

Abstract
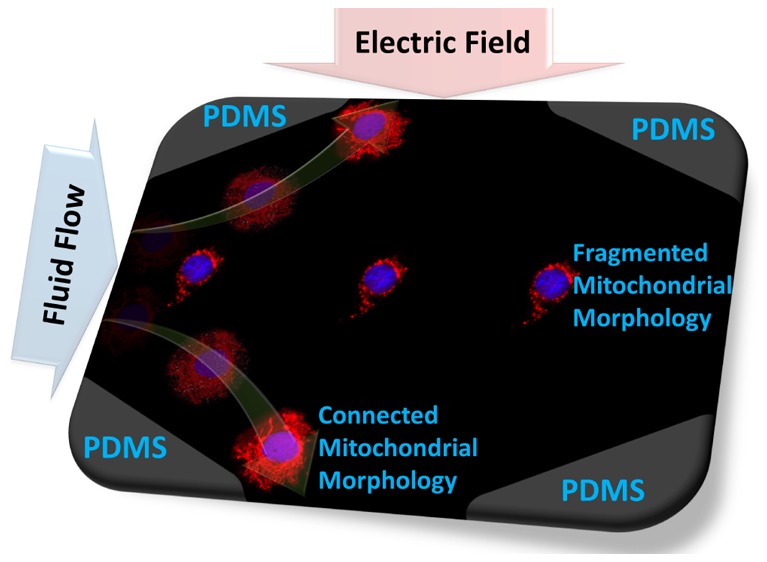
Mitochondrial dynamics play an important role within several pathological conditions, including cancer and neurological diseases. For the purpose of identifying therapies that target aberrant regulation of the mitochondrial dynamics machinery and characterizing the regulating signaling pathways, there is a need for label-free means to detect the dynamic alterations in mitochondrial morphology. We present the use of dielectrophoresis for label-free quantification of intracellular mitochondrial modifications that alter cytoplasmic conductivity, and these changes are benchmarked against label-based image analysis of the mitochondrial network. This is validated by quantifying the mitochondrial alterations that are carried out by entirely independent means on two different cell lines: human embryonic kidney cells and mouse embryonic fibroblasts. In both cell lines, the inhibition of mitochondrial fission that leads to a mitochondrial structure of higher connectivity is shown to substantially enhance conductivity of the cell interior, as apparent from the significantly higher positive dielectrophoresis levels in the 0.5–15 MHz range. Using single-cell velocity tracking, we show ∼10-fold higher positive dielectrophoresis levels at 0.5 MHz for cells with a highly connected versus those with a highly fragmented mitochondrial structure, suggesting the feasibility for frequency-selective dielectrophoretic isolation of cells to aid the discovery process for development of therapeutics targeting the mitochondrial machinery.
Mitochondria, which are key regulators of metabolism and cell death within eukaryotic cells, undergo constant cycles of fusion and fission, thereby allowing the cell to quickly adapt to environmental conditions for promoting cellular health.1−3 Mutation and aberrant regulation of the mitochondrial fusion and fission machinery is associated with a number of human diseases,4−6 including Parkinson’s disease, Alzheimer’s disease, and diabetes,7 as well as in physiological processes whose dysregulation are classical hallmarks of human cancer.4 Since increased mitochondrial fission can promote glycolysis,8 it has been postulated that tumors may increase mitochondrial fission activity to promote the metabolic shifts9 that create the molecular building blocks required for rapid proliferation.10 Hence, analysis of phenotypes caused by mitochondria shaping proteins can help uncover new diagnostic and therapeutic strategies for disease states, especially in conjunction with tools that monitor specificity of the subcellular alterations.
Activation of the Ras-MAPK (mitogen-activated protein kinase) pathway promotes phosphorylation of the mitochondrial fission GTPase Drp1 (dynamin-related protein 1), which subsequently induces mitochondrial fission. On the other hand, mitofusin proteins 1 and 2 (MFN1/2) on the outer mitochondrial membrane are responsible for reversing the mitochondrial fission pathway.11 In recent work,12 we demonstrated that mitochondrial fission was required for tumor growth in a xenograft model of pancreatic cancer, since its inhibition through shRNA-mediated knockdown of Drp1 was shown to block tumor growth. It has also been shown that mitochondrial fission promotes maintenance of stem cells in glioma and that inhibition of Drp1-dependent mitochondrial fission can effectively block tumor growth in a mouse model of gliomagenesis.13 Despite the wealth of data linking mitochondrial fission to tumor growth, the discovery of therapeutics targeting the mitochondrial machinery has been limited. One reason is a lack of robust methods to analyze dynamic changes in mitochondrial morphology. Genetic and pharmacological screens are powerful tools to identify novel signaling pathways and new inhibitors, but they require unbiased and quantitative readouts. Previous quantification attempts based on high-content image analysis require stains or markers to identify mitochondria and also require the fixing of cells for optimal images.14 Apart from being time-consuming, this methodology cannot be easily applied to nonadherent cell types such as immune cells and is not capable of isolating live cell populations based on their mitochondrial structure, especially from heterogeneous samples, for downstream quantitative analysis.
