, 19 family members have been identified, and some members have been implicated in cancer progression, such as ADAMTS8 5, ADAMTS9 6, ADAMTS15 7 and so on. ADAMTS18, a novel member of ADAMTS family which we are focusing on, located at 16q23.1 in the human genome 8, was identified in 2002 9.
Previous studies have reported ADAMTS18 possessing tumor‐suppressing activities, which were reversed by promoter hypermethylation in several malignancies, including nasopharyngeal, esophageal squamous cell, hepatocellular, cervical carcinomas 8. Subsequent studies have shown the prevalence of hypermethylation and low expression of ADAMTS18 in gastric, colorectal, pancreatic, and clear cell renal cell carcinomas 10, 11. Based on these findings, we have identified ADAMTS18 as a candidate TSG. However, studies on its roles in tumor suppression and related molecular mechanisms in breast cancer remain unclear. Here, we evaluated the expression and methylation status of ADAMTS18 in breast cancer cell lines and primary tumors, and further assessed its biological functions. We also explored relevant mechanisms of ADAMTS18 in breast tumorigenesis.
Materials and Methods
Cell lines and samples
Several breast tumor cell lines (BT549, MB231, MB468, MCF7, SK‐BR‐3, T47D, YCC‐B2, YCC‐B3, and ZR‐75‐1) were used as described previously 12, 13. All samples used in these studies were obtained from the tissue bank of the First Affiliated Hospital of Chongqing Medical University 14, including normal breast tissues and paired primary breast tumor tissues. This study was approved by the Institutional Ethics Committees of the First Affiliated Hospital of Chongqing Medical University (approval notice # 2010/2012(23)).
Cell lines were treated with 10 mmol/L 5‐aza‐2′‐deoxycytidine (Sigma‐ Aldrich, St Louis, MO) for 3 days and further treated with 100 nmol/L trichostatin A (TSA; Cayman Chemical Co., Ann Arbor, MI) for an additional 24 h.
Establishment of stable cell lines
ADAMTS18 full‐length cDNA‐expressing vector pcDNA3.1 (+)‐ADAMTS18 was generated as previously described and the sequence was verified 8. To establish stably transfected tumor cells with ADAMTS18 expression, cells (BT549, MB231 and MCF7) were transfected with ADAMTS18 expression plasmids using Lipofectamine™ 2000 (Invitrogen), and further screened a monoclonal cell population by G418.
Semiquantitative RT‐PCR and quantitative RT‐PCR (qRT‐PCR)
Semiquantitative RT‐PCR was performed as described previously 15. RT products were amplified with 32 cycles for ADAMTS18, and 23 cycles for β‐actin, using Go‐Taq® (Promega, Madison, WI). SYBR® Green Master Mix (Applied Biosystems, Grand Island, NY) was used for qPCR analyses as measured by the 7500 Real‐Time PCR System (Applied Biosystems) as described previously 16, 17.
DNA bisulfite treatment and methylation‐specific PCR (MSP)
Genomic DNA was extracted from tumors and normal tissues using a QIAamp DNA Mini Kit (Qiagen, Hilden, Germany). DNA bisulfite treatment and methylation‐specific PCR (MSP) analysis were performed as described previously 18. Bisulfite‐treated DNA was amplified by MSP using ADAMTS18 methylated or unmethylated primer set (Table S2) by AmpliTaq‐Gold DNA polymerase (Applied Biosystems, Foster City, CA), with an annealing temperature of 60°C for methylation detection, and 58°C for unmethylation detection.
Cell viability assay
Cells were replated into 96‐well plates after being transfected with ADAMTS18 expression vector and empty vector for 48 h, and further measured using the Cell Counting Kit‐8 (CCK‐8, Beyotime, Shanghai, China) as described previously 17, 19.
Colony formation assay
Monolayer culture was used for colony formation assay. Cells were selected for ~14 days with presence of G418, after transfection for 48 h. Surviving colonies (≥50 cells/colony) were counted and stained with gentian violet. All experiments were performed in triplicate wells, three times.
Migration and invasion assays
Cell motility and invasive abilities were assessed by Transwell® and Matrigel™ invasion assays (Corning Life Sciences, Bedford, MA) as described previously 12, 13., Cells were migrated and invaded to the lower side of the membrane, and further stained with 0.1% crystal violet. Cells were then counted in five microscopic fields, and the mean values were counted.
Wound healing assay
Wound healing assay was performed as described previously 17, 19. A mechanical wound was created after scratching with a pipette tip, and images were taken at different time points. The distance between the wound edges was measured and quantified.
