

Abstract
We report a 36-year-old Caucasian male identified with distal partial trisomy 15q and partial monosomy 16p from an unbalanced chromosome translocation detected by microarray and FISH analysis. He had a history of developmental delay and intellectual disability, chronic anemia, tall and slender stature, thoracic scoliosis and lumbar lordosis, and dysmorphic features. The distal partial trisomy 15q included the insulin-like growth factor 1 receptor gene involved with growth, while genes in the distal partial monosomy 16p region are involved with alpha hemoglobin production, intellectual disability, dysmorphic features, and acromegaly. The chromosome derivative found in our patient contains genes known to play a role in his phenotype.
Keywords: Autism, Dysmorphic features, Intellectual disability, Monosomy 16p13.3, Trisomy 15q26, Unbalanced chromosome translocation
Distal partial trisomy 15q is a rare chromosomal abnormality typically arising from unbalanced translocations producing a duplication of 15q and reported in at least 28 cases [Jones, 2006; Chen et al., 2011]. The break-points for the duplication of 15q may vary, but the general phenotype remains similar, including pre- and post-natal overgrowth and mild to profound intellectual disability [Jones, 2006; Su et al., 2010; Chen et al., 2011], downslanting palpebral fissures and elongated face, a broad nasal bridge and long philtrum, a high-arched palate, micrognathia, joint defects with slender fingers/toes, and hypotonia [Christofolini et al., 2014]. Tall stature and long fingers have also been reported in individuals with tetrasomy 15q [Blennow et al., 1994; Rowe et al., 2000].
Partial monosomy 16p13 is a chromosomal abnormality that is either de novo or inherited from a parent with a balanced translocation. Many patients with deletions involving chromosome 16p13 often include the CREBBP gene associated with Rubinstein-Taybi syndrome [Jones, 2006]. However, few patients have been described with deletions telomeric to the CREBBP gene. For example, Nelson et al. [2010] described a patient with a 555-kb deletion of the 16p13.3 band telomeric to the CREBBP gene with key features of tracheobronchomalacia, metopic craniosynostosis, hypospadias with chordee, torticollis, strabismus, fifth-finger clinodactyly, hallux valgus deformity, and global developmental delay. Herein, we describe an adult male with the rare occurrence of a distal partial trisomy 15q and partial monosomy 16p13 resulting from an unbalanced chromosome translocation telomeric to the CREBBP gene and presenting with clinical features representing both chromosomal abnormalities.
Clinical Report
Our patient is a 36-year-old Caucasian male who presented for genetic evaluation and an unbanded karyotype analysis performed over 35 years ago showing an abnormal elongated chromosome 16. Briefly, the patient was the product of an unremarkable full-term pregnancy, weighing 4.27 kg (90th percentile) with a length of 54.6 cm (95th percentile). His developmental milestones were delayed during infancy requiring therapeutic services and interventions. He was later diagnosed with severe intellectual disability requiring special services throughout childhood into adulthood. Other features noted were autism, obsessive compulsive disorder, mild mid-frequency hearing loss of the right ear, and involuntary facial, motor, and vocal tics consistent with the diagnosis of Tourette syndrome. He was placed in special education classes at an early age and graduated from high school. He then attended and graduated from an extended education program that teaches life skills. His receptive and comprehension skills are better than his expressive skills by history. He has very good memory recall. He continues to learn and to gain life skills.
His health concerns during childhood and later in adulthood included colitis, hypochromic microcytic anemia, subclinical hypothyroidism, hypoglycemia, seizures, flat feet, and thoracic scoliosis and lumbar lordosis. He reportedly had a tall, slender appearance with low muscle tone and hypersensitivity to loud noises. There were no cardiac, liver, genitourinary, or kidney concerns.
During the physical examination at 36 years of age, he was a pleasant, tall Caucasian male with limited speech; a long, narrow face; medial flaring of broad eyebrows; downslanting palpebral fissures; maxillary flatness; large protruding/prominent ears; a narrow, high-arched palate; mild pectus excavatum with a small, assymetric rib cage and narrow anterior-posterior chest diameter; thoracic scoliosis and lumbar lordosis; tapering fingers with increased mobility; flat feet; bilateral hallux valgus, and mottled soft skin. Frontal and profile facial views of the patient are seen in figure 1a. He had generalized hypotonia but with normal coordination. No edema or tenderness, leg length asymmetry, transverse palmar creases, cutaneous lesions, abdominal masses, or respiratory distress were present. His height was 180 cm (70th percentile), weight was 63.19 kg (25th percentile), head circumference was 53.0 cm (2nd percentile), inner canthal distance was 3.1 cm (60th percentile), outer canthal distance was 8.0 cm (10th percentile), palpebral fissure length was 2.6 cm (25th percentile), ear length was 6.8 cm (85th percentile), total hand length was 20.9 cm (>97th percentile), and middle finger length was 8.3 cm (97th percentile). Full frontal and profile views of the patient are seen in figure 1b, and a view of his hands in figure 1c. The patient’s family history was unremarkable. An X-ray scoliosis survey (2 views) was obtained and showed an S-shaped thoracic scoliosis with a 29° right-sided thoracic scoliosis centered over the T7–T8 level and a 22° left-sided upper thoracic scoliosis. There was forward inclination of the thoracic and lumbar spine with reversal of thoracic kyphosis and moderate cervical kyphosis (fig. 1d). His serum iron and ferretin levels were normal. His complete blood count showed low hemoglobin, MCV, MCH, and MCHC levels and a high RDW level, while other complete blood count levels were within normal range. His glucose level was low, but his liver function, thyroid and lipid levels were normal.