In this work, we explore a label-free approach based on cell electrophysiology to quantify alterations to mitochondrial structure induced by Drp1 and MFN1/2, which are independently quantified by mitochondrial image analysis of the respective labeled cells. Electrophysiology-based methods are of interest since they can characterize the mitochondrial alterations, as well as enable frequency-selective dielectrophoretic isolation of cells with a particular mitochondrial morphology for downstream analysis. Dielectrophoresis (DEP) causes the frequency-selective translation of polarized bioparticles under a spatially nonuniform electric field, either toward the high-field region by positive DEP (pDEP) for highly polarizable particles versus the media, or away from high-field region by negative DEP (nDEP) for particles within highly polarizable media.15,16 The DEP frequency spectra can be fit using a standard shell dielectric model to compute conductivity of cell interior,17 which strongly depends on the maximum level of pDEP of a cell in the megahertz (MHz) range, at a given media conductivity.18−20 Herein, we show a significant enhancement in cellular pDEP levels in the 0.5–15 MHz range after genetic manipulations that inhibit mitochondrial fission, as validated using independent mitochondrial modification methods that are carried out on two different cell lines: Drp1 knockdown on human embryonic kidney (HEK) cells and Drp1 knockout on mouse embryonic fibroblasts (MEFs). On the basis of this, we infer that significant alterations in intracellular mitochondrial structure can be identified and quantified, suggesting feasibility for utilizing label-free dielectrophoretic methods to selectively isolate cells based on their mitochondrial morphology.
Materials and Methods
Cell Lines
Generation of HEK cells expressing HRasG12V plus Drp1 shRNA or shScramble control were previously described.12 Mouse embryonic fibroblasts (MEFs) with Mfn1/2 knockout (MfnKO MEFs) were purchased from American Type Culture Collection (ATCC). To generate Drp1 knockout MEFs, Drp1flox/flox mice21 were bred to TP53flox/flox mice.22 MEFs were generated from Drp1flox/flox; TP53flox/flox embryos and subsequently infected with adeno-associated-CMV-Cre-GFP (AAV-CMV-Cre-GFP, University of North Carolina at Chapel Hill Vector Core). Single-cell clones were isolated and recombination of both alleles of Drp1 and p53 was confirmed by polymerase chain reaction (PCR).
Cell Imaging for Analysis of Mitochondrial Morphology
The described HEK and MEF cell lines were plated on glass microslides a day prior to visualization of their mitochondria by one of the following methods: (1) cells were fixed, permeabilized, and incubated with α-Tom20 primary antibody (Santa Cruz Biotechnology) in conjunction with an α-rabbit Alexa-488 secondary antibody (Life Technologies); (2) cells were engineered to stably express mitochondria-targeted YFP23 (BD Biosciences). The Tom20 antibody has been extensively validated to specifically recognize mitochondrial features.24 The alternative staining method is based on a vector expressing YFP that is fused to the N-terminal mitochondrial targeting sequence of subunit VIII of human cytochrome c oxidase (a mitochondrial transmembrane protein). A Zeiss LSM 700 confocal microscope with 63× oil objective was used for mitochondrial imaging, with image analysis presented in the Results and Discussion section. Flow cytometry of suspended cells (see Supporting Information sections S1 and S2) was used to confirm that there were no substantial alterations to the size of HEK cells after genetic alteration of their mitochondrial morphology.
Dielectrophoretic Spectral Measurements
DEP spectral analysis was performed with unlabeled cells (∼106/mL). Prior to the DEP experiments, the cell media was replaced with 8.8% sucrose water, with media conductivity of 150 ± 5% mS/m, as adjusted by its own culture media. Viability of cells within this altered media was verified over a period of 1 h, as assessed by trypan blue exclusion and by stability of their mitochondrial morphologies by immunofluorescence (see Supporting Information section S1). DEP spectral measurements were conducted on a 3DEP dielectrophoretic analyzer (DepTech, Uckfield, U.K.) using a recording interval set to 30 s at 10 Vpp, with data collected over 20 points between 100 kHz and 45 MHz. In this 3DEP reader, the electric field is applied to gold-plated conducting electrode stripes inside the wall of each well, with the DEP response measured at 20 different frequencies that are applied individually within each well. The relative DEP force at each frequency is obtained by analyzing spatiotemporal variations in light intensity from particle scattering using particular bands in each of the 20 wells, after normalization to the background at zero field (time = 0), after accounting for the field profile.25,26 For each batch of cells, the relative DEP force at each frequency was obtained using two independent measurements and this process was repeated for five separate batches of the same cell type to ensure reproducibility, as per the error bars. The maximum pDEP force level in the megahertz range for each cell line (HEK and MEFs) was used as the basis to normalize all other measured DEP force levels for the respective cell line. This assumes that the maximum pDEP level for each cell line occurs when the cell achieves its maximum polarization level for a particular modification.