Tail vein assay of cancer metastasis
ADAMTS18‐expressing MB231 cells or control cells (2.5 × 106 cells) were injected into the lateral tail vein of female Balb/c nude mice in a volume of 0.15 mL as described previously 20, with four mice per group (control group: #3647, #3648, #3682, and #3683; experimental group: #3649, #3684, #3685, and #3698). After 8 weeks, two groups of mice were killed, and metastatic nodules were counted on the lung surface, which were further confirmed by histopathological analysis.
Western blot
Protein Extraction Reagent (Thermo Scientific, Rockford, IL) was used for protein extraction. Western blot analysis was performed as described previously 12, 13, 16, 19.Antibodies included: ADAMTS18 (ab135728), occludin (ab31721, Abcam, Cambridge, UK); AKT (#4691), p‐AKT (#4060), p‐P65 (#3033), E‐cadherin (#3195), vimentin (#5741), Snail (#3879, Cell Signaling Technology, Danvers, MA, USA); β‐actin (LK‐ab008‐100; Liankebio, China).
Statistical analysis
Statistical analysis was performed using SPSS 16.0 software (SPSS, Chicago, IL, USA). The Wilcoxon rank‐sum test was used for evaluating ADAMTS18 expression. Fisher's exact tests were used to analyse correlation of ADAMTS1 methylation with clinicopathological parameters. The two‐tailed t‐test was used to assess migration data. Data are reported as values of means ± SD. P < 0.05 was considered as statistically significant.
Results
ADAMTS18 is downregulated in breast cancer cells and tissues
Initial evidence has indicated that ADAMTS18 gene expression is decreased in many human cancer cells 8, 10. We first evaluated ADAMTS18 expression in a panel of breast cancer cell lines and normal breast tissues using semiquantitative RT‐PCR. We found that ADAMTS18 was readily expressed in normal breast tissues, but frequently silenced or decreased in all nine breast tumor cell lines studied (Fig. 1A). ADAMTS18 expression in primary breast tumors and surgical‐margin tissues was further examined by qRT‐PCR. Results showed that ADAMTS18 was obviously decreased (31/35 of the samples) in breast cancer tissues, as compared with surgical‐margin tissues (P < 0.05) (Fig. 1B). Moreover, through analyzing the online Oncomine database, ADAMTS18 was significantly downregulated in breast cancer tissues, compared with normal breast tissues (Fig. 1C).
Figure 1.
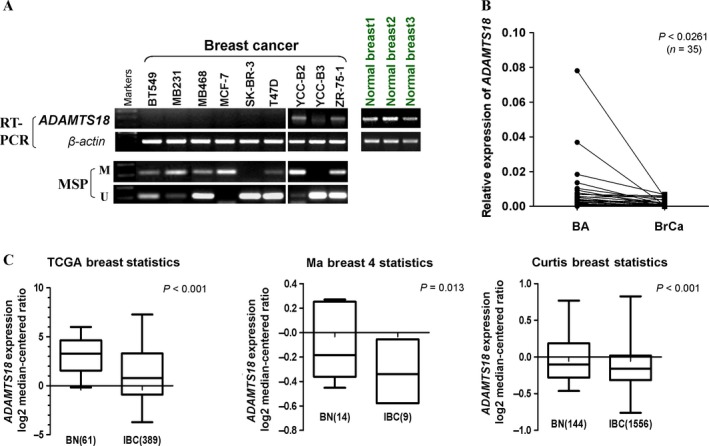
ADAMTS18 is frequently downregulated in breast cancer cell lines and breast cancer specimens. (A) The expression profile of ADAMTS18 mRNA in breast cancer cell lines and normal breast tissues by semiquantitative RT‐PCR, using β‐actin expression as an internal control. (B) ADAMTS18 mRNA levels were significantly decreased in primary breast cancer as compared with adjacent noncancer tissues as determined by quantitative real‐time PCR (n = 35). ADAMTS18 mRNA levels were normalized with the β‐actin mRNA level. (P < 0.05). (C) ADAMTS18 mRNA expression levels was assayed from the Oncomine database tool (http://www.oncomine.org). BN, Normal Breast tissues; BrCa, breast cancer; IBC, Invasive Breast Carcinoma.
Promoter methylation contributes to ADAMTS18 suppression in breast cancer
We next examined whether promoter methylation leads to ADAMTS18 suppression in breast cancer. MSP showed that ADAMTS18 was methylated in breast tumor cell lines with its silencing or downregulation (Fig. 1A). We further evaluated whether promoter methylation directly regulates ADAMTS18 gene expression. We treated four cell lines (BT549, MB231, MB468, and MCF7) with demethylating agent Aza and histone deacetylase inhibitor TSA. After treatment, ADAMTS18 expression was restored to different degrees. Meanwhile, MSP showed that the CGI (CpG island) was demethylated to some extent in the presence of drug (Fig. 2A). These results suggest that promoter methylation mechanism is responsible for ADAMTS18 silencing in breast cancer cells.