Fig. 1.
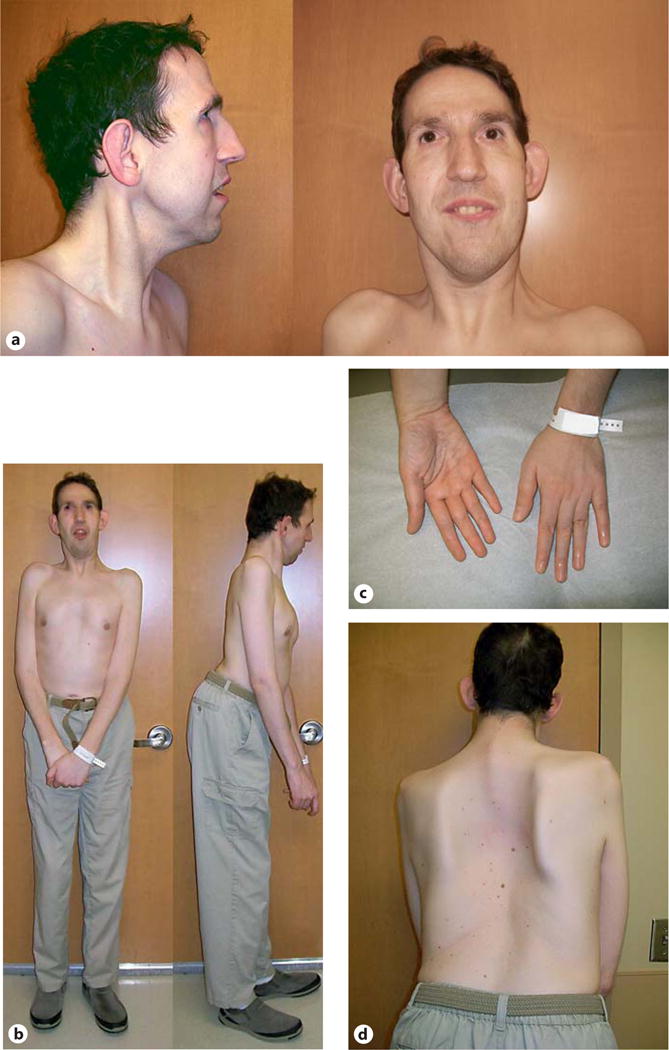
Multiple views (a–d) of our male patient at 36 years of age with distal partial trisomy 15q26 and partial monosomy 16p13.3.
Cytogenetic and Molecular Studies
Due to the history of an abnormal chromosome 16 finding based on early cytogenetic testing and dysmorphic features, a genome-wide copy number analysis was undertaken using an Illumina single nucleotide polymorphism (SNP) microarray containing >845,000 SNP markers, covering both coding and noncoding human genome sequences (CombiMatrix Diagnostics Laboratory, Irvine, Calif., USA). Genomic imbalances were reported using UCSC hg19 (NCBI build 37, Feb. 2009), and the array showed a 7.8-Mb duplication involving chromosome region 15q26.2q26.3 (hg19 coordinates chr15: 94,710,928–102,531,392) and a 1.9-Mb deletion involving chromosome region 16p13.3 (hg19 coordinates chr16: 0–1,930,412). The 15q26.2q26.3 duplication contained 41 genes, including IGF1R, SYNM, MEF2A, ADAMTS17, CERS3, ALDH1A3, and CHSY1 (fig. 2). The 16p13.3 deletion contained 88 genes, including HBA2, HBA1, AXIN1, CCDC78, LMF1, SOX8, SSTR5, CACNA1H, BAIAP3, CLCN7, IFT140, and IGFALS (fig. 3).
Fig. 2.