Fitting DEP Spectra Using a Multishell Dielectric Model
In order to discern the particular subcellular regions influenced by the mitochondrial alterations carried out herein, the acquired DEP spectra over the 0.1–40 MHz range was fit to a standard three-shell or four-layer dielectric model of the cell. The layers from cell interior to its envelope include the nucleus, nuclear envelope, cytoplasm, and cell membrane17 (Figure 1). The time averaged DEP force (FDEP) on spherical particles is given by
| 1 |
Here, εm is the permittivity of the medium surrounding the cell, R is the radius of the cell, ω is the radian frequency of the applied field, and E is the electric field in the region where the cell is located. The Clausius–Mossotti factor (Ki(ω)) is a frequency-dependent measure of cell polarizability determining force direction as
| 2 |
Here, εm* and εc′* are the complex permittivity of the medium and effective permittivity of cell, respectively. In each case, the complex permittivity depends on the respective permittivity (ε) and conductivity (σ) values as per the following frequency dispersion:
| 3 |
The effective permittivity of cell (εc′*) is calculated in the following iterative manner. First, the innermost layer of the cell, i.e., the nucleus, and its surrounding envelope are approximated and replaced by a sphere of the same size (nucleus radius plus thickness of the nucleus membrane). The effective permittivity is expressed in terms of the respective complex permittivities, εnucleus* and εnucEnvelope*, while “a” is the ratio of external (nucleus plus envelope) to internal (nucleus) radius of the combined layers:
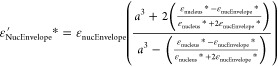 |
4 |
Next, the same process is repeated until the whole cell is replaced by a single sphere to consider the four-layer spherical model (including nucleus, nuclear envelope, cytoplasm, and cytoplasmic membrane). In this manner, the effective permittivity of the nucleus layer in eq 4 is combined with its surrounding cytoplasm to give effective cytoplasmic permittivity and with cell membrane to give effective cell permittivity. On the basis of this, eq 2 is used to fit the experimental DEP response of each cell modification. Parameters of the closest fit to the data are used to represent dielectric properties of cell elements.
Figure 1.
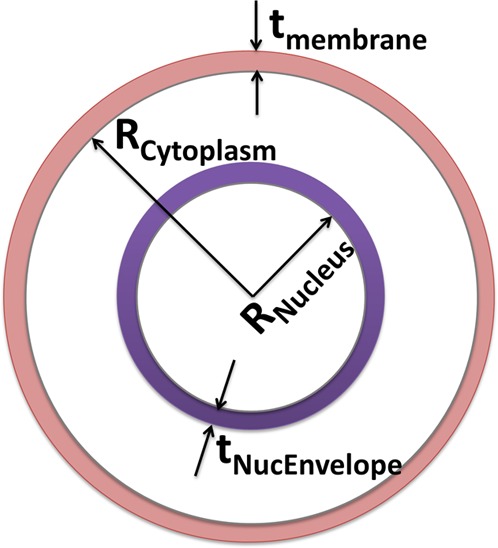
Geometry of the four-layer dielectric model (each layer with permittivity or ε and conductivity or σ) that is used to fit the DEP spectra, including membrane (σmem and εmem), cytoplasm (σcyto and εcyto), nuclear envelope (σNucEnvelope and εnucEnvelope), and nucleus (σnucleus and εnucleus).
Velocity Tracking To Quantify DEP Force
To quantify pDEP levels for cells with modified mitochondria, we used velocity tracking18 on an electrodeless dielectrophoresis platform.27,28 The lowest frequency showing obvious differences in pDEP between cells with differing mitochondrial morphology was chosen (0.5 MHz). Standard PDMS [poly(dimethylsiloxane)] micromolding methods were used to fabricate diamond-shaped posts that serve as sharp lateral constrictions in the channel (1000–70 μm). This electrodeless DEP device was bonded using oxygen plasma treatment to a glass coverslip for easy microscopic imaging of DEP behavior. A flow field was used to place cells in the uniform field area. An orthogonal electric field was then applied between Pt electrodes (100 Vpp/cm) at the frequency of interest, using a power amplifier.29 The trajectory of unlabeled polarized cells (105/mL) from the region just outside the diamond post (i.e., region of “no field gradient”) to the tip of the diamond post (i.e., region of “highest field gradient”) was recorded as high frame per second movies. On the basis of Newton’s second law for a particle of mass m, the net dielectrophoretic force (FDEP) on the accelerated particle of radius R, within a medium of viscosity η, can be determined by tracking displacement (x) as a function of time (t) to obtain (dx/dt) and (d2x/dt2) for each genetic modification on the respective cell types, as per
| 5 |
In this manner, FDEP levels under pDEP at the frequency of interest can be used to quantify mitochondrial alterations to HEK cells.
Results and Discussion
Images Analysis to Quantify Mitochondrial Structure
In order to separate mitochondrial features from those of the nucleus, we use color channeling, since they are differentially stained (see Supporting Information section S3). This is achieved by maintaining one channel with each of RGB colors at a particular time and detecting objects with that specific color, followed by a decision tree to separate the features (Figure 2, part a vs b). Following this, multiple preprocessing steps, including median, top-hat, and adaptive wiener filtering are applied to remove noise and other imperfections in the image. After preprocessing, an adaptive thresholding method is used for segmentation of the mitochondrial network, to identify individual mitochondria, as shown in Figure 2c. This structure is then used to identify branches (individual mitochondrion) and branch points (mitochondrial joints). Each branch is used as a mask to perform measurements such as length, width, and area on each individual mitochondrion (Figure 2, part c vs d). The connectedness factor (Cf) is a measure of connectivity of the mitochondrial network based on its comparison to an ideal network, as given by
| 6 |
Here, Bi is the number of branches in each connected group in the mitochondrial network of the cell, with the connected group representing a group of two or more mitochondria that are connected together, and ni is the number of branch points in the same connected group. Bideal is the number of the branches within the fully connected ideal network, as given by
| 7 |
As per the red dashed line in Figure 2d, the strength of a particular connection for each mitochondrial component of the cell is defined as Bini, where Bi is the number of branches in ith component in the mitochondrial network of the cell and ni is the number of branch points in the same component.