Figure 2.
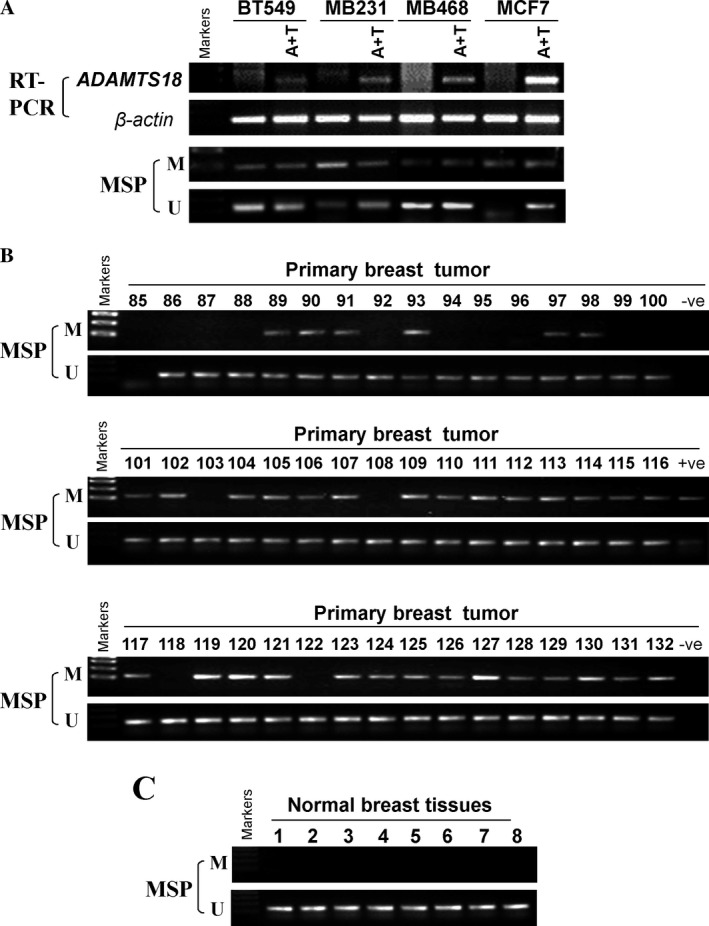
Promoter methylation of ADAMTS18 in breast cancer. (A) ADAMTS18 mRNA expression was restored after treatment with demethylation reagent, Aza and histone deacetylase inhibitor, TSA in four breast cancer cell lines (A+T refers to addition of Aza and TSA in culture media). Demethylation was confirmed by MSP (methylation‐specific PCR). (B) Promoter methylation of ADAMTS18 in primary breast cancer was determined. (C) MSP analysis of ADAMTS18 methylation in normal tissues. M, methylated DNA; U, unmethylated DNA.
To further evaluate ADAMTS18 promoter methylation in primary breast tumors, we examined ADAMTS18 methylation in 48 primary breast tumor samples and eight normal breast tissue samples. ADAMTS18 methylation was observed in 34 out of 48 (70.8%) primary tumors, but not in normal breast tissues (Fig. 2B, C and Table 1), indicating methylation‐mediated ADAMTS18 inactivation in breast cancer. We further analyzed the association between ADAMTS18 methylation and clinicopathological features, including age, tumor size, tumor grade, lymph node metastasis, ER status, PR status, HER2 status, P53 status, and Ki67 status. No significant correlation of ADAMTS18 methylation with any clinicopathological features (Table S1) was observed.
Table 1.
Promoter methylation status of ADAMTS18 in primary breast tumors
| Tissue | Samples (number) | ADAMTS18 promoter | Frequency of methylation | P value | |
|---|---|---|---|---|---|
| methylated | Unmethylated | ||||
| Breast tumor | 48 | 34 | 14 | 70.8% | 0.0002 |
| Normal breast | 8 | 0 | 8 | 0% | |
ADAMTS18 is an antimetastatic tumor suppressor for breast cancer cells
We further assessed tumor‐suppressive functions of ADAMTS18. RT‐PCR and western blot confirmed ADAMTS18 expression in ADAMTS18‐transfected breast tumor cells (Fig. 3A, B). Colony formation and CCK8 assays showed that overexpression of ADAMTS18 had no detectable effect on the proliferation of breast cancer MB231 cells (P > 0.05) (Fig. 3C, D).
Figure 3.
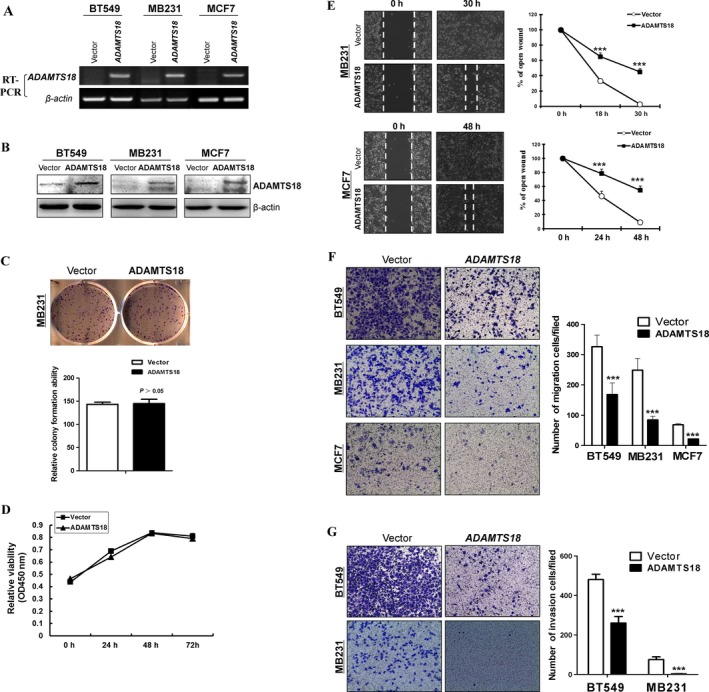
Effects of ectopic ADAMTS18 on migration and invasion of breast cancer cells. (A) RT‐PCR analyses of ADAMTS18 mRNA expression in three ADAMTS18 overexpressing cell lines (BT549, MB231, and MCF7). (B) Western blotting analyses of ADAMTS18 expression in ADAMTS18 overexpressing BT549, MB231, and MCF7 cells. (C) Representative colony formation assay and quantitative analysis. The numbers of G418‐resistant colonies in vector‐transfected controls were set to 100%, values are expressed as the mean ± SD from three experiments. (D) CCK‐8 assay for cell proliferation on vector‐ and ADAMTS18‐transfected MB231 cells. (E) Migration of ADAMTS18‐transfected or empty vector‐transfected tumor cells by scratch wound healing assays. The width of the remaining open wound was measured in relation to time 0 h. Original magnification, 100×. *P < 0.05, **P < 0.01, and ***P < 0.001. (F) Significant reduction in cell migration was observed in cells expressing ADAMTS18 compared with control cells. Representative images of migrated BT549, MB231, and MCF7 cells in each group are shown. Migrating cells at the lower surface of the Transwell® chamber were counted. (E) Effects of ADAMTS18 overexpression on invasive ability of BT549 and MB231 cells by the Boyden chamber invasion assay. (G) Cells at the lower surface of the Transwell® chamber were counted. Original magnification, 100×. *P < 0.05, **P < 0.01, and ***P < 0.001.
Scratch wound healing assays showed that ADAMTS18‐transfected cells showed slower closure of the scratched wound in MB231 and MCF7 cell lines (P < 0.001) (Fig. 3E). In addition, we measured cell migration and invasion using Transwell® assays (with or without Matrigel) in ADAMTS18‐transfected and the respective control cells. Results showed that ADAMTS18 overexpression significantly decreased cell migration in BT549, MB231, MCF7, and invasion in BT549 and MB231 (P < 0.001), confirming the antimetastatic properties of ADAMTS18 (Fig. 3F, G).
To evaluate tumor metastatic ability of ADAMTS18 in vivo, two groups of the Balb/c nude mice were randomly injected intravenously in the tail vein with ADAMTS18‐transfected or vector (Vec)‐transfected MB231 cells. As shown in Figure 4, the ADAMTS18 overexpression group mice had a significantly small number and sparse lung nodules than the control group (P < 0.001). Hematoxylin and eosin (H&E) staining confirmed that the nodules selected randomly on the surfaces of mice lungs were metastatic tumors. Histological analyses further confirmed that ADAMTS18 suppresses lung metastasis of MB231 cells in nude mice (Fig. 4C).
Figure 4.