Figure 2.

Methodology used to measure mitochondrial structure through computing a connectedness factor. (a) Color channeling on the image of the stained cell followed by a decision tree is used to separate out the mitochondrial features (b). Preprocessing steps followed by adaptive thresholding is used for segmentation of the mitochondrial network to identify individual mitochondria (c), for identifying branches (individual mitochondrion) and branch points (mitochondrial joints). Each branch is used as a mask to perform measurements such as length, width, and area on each individual mitochondrion so that the connectedness factor can be computed (eq 6), as indicated by the red dashed line in panel d.
Genetic Manipulation of Mitochondrial Morphology
HRas is a proto-oncogene whose activation results in many downstream physiological changes, including altered metabolism, increased proliferation, blunted apoptosis, and changes in gene expression.30 In order to generate cell lines that exhibit significant differences in mitochondrial morphology, but with minimal changes to other aspects of cellular physiology, we carried out the following.
First, we stably expressed a constitutively active version of HRas called HRasG12V in immortalized HEK cells, which we previously demonstrated to result in a highly fragmented mitochondrial network.12 Next, we stably expressed shRNA targeting either Drp1 (HEK-RAS-12V-shDrp) or a scramble control sequence (HEK-RAS-12V-shScramble). As expected, shRNA-mediated knockdown of Drp1 results in a complete reversal of the mitochondrial fragmentation, indicating an extremely interconnected phenotype (92% connectedness, Figure 3i). Conversely, expression of the scramble control sequence had no effect on the HRas-induced mitochondrial fission, thereby causing these cells to maintain a highly fragmented phenotype (25% connectedness, Figure 3ii). Importantly, because we are directly targeting the mitochondrial fission machinery (Drp1) in cells that start with a fragmented phenotype, this approach allows us to generate two extreme mitochondrial phenotypes (i.e., highly fragmented and highly connected) with minimal differences in all other aspects of cellular physiology. Furthermore, we compare these “extreme” mitochondrial morphologies using a different cell type: mouse embryonic fibroblasts (MEFs) generated by a different method, i.e., genetic knockout of the mitochondrial fission (Drp1)21 and fusion (Mfn1/2)31 machinery, respectively. Since Drp1 and MFN1/2 primarily influence mitochondrial dynamics, their deletion should induce minimal changes in overall cellular physiology, aside from mitochondrial fission and fusion.32 We generated immortalized Drp1–/– MEFs by infecting cells isolated from Drp1flox/flox; TP53flox/flox embryos with adenoviral Cre recombinase. As expected, due to their inability to perform mitochondrial fission, these cells exhibit extreme mitochondrial connectedness (91% connectedness, Figure 3iii). To generate the opposite phenotype, we obtained immortalized Mfn1/Mfn2 double knockout cells that are deficient in mitochondrial outer membrane fusion.31 As expected, these MEFs exhibit highly fragmented mitochondrial network, due to unopposed mitochondrial fission (17% connectedness, Figure 3iv). Notably, both sets of MEFs are from the same strain of inbred mice (129/Sv) and both are p53 deficient (Mfn1/2–/– MEFs express SV40 Large T-antigen, which inhibits p5333). Hence, these cells should exhibit minimal changes in overall physiology, independent of mitochondrial morphology. In this manner, using two different cell types and two separate methods to alter mitochondrial morphology, we envision that the measured alterations to cellular physiology arise solely due to mitochondrial phenotype, rather than due to off-target changes associated with our method.
Figure 3.

Fluorescent images of fixed cells using anti-Tom20-labeling to reveal mitochondrial (green and red for HEK and MEFs, respectively) features (nucleus labeled blue). For mitochondrial features, the described image analysis method is used to compute the number of branches, branch area (in pixels), and connectedness factor as per eq 6, as indicated for each cell type. Note that, while the respective fixed cells appear to differ in size due to well-known interfacial interactions with the substrate, the size variations of suspended cells are minimal based on flow cytometry data (Supporting Information section).