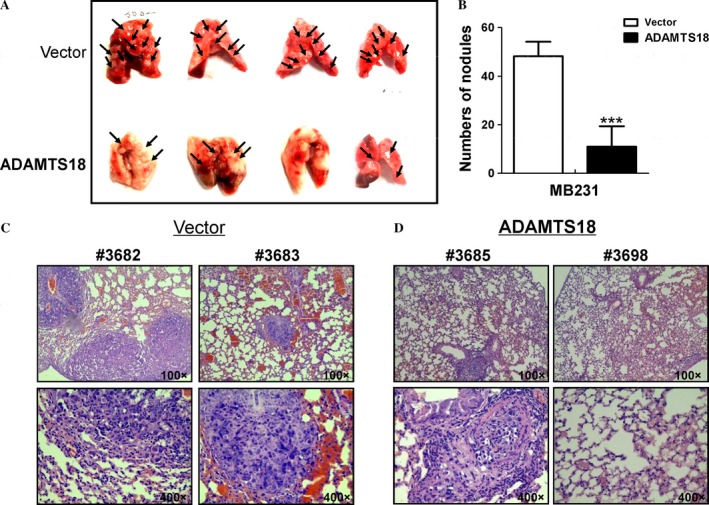
ADAMTS18 inhibits breast cancer cells' metastasis in vivo. (A) Representative images of lungs with metastatic nodules 2 months after injection of control and ADAMTS18 overexpressed MB231 stable cell lines. Arrows indicate metastatic nodules. (B) The number of lung metastatic nodules was counted under a dissecting microscope. Compared with the control group, a statistically dramatic decrease in the number of the lung metastases was seen in the ADAMTS18 overexpressing cell group. (C) Hematoxylin and eosin (H&E) staining of sections of lungs 2 months after injection of ADAMTS18 overexpressing MB231 cells into Balb/c nude mice through their tail veins. Upper panel, magnification, 100×; lower panel, magnification, 400×. *P < 0.05, **P < 0.01, and ***P < 0.001.
ADAMTS18 suppresses epithelial to mesenchymal transition of breast cancer cells
Epithelial to mesenchymal transition (EMT) plays a crucial role in tumor cell metastasis. Cell morphology assay showed cobblestone‐like changes in ADAMTS18‐expressing BT549 and MB231 cells (Fig. 5A), suggesting the epithelial phenotype conversion. EMT‐related proteins were further examined. Consistent with the morphological changes, our results showed that vimentin and Snail proteins were decreased and epithelial marker occludin was increased in ADAMTS18‐transfected BT549 and MB231 cells compared to the control cells, while E‐cadherin and occludin was increased and Snail was decreased in ADAMTS18‐expressing MCF7 cells (Fig. 5B). These data demonstrate that ADAMTS18 suppresses EMT of breast tumor cells.
Figure 5.
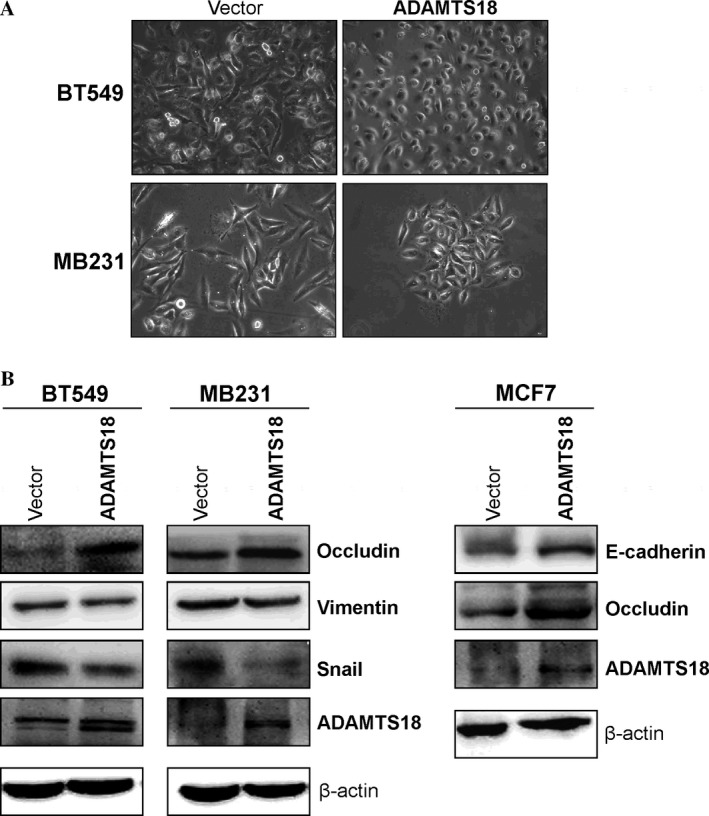
ADAMTS18 inhibited tumor cell EMT. (A) Morphological changes of BT549 and MB231 cells after ADAMTS18 transfection as seen by phase contrast microscopy. Original magnification, 100×. (B) Western blots showing the expression of E‐cadherin, occludin, vimentin, and Snail in ADAMTS18 or vector‐transfected cells. β‐actin was used as a loading control.
ADAMTS18 suppresses AKT and NF‐kB signaling pathways in breast cancer cells
To further explore molecular mechanisms responsible for ADAMTS18‐mediated EMT suppression, we analyzed the effects on AKT and NF‐kB signaling pathways 21. As expected, ADAMTS18‐transfected BT549, MB231, MCF7 cells—all displayed lower levels of phosphorylated AKT and phosphorylated p65 than the control cells (Fig. 6A). These findings suggest that ADAMTS18 may act as an antagonist of AKT and NF‐KB signaling pathway during cell EMT progression in breast cancer (Fig. 6B).
Figure 6.
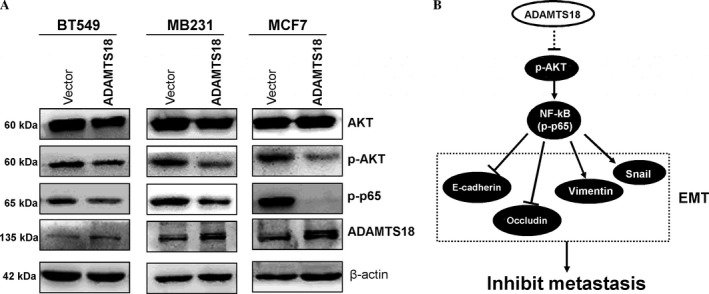
Ectopic expression of ADAMTS18 disrupts AKT and NF‐κB signaling. (A) Western blotting was performed in ADAMTS18 or vector‐transfected BT549, MB231, and MCF7 cells, using antibodies against AKT, phospho‐AKT, and phospho‐P65. β‐actin was used as a control. (B) Proposed mechanism of tumor‐suppressive function of ADAMTS18 through suppression the AKT and NF‐κB signaling pathway.
Discussion
In this study, we found frequent downregulation or silencing of ADAMTS18 by promoter methylation in breast cancer cells and primary tissues. ADAMTS18 is methylated in 70.8% of primary breast cancers, but not in normal breast tissues, indicating tumor‐specific methylation. Demethylation treatments could restore ADAMTS18 expression in breast cancer cells, indicating that promoter methylation directly mediates its silencing. Our data show that ectopic expression of ADAMTS18 inhibited the migration and invasion of breast cancer cell lines through downregulation of AKT and NF‐κB activity with no significant effect on cell proliferation. ADAMTS18 suppressed tumor metastasis of breast cancer in vivo. These results suggest that ADAMTS18 functions a tumor suppressor through suppressing migration and invasion in breast cancer.
Epigenetic inactivation includes promoter methylation and histone modification, which further regulates cancer gene expression 22, 23. ADAMTS18 was downregulated and methylated in clear cell renal cell, gastric, colorectal, and pancreatic carcinomas 10, 11. However, another ADAMTS18 family member, ADAMTS8 was frequently decreased by promoter methylation in common carcinoma cell lines 24. ADAMTS9 was reduced by promoter methylation in gastric cancer cells, and ADAMTS15 was genetically inactivated in colon cancer. Here, we found promoter methylation and downregulation of ADAMTS18 in primary breast cancer tissues and cell lines. There was no methylation detected in SK‐BR‐3 and YCC‐B3 breast cancer cell lines with silenced ADAMTS18, suggesting that other mechanisms like histone modifications mediated ADAMTS18 silencing. We found that ADAMTS18 promoter methylation was observed in 34 out of 48 (70.8%) primary breast carcinoma sample. However, previous study 8 presented that ADAMTS18 methylation in breast carcinoma was 24% (5/21). The reason caused the difference may be different sample sources from Hong Kong and mainland, or limited sample sizes, which needs further investigation by a large sample‐sized study. In our previous study, we also found frequent methylation of ADMTS18 in other common tumors like 70% of NPC, 52% of ESCC, and 63% of cervical cancer 8. Thus, development of quantitative detection method for ADAMTS18 methylation will be our next study.
Although, as demonstrated here and in other studies, ADAMTS18 is downregulated in multiple cancers, few studies have been reported concerning its related mechanism in tumorigenesis. Our study appears to be the first to reveal its underlying mechanism. We found that the inhibition of tumor migration and invasion by ADAMTS18 are associated with deregulation of AKT and NF‐κB signaling pathway, in which AKT pathway was in line with the findings in ADAMTS9, another member of the same subgroup 6. ADAMTS9 functions as a tumor suppressor through decreasing cell proliferation and inhibiting angiogenesis via regulating AKT/mTOR pathway.
AKT signaling also plays a crucial role in EMT, further leading to tumor migration and invasion 25. Alternatively, AKT can liberate NF‐κB (p‐P65) to enter into the nucleus and upregulate Snail and Slug expression to promote EMT 26, 27, 28. Further investigation of the exact mechanism by which ADAMTS18 can act on AKT during the course of multistep tumor progression is needed.
In summary, we demonstrate that ADAMTS18 silencing in breast cancer is significantly correlated with promoter CpG methylation. ADAMTS18 acts as an antagonist of AKT and NF‐κB signaling, further suppressing EMT and metastasis of breast cancer cells. Our study may help to establish new cancer gene therapeutic strategies for breast cancer.