Mitochondrial Phenotype-Induced DEP Alterations
Modifications to the mitochondrial structure influence cellular physiology, which is measured in this work, based on alterations to cytoplasmic electrophysiology. Each subcellular region dominates the DEP-induced cell polarizability response over a particular frequency range, depending on the contributions to its electrophysiology by the net conductivity and permittivity dispersion.34 Hence, modifications to the mitochondrial structure can be selectively probed based on alterations to the DEP frequency response in the frequency region wherein the electrophysiology due to mitochondrial structure dominates the net polarizability dispersion. It is also noteworthy that a critical level of media conductivity (σm) is required for optimal distinction of alterations in DEP spectra for particular cell types with differing mitochondrial connectedness. For instance, low σm levels enhance the contrast for discerning alterations in crossover to pDEP behavior due to morphological modifications to the cell envelope,35−37 which influence the respective permittivity values. Similarly, as per the spectral simulations and data in Supporting Information section S4, relatively higher σm levels that are closer to the conductivity of the cytoplasm (σcyto) wherein the mitochondria are situated should enable distinctions in mitochondrial phenotype. In this manner, alterations in the cell DEP response can be indicative of modifications to mitochondrial features, as long as the appropriate σm level and DEP frequency range are chosen for these comparisons. On the basis of this, the spectra are measured at σm of 0.15 S/m, since it is close to the anticipated range for σcyto (0.5–1 S/m), while being low enough for pDEP behavior (i.e., σcyto > σm). Comparing the DEP response of HEK-RAS-12V-shDrp (Figure 4a, part i) that exhibits a highly connected mitochondrial structure (see Figure 3i) to that of HEK-RAS-12V-shScramble (Figure 4a, part ii) that exhibits highly fragmented mitochondria (see Figure 3ii), the chief differences are in the 0.5–15 MHz region, as verified by a t test at each frequency to confirm well-separated mean values at 95% significance level. The DEP-well device used to measure these responses is shown in Figure 4b. Similarly, comparing the DEP response of MEF-Drp-KO cells (Figure 4c, part iii) that exhibit a highly connected mitochondrial structure (see Figure 3iii) versus MEF-Mfn-KO cells (Figure 4c, part iv) that exhibit a highly fragmented mitochondrial structure (see Figure 3iv), it is apparent that the differences are in the 0.5–15 MHz region (verified by a t test at each frequency to confirm separation at 95% significance level). In Supporting Information section S5, we present additional controls comparing wild-type (WT) and knockout (KO) MEF cell lines with Drp1 and Mfn modifications. Interestingly, the mitochondrial connectedness from image analysis, as well as based on pDEP levels in 0.5−15 MHz region, show that the phenotypes for WT cells lie between those of the respective KO cells. The differences in mitochondrial features between Mfn-WT and Mfn-KO are slightly larger versus the respective differences between Drp-KO and Drp-WT, which may be attributed to the differing rates of the fusion versus fission processes. This consistency further strengthens our inference that the pDEP measurements are truly representative of differences in mitochondrial morphology and not another aspect of cell physiology. It is noteworthy that DEP spectra in the 0.5–15 MHz frequency range are dominated by electrophysiology of the cell interior, wherein the mitochondria are situated. Hence, the pDEP levels for cells with a highly connected mitochondrial structure are consistently higher than those of cells with a highly fragmented mitochondrial structure, considering both cell types (HEK and MEFs) and independent of the method used to generate the respective mitochondrial phenotype (knockdown vs knockout). Figure 4d shows some sample images in the DEP-well device that are used to quantify varying DEP levels versus frequency, based on the spatiotemporal profiles of light scattering induced by DEP motion. Using a three-shell or four-layer dielectric model (Materials and Methods section), the respective spectra were fit to obtain dielectric properties for each subcellular region of interest (Table 1). Comparing the fitted dielectric parameters, it is apparent that an enhancement of mitochondrial connectivity increases conductivity of the cell interior, as reflected by that of the cytoplasm (σcyto) and nucleus envelope (σnucEnvelope). While Drp1 knockdown in HEK cells causes ∼70% higher σcyto levels and 3-fold higher σnucEnvelope levels, Drp1 knockout in MEFs causes ∼60% higher σcyto levels and 3-fold higher σnucEnvelope levels. Considering the two parameters, the influence of σcyto would likely dominate over that of σnucEnvelope, since σcyto is significantly higher (nearly 100-fold higher than σnucEnvelope). Hence, increasing σcyto by 50–70% would substantially increase pDEP levels in the 0.5–15 MHz, whereas a 3-fold rise in the substantially lower conductivity parameter (σnucEnvelope) causes only gradual alterations to slope of the DEP spectra in the 0.5–2 MHz region, especially at the chosen σm of 0.15 S/m (≫σnucEnvelope).
Figure 4.

Dielectrophoretic frequency spectra of (a) oncogenic HRas-modified HEK cells expressing Drp1 shRNA (i) vs scramble control shRNA (ii) and (c) MEFs lacking Drp1 (iii) vs those lacking mitochondrial fusion GTPases Mfn1 and Mfn2 (iv). Respective fixed cell images are in Figure 3. DEP spectra were measured at σm of 0.15 S/m in a 3DEP reader using 20 individual wells with ring electrodes (b) to measure relative DEP force levels based on spatiotemporal variations in light scattering, as per example images shown in panel d for nDEP at 150 kHz, no DEP at 300 kHz, and pDEP at 3 MHz field frequencies.
Table 1. Fitted Dielectric Parameters to DEP Spectraa.
| parameter (unit) | shScramble (HEK) (Ct = 25%) | shDRP (HEK) (Ct = 95%) | Mfn-KO (MEF) (Ct = 17%) | DRP-KO (MEF) (Ct = 91%) |
|---|---|---|---|---|
| εmem | 14 | 14 | 13 | 13 |
| σmem (S/m) | 0.1 × 10–6 | 0.1 × 10–6 | 0.1 × 10–5 | 0.1 × 10–5 |
| εcytoplasm | 60 | 60 | 60 | 65 |
| σcytoplasm (S/m) | 0.30 | 0.52 | 0.35 | 0.56 |
| εnucEnvelope | 25 | 25 | 25 | 25 |
| σnucEnvelope (S/m) | 0.9 × 10–3 | 3 × 10–3 | 1.3 × 10–3 | 3 × 10–3 |
| εnucleus | 60 | 60 | 60 | 60 |
| σnucleus (S/m) | 1.4 | 1.4 | 1.5 | 1.4 |
See Supporting Information section S6.