Conflicts of Interest
The authors declare no conflict of interest.
Supporting information
Table S1. ADAMTS18 methylation and clinicopathologic features of breast tumors
Table S2. List of primers used in this study
Acknowledgments
This study was supported by grants from National Natural Science Foundation (#81372898, #81172582, #31420103915, #81572327) and VC special fund from The Chinese University of Hong Kong. We thank Prof. Qian Tao for his guidance for the project and comments for the manuscript and Prof. Kathleen Kelly (NCI, NIH, Maryland) for some cell lines.
Cancer Medicine 2017; 6(6):1399–1408
Contributor Information
Lili Li, Email: lili_li@cuhk.edu.hk.
Guosheng Ren, Email: rengs726@126.com.
References
- 1. Torre, L. A. , Bray F., Siegel R. L., Ferlay J., Lortet‐Tieulent J., and Jemal A.. 2015. Global cancer statistics, 2012. CA Cancer J. Clin. 65:87–108. [DOI] [PubMed] [Google Scholar]
- 2. de van Vijver, M. J. , He Y. D., van't Veer L. J. , H. Dai , Hart A. A., Voskuil D. W., et al. 2002. A gene‐expression signature as a predictor of survival in breast cancer. N. Engl. J. Med. 347:1999–2009. [DOI] [PubMed] [Google Scholar]
- 3. Kuno, K. , Kanada N., Nakashima E., Fujiki F., Ichimura F., and Matsushima K.. 1997. Molecular cloning of a gene encoding a new type of metalloproteinase‐disintegrin family protein with thrombospondin motifs as an inflammation associated gene. J. Biol. Chem. 272:556–562. [DOI] [PubMed] [Google Scholar]
- 4. Wei, J. , Liu C. J., and Li Z.. 2014. ADAMTS‐18: a metalloproteinase with multiple functions. Frontiers in bioscience (Landmark edition) 19:1456–1467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Georgiadis, K. E. , Hirohata S., Seldin M. F., and Apte S. S.. 1999. ADAM‐TS8, a novel metalloprotease of the ADAM‐TS family located on mouse chromosome 9 and human chromosome 11. Genomics 62:312–315. [DOI] [PubMed] [Google Scholar]
- 6. Du, W. , Wang S., Zhou Q., Li X., Chu J., Chang Z., et al. 2013. ADAMTS9 is a functional tumor suppressor through inhibiting AKT/mTOR pathway and associated with poor survival in gastric cancer. Oncogene 32:3319–3328. [DOI] [PubMed] [Google Scholar]
- 7. Viloria, C. G. , Obaya A. J., Moncada‐Pazos A., Llamazares M., Astudillo A., Capella G., et al. 2009. Genetic inactivation of ADAMTS15 metalloprotease in human colorectal cancer. Cancer Res. 69:4926–4934. [DOI] [PubMed] [Google Scholar]
- 8. Jin, H. , Wang X., Ying J., Wong A. H., Li H., Lee K. Y., et al. 2007. Epigenetic identification of ADAMTS18 as a novel 16q23.1 tumor suppressor frequently silenced in esophageal, nasopharyngeal and multiple other carcinomas. Oncogene 26:7490–7498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Cal, S. , Obaya A. J., Llamazares M., Garabaya C., Quesada V., and Lopez‐Otin C.. 2002. Cloning, expression analysis, and structural characterization of seven novel human ADAMTSs, a family of metalloproteinases with disintegrin and thrombospondin‐1 domains. Gene 283:49–62. [DOI] [PubMed] [Google Scholar]
- 10. Li, L. , Tao Q., Jin H., van Hasselt A., Poon F. F., Wang X., et al. 2010. The tumor suppressor UCHL1 forms a complex with p53/MDM2/ARF to promote p53 signaling and is frequently silenced in nasopharyngeal carcinoma. Clin. Cancer Res. 16:2949–2958. [DOI] [PubMed] [Google Scholar]
- 11. Xu, B. , Zhang L., Luo C., Qi Y., Cui Y., Ying J. M., et al. 2015. Hypermethylation of the 16q23.1 tumor suppressor gene ADAMTS18 in clear cell renal cell carcinoma. Int. J. Mol. Sci. 16:1051–1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Xiang, T. , Fan Y., Li C., Li L., Ying Y., Mu J., et al. 2016. DACT2 silencing by promoter CpG methylation disrupts its regulation of epithelial‐to‐mesenchymal transition and cytoskeleton reorganization in breast cancer cells. Oncotarget 7:70924–70935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Yin, X. , Xiang T., Li L., Su X., Shu X., Luo X., et al. 2013. DACT1, an antagonist to Wnt/beta‐catenin signaling, suppresses tumor cell growth and is frequently silenced in breast cancer. Breast Cancer Res. 15:R23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Xiang, T. , Li L., Yin X., Zhong L., Peng W., Qiu Z., et al. 2013. Epigenetic silencing of the WNT antagonist Dickkopf 3 disrupts normal Wnt/beta‐catenin signalling and apoptosis regulation in breast cancer cells. J. Cell Mol. Med. 17:1236–1246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ying, J. , Li H., Seng T. J., Langford C., Srivastava G., Tsao S. W., et al. 2006. Functional epigenetics identifies a protocadherin PCDH10 as a candidate tumor suppressor for nasopharyngeal, esophageal and multiple other carcinomas with frequent methylation. Oncogene 25:1070–1080. [DOI] [PubMed] [Google Scholar]
- 16. Xiang, T. , Li L., Fan Y., Jiang Y., Ying Y., Putti T. C., et al. 2010. PLCD1 is a functional tumor suppressor inducing G(2)/M arrest and frequently methylated in breast cancer. Cancer Biol. Ther. 10:520–527. [DOI] [PubMed] [Google Scholar]
- 17. Yin, X. , Xiang T., Mu J., Mao H., Li L., Huang X., et al. 2016. Protocadherin 17 functions as a tumor suppressor suppressing Wnt/beta‐catenin signaling and cell metastasis and is frequently methylated in breast cancer. Oncotarget 7:51720–51732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Tao, Q. , Huang H., Geiman T. M., Lim C. Y., Fu L., Qiu G. H., et al. 2002. Defective de novo methylation of viral and cellular DNA sequences in ICF syndrome cells. Hum. Mol. Genet. 11:2091–2102. [DOI] [PubMed] [Google Scholar]
- 19. Li, C. , Tang L., Zhao L., Li L., Xiao Q., Luo X., et al. 2015. OPCML is frequently methylated in human colorectal cancer and its restored expression reverses EMT via downregulation of smad signaling. Am. J. Cancer Res. 5:1635–1648. [PMC free article] [PubMed] [Google Scholar]
- 20. Feng, X. , Wu Z., Wu Y., Hankey W., Prior T. W., Li L., et al. 2011. Cdc25A regulates matrix metalloprotease 1 through Foxo1 and mediates metastasis of breast cancer cells. Mol. Cell. Biol. 31:3457–3471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Iwatsuki, M. , Mimori K., Yokobori T., Ishi H., Beppu T., Nakamori S., et al. 2010. Epithelial‐mesenchymal transition in cancer development and its clinical significance. Cancer Sci. 101:293–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Jones, P. A. , and Baylin S. B.. 2007. The epigenomics of cancer. Cell 128:683–692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Baylin, S. B. , and Ohm J. E.. 2006. Epigenetic gene silencing in cancer ‐ a mechanism for early oncogenic pathway addiction? Nat. Rev. Cancer 6:107–116. [DOI] [PubMed] [Google Scholar]
- 24. Choi, G. C. , Li J., Wang Y., Li L., Zhong L., Ma B., et al. 2014. The metalloprotease ADAMTS8 displays antitumor properties through antagonizing EGFR‐MEK‐ERK signaling and is silenced in carcinomas by CpG methylation. Mol. Cancer Res. 12:228–238. [DOI] [PubMed] [Google Scholar]
- 25. Larue, L. , and Bellacosa A.. 2005. Epithelial‐mesenchymal transition in development and cancer: role of phosphatidylinositol 3′ kinase/AKT pathways. Oncogene 24:7443–7454. [DOI] [PubMed] [Google Scholar]
- 26. Sizemore, N. , Lerner N., Dombrowski N., Sakurai H., and Stark G. R.. 2002. Distinct roles of the Ikappa B kinase alpha and beta subunits in liberating nuclear factor kappa B (NF‐kappa B) from Ikappa B and in phosphorylating the p65 subunit of NF‐kappa B. J. Biol. Chem. 277:3863–3869. [DOI] [PubMed] [Google Scholar]
- 27. Julien, S. , Puig I., Caretti E., Bonaventure J., Nelles L., van Roy F., et al. 2007. Activation of NF‐kappaB by Akt upregulates Snail expression and induces epithelium mesenchyme transition. Oncogene 26:7445–7456. [DOI] [PubMed] [Google Scholar]
- 28. Saegusa, M. , Hashimura M., Kuwata T., and Okayasu I.. 2009. Requirement of the Akt/beta‐catenin pathway for uterine carcinosarcoma genesis, modulating E‐cadherin expression through the transactivation of slug. Am. J. Pathol. 174:2107–2115. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. ADAMTS18 methylation and clinicopathologic features of breast tumors
Table S2. List of primers used in this study