Single-Cell Quantification of pDEP Trapping Force Levels
The application of DEP toward frequency-selective isolation of cells based on their electrophysiology due to a particular mitochondrial structure requires that there be significant differences in their trapping force levels at optimal frequencies. While the DEP-well device measures the force spectra over a wide frequency range (0.1–20 MHz) by quantifying spatiotemporal alterations in light scattering due to the DEP motion of particles, it is an indirect method to measure DEP force on particle ensembles, without considering the efficacy of trapping forces for the purpose of DEP isolation at a particular flow rate. Hence, in order to quantify the differences in pDEP force levels between cells of differing mitochondrial structure, we use velocity tracking methods to directly measure force levels with single-cell sensitivity (details in the Materials and Methods section).
Specifically, we choose an electrodeless DEP device configuration (Figure 5a) for these measurements, wherein the electric field is applied orthogonal to fluid flow. This device is also significant to assess feasibility for selective cell isolation based on mitochondrial structure, since the spatial field nonuniformities created by constrictions due to insulating posts can enable frequency-selective isolation of cells at the constriction tips, based on magnitude of their pDEP levels versus orthogonal drag force due to flow. Specifically, we choose to compare HRas-modified HEK cells expressing shDrp versus those expressing a shScramble sequence, to verify significant differences in pDEP for cells with a highly connected versus a highly fragmented mitochondrial structure. While significant differences in pDEP levels are obvious for the respective spectra over the 0.5–15 MHz range in Figure 4a, we choose 0.5 MHz for these measurements, since power of the amplifier circuit that is used to generate the field over a wide spatial extent degrades at higher frequencies. On the basis of the images in Figure 5b, rapid pDEP trapping is apparent in Figure 5b, parts i–iii (video in the Supporting Information) for shDrp-expressing cells with a highly connected mitochondrial structure exhibiting pDEP levels of 1 nN ± 15% based on 10 independent single-cell measurements. The analogous measurement with HEK cells expressing oncogenic HRasG12V plus a scramble control, as per pDEP trapping images in Figure 5b, parts iv–vi, shows significantly lower pDEP levels that are measured at 0.1 nN ± 25% (based on 10 independent single-cell measurements). These results based on direct pDEP measurements on single cells validate the spectral distinctions of Figure 4. Assuming a steady drop in pDEP force across constriction region of the device, a flow rate of ∼1.35 μL/min is sufficient to ensure that the fluid velocity on the cells is significantly higher than the drag force due to FpDEP at the 0.1 nN level, while being lower than the drag force due to FpDEP at the 1 nN level (see Supporting Information section S7 for parameters used for this calculation). On the basis of this flow rate, a starting concentration of 105 cells/mL can be enriched for cells with the higher FpDEP, at a rate of ∼100 cells/min, to enable frequency-selective cell isolation at 0.5 MHz based on the cytoplasmic conductivity due to its mitochondrial features.
Figure 5.
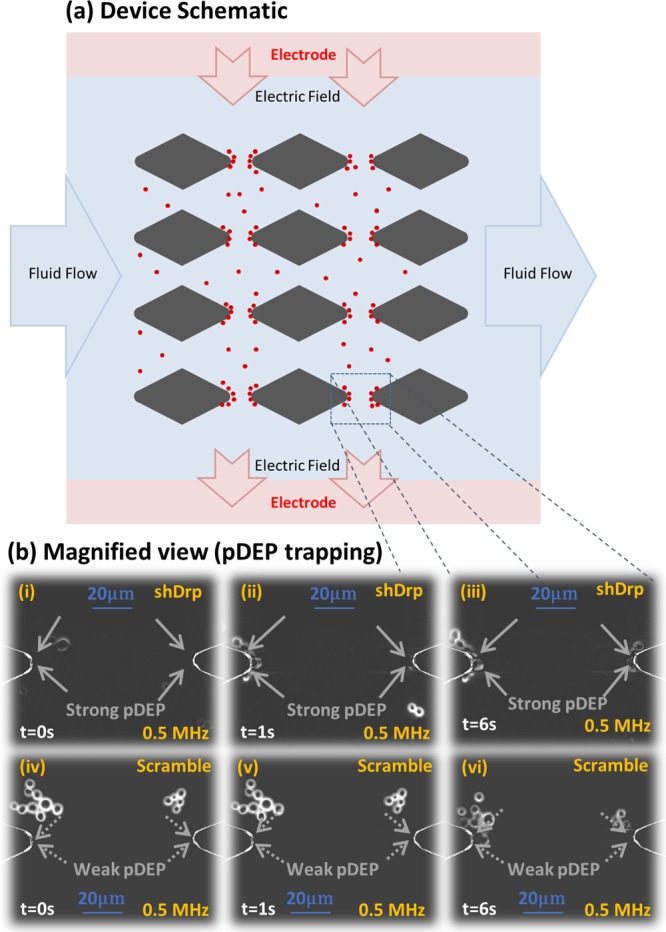
(a) Microfluidic electrodeless dielectrophoresis device using insulating posts of PDMS (gray) to enable frequency-selective cell trapping, with the electric field orthogonal to the inertial flow. (b) Field (100 Vpp/cm, 0.5 MHz in σm of 0.15 S/m) is applied at t = 0 and shows substantial pDEP levels for cells Ras-expressing HEK cells with shDrp modification (1 nN ± 15%) vs with shScramble modification (0.1 nN ± 25%), presumably due to their differences mitochondrial morphology. See the DEP movie in the Supporting Information.
Conclusions
Alterations in connectivity of the intracellular mitochondrial network after select genetic modifications can be quantified based on changes in the intracellular cytoplasmic conductivity, which is determined by fitting dielectrophoretic spectra of the respective cells to a standard shell dielectric model. This label-free DEP quantification method is highly consistent with image analysis methods to quantify the mitochondrial network of labeled cells. Specifically, direct inhibition of mitochondrial fission through shRNA-mediated knockdown of Drp1 increases the cytoplasmic conductivity by ∼70% in human embryonic kidney cells and a full genetic knockout of Drp1 in mouse embryonic fibroblasts causes a 60% rise in cytoplasmic conductivity over cells deficient for mitochondrial fusion. Utilizing a frequency of 0.5 MHz, we demonstrate that human embryonic kidney cells with a highly connected mitochondrial network exhibit ∼10-fold higher trapping forces under positive dielectrophoresis versus those with a highly fragmented mitochondrial network. On the basis of this, we envision a label-free platform for frequency-selective cell isolation to transform the discovery process, both for small-molecule modulators of the mitochondrial dynamics machinery and for novel signaling pathways that regulate the machinery under a variety of physiological conditions.
Acknowledgments
We acknowledge Seed Grant support from University of Virginia’s Cancer Center. D.F.K. acknowledges support from Research Scholar Grant of the American Cancer Society and 1R01CA200755-01 from the National Cancer Institute.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.analchem.6b04666.
DEP movie (MPG)
Stability of cellular mitochondrial morphology in DEP buffer (Figures S1 and S2), cell size comparisons by flow cytometry after modifications (Figures S3 and S4), DEP simulations and measurements to optimize media conductivity (Figures S5 and S6), and additional wild-type controls and fitted geometric and dielectric parameters (Table S1) (PDF)
Author Contributions
The manuscript was written through contributions of all authors, who have given approval to this submitted version.
The authors declare no competing financial interest.
Supplementary Material
References
- Kasahara A.; Scorrano L. Trends Cell Biol. 2014, 24, 761–770. 10.1016/j.tcb.2014.08.005. [DOI] [PubMed] [Google Scholar]
- Chen H. C.; Chan D. C. Hum. Mol. Genet. 2005, 14, R283–R289. 10.1093/hmg/ddi270. [DOI] [PubMed] [Google Scholar]
- Ferree A.; Shirihai O. Mitochondrial Oxidative Phosphorylation: Nuclear-Encoded Genes, Enzyme Regulation, and Pathophysiology 2012, 748, 13–40. 10.1007/978-1-4614-3573-0_2. [DOI] [Google Scholar]
- Hanahan D.; Weinberg R. A. Cell 2011, 144, 646–674. 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- Hoppins S.; Nunnari J. Science 2012, 337, 1052–1054. 10.1126/science.1224709. [DOI] [PubMed] [Google Scholar]
- Scorrano L. Eur. J. Clin. Invest. 2013, 43, 886–893. 10.1111/eci.12135. [DOI] [PubMed] [Google Scholar]
- Westermann B. Nat. Rev. Mol. Cell Biol. 2010, 11, 872–884. 10.1038/nrm3013. [DOI] [PubMed] [Google Scholar]
- Westermann B. Biochim. Biophys. Acta, Bioenerg. 2012, 1817, 1833–1838. 10.1016/j.bbabio.2012.02.033. [DOI] [PubMed] [Google Scholar]
- Ferreira L. M. Exp. Mol. Pathol. 2010, 89, 372–380. 10.1016/j.yexmp.2010.08.006. [DOI] [PubMed] [Google Scholar]
- Vander Heiden M. G.; Cantley L. C.; Thompson C. B. Science 2009, 324, 1029–1033. 10.1126/science.1160809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffin E. E.; Detmer S. A.; Chan D. C. Biochim. Biophys. Acta, Mol. Cell Res. 2006, 1763, 482–489. 10.1016/j.bbamcr.2006.02.003. [DOI] [PubMed] [Google Scholar]
- Kashatus J. A.; Nascimento A.; Myers L. J.; Sher A.; Byrne F. L.; Hoehn K. L.; Counter C. M.; Kashatus D. F. Mol. Cell 2015, 57, 537–551. 10.1016/j.molcel.2015.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie Q.; Wu Q. L.; Horbinski C. M.; Flavahan W. A.; Yang K. L.; Zhou W. C.; Dombrowski S. M.; Huang Z.; Fang X. G.; Shi Y.; Ferguson A. N.; Kashatus D. F.; Bao S. D.; Rich J. N. Nat. Neurosci. 2015, 18, 501–510. 10.1038/nn.3960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serasinghe M. N.; Wieder S. Y.; Renault T. T.; Elkholi R.; Asciolla J. J.; Yao J. L.; Jabado O.; Hoehn K.; Kageyama Y.; Sesaki H.; Chipuk J. E. Mol. Cell 2015, 57, 521–536. 10.1016/j.molcel.2015.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pohl H. A.Dielectrophoresis: The Behavior of Neutral Matter in Nonuniform Electric Fields; Cambridge University Press: Cambridge, U.K., 1978. [Google Scholar]
- Morgan H.; Green N. G.. AC Electrokinetics: Colloids and Nanoparticles; Research Studies Press: Philadelphia, PA, 2003. [Google Scholar]
- Jones T. B.Electromechanics of Particles; Cambridge University Press: Cambridge, U.K., 1995; p 265 [Google Scholar]
- Su Y. H.; Tsegaye M.; Varhue W.; Liao K. T.; Abebe L. S.; Smith J. A.; Guerrant R. L.; Swami N. S. Analyst 2014, 139, 66–73. 10.1039/C3AN01810E. [DOI] [PubMed] [Google Scholar]
- Su Y.-H.; Warren C. A.; Guerrant R. L.; Swami N. S. Anal. Chem. 2014, 86, 10855–10863. 10.1021/ac5029837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gagnon Z. R. Electrophoresis 2011, 32, 2466–2487. 10.1002/elps.201100060. [DOI] [PubMed] [Google Scholar]
- Wakabayashi J.; Zhang Z. Y.; Wakabayashi N.; Tamura Y.; Fukaya M.; Kensler T. W.; Iijima M.; Sesaki H. J. Cell Biol. 2009, 186, 805–816. 10.1083/jcb.200903065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonkers J.; Meuwissen R.; van der Gulden H.; Peterse H.; van der Valk M.; Berns A. Nat. Genet. 2001, 29, 418–425. 10.1038/ng747. [DOI] [PubMed] [Google Scholar]
- Karbowski M.; Lee Y.-J.; Gaume B.; Jeong S.-Y.; Frank S.; Nechushtan A.; Santel A.; Fuller M.; Smith C. L.; Youle R. J. J. Cell Biol. 2002, 159, 931–938. 10.1083/jcb.200209124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Likić V. A.; Perry A.; Hulett J.; Derby M.; Traven A.; Waller R. F.; Keeling P. J.; Koehler C. M.; Curran S. P.; Gooley P. R.; Lithgow T. J. Mol. Biol. 2005, 347, 81–93. 10.1016/j.jmb.2004.12.057. [DOI] [PubMed] [Google Scholar]
- Broche L. M.; Hoettges K. F.; Ogin S. L.; Kass G. E. N.; Hughes M. P. Electrophoresis 2011, 32, 2393–2399. 10.1002/elps.201100063. [DOI] [PubMed] [Google Scholar]
- Rohani A.; Varhue W.; Su Y. H.; Swami N. S. Biomicrofluidics 2014, 8, 052009. 10.1063/1.4897283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou C.-F.; Tegenfeldt J. O.; Bakajin O.; Chan S. S.; Cox E. C.; Darnton N.; Duke T.; Austin R. H. Biophys. J. 2002, 83, 2170–2179. 10.1016/S0006-3495(02)73977-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cummings E. B.; Singh A. K. Anal. Chem. 2003, 75, 4724–4731. 10.1021/ac0340612. [DOI] [PubMed] [Google Scholar]
- Farmehini V.; Rohani A.; Su Y.-H.; Swami N. Lab Chip 2014, 14, 4183–4187. 10.1039/C4LC00801D. [DOI] [PubMed] [Google Scholar]
- Young T.; Mei F.; Liu J.; Bast R. C.; Kurosky A.; Cheng X. Oncogene 2005, 24, 6174–6184. 10.1038/sj.onc.1208753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H. C.; Detmer S. A.; Ewald A. J.; Griffin E. E.; Fraser S. E.; Chan D. C. J. Cell Biol. 2003, 160, 189–200. 10.1083/jcb.200211046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva Ramos E.; Larsson N. G.; Mourier A. Biochim. Biophys. Acta, Bioenerg. 2016, 1857, 1277–1283. 10.1016/j.bbabio.2016.04.002. [DOI] [PubMed] [Google Scholar]
- Ali S. H.; DeCaprio J. A. Semin. Cancer Biol. 2001, 11, 15–22. 10.1006/scbi.2000.0342. [DOI] [PubMed] [Google Scholar]
- Castellarnau M.; Errachid A.; Madrid C.; Juarez A.; Samitier J. Biophys. J. 2006, 91, 3937–3945. 10.1529/biophysj.106.088534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su Y.-H.; Rohani A.; Warren C. A.; Swami N. S. ACS Infect. Dis. 2016, 2, 544–551. 10.1021/acsinfecdis.6b00050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gascoyne P. R.; Shim S.; Noshari J.; Becker F. F.; Stemke-Hale K. Electrophoresis 2013, 34, 1042–1050. 10.1002/elps.201200496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohani A.; Varhue W.; Su Y. H.; Swami N. S. Electrophoresis 2014, 35, 1795–1802. 10.1002/elps.201400021. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.