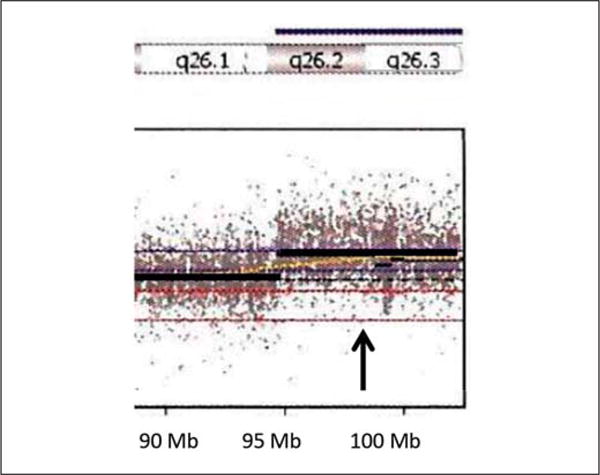
Duplication (arrow) of chromosome 15q26.2q26.3 (94,710,928–102,531,392 bp) found with microarray analysis. Genes included within this duplication are MCTP2, LOC400456, LOC145820, NR2F2, MIR1469, SPATA8, LOC91948, ARRDC4, FAM169B, MIR4714, IGF1R, PGPEP1L, SYNM, TTC23, HSP90B2P, LRRC28, MEF2A, LYSMD4, DNM1P46, ADAMTS17, FLJ42289, CERS3, PRKXP1, LINS, ASB7, ALDH1A3, LRRK1, CHSY1, VIMP, SNRPA1, LOC100507472, PCSK6, TM2D3, TARSK2, OR4F6, OR4F15, OR4F13P, OR4F4, FAM138E, WASH3P, and DDX11L9.
Fig. 3.
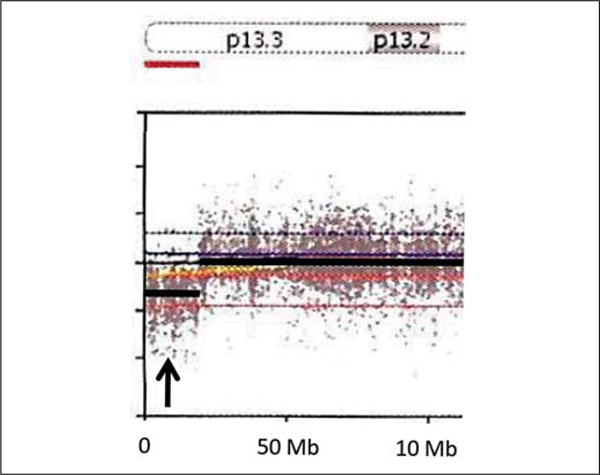
Deletion (arrow) of chromosome 16p13.3 (0–1,930,412 bp) found with microarray analysis. Genes included within this deletion are DDX11L10, POLR3K, SNRNP25, RHBDF1, MPG, NPRL3, HBZ, HBM, HBA2, HBA1, BHQ1, LUC7L, ITFG3, RGS11, ARHGDIG, MIR5587, SOLH, MIR3176, C16orf11, NHLRC4, PIGQ, RAB40C, WFIKKN1, C16orf13, FAM195A, WDR90, RHOT2, RHBCL1, STUB1, JMJD8, WDR24, FBXL16, METRN, FAM173A, CCDC78, HAGHL, NARFL, MSLN, MIR662, RPUSD1, CHTF18, GNG13, PRR25, LMF1, SOX8, SSTR5-AS1, SSTR5, C1QTNF8, CACNA1H, TPSG1, TPSB2, TPSAB1, TPSD1, UBE21, BAIAP3, TSR3, GNPTG, UNKL, C16orf91, CCDC154, CLCN7, PTX4, TELO2, TMEM204, IFT140, CRAMP1L, HN1L, MIR3177, MAPK8IP3, NME3, MRPS34, EME2, SPSB3, NUBP2, IGFALS, HAGH, FAHD1, MEIOB, and LINC00254.
Based on the microarray results, FISH was performed on synchronized peripheral blood cells using homebrew BAC DNA probes specific to 15q26.3 (RP11-123O9) and 16p13.3 (CTD-2608C14) with 16p11.2 (RP11-301D18) as a control in 2 separate assays (CombiMatrix Diagnostics Laboratory). Metaphase and interphase FISH analyses were consistent with an unbalanced derivative chromosome 16 arising from the translocation of the terminal long arm of chromosome 15 at 15q26.2 to the terminal short arm of chromosome 16 at 16p13.3, leading to partial trisomy 15q26 and partial monosomy 16p13.3. A normal chromosome 16 and a homologous pair of chromosome 15 were observed (fig. 4). These chromosome results would be consistent with the abnormal chromosome findings reported by the mother and performed over 35 years ago which identified extra material of unknown origin on chromosome 16. Chromosome studies on the mother were normal, but the father was not available for testing.
Fig. 4.
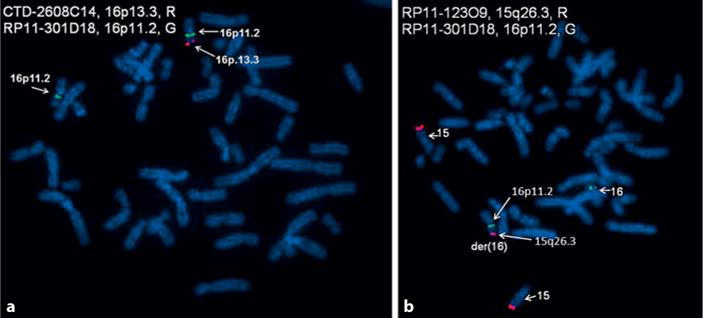
FISH metaphase images from our patient. a FISH image showing the deleted region of 16p13.3 with loss of the 16p13.3 probe on the derivative chromosome 16. b FISH image showing 3 copies of a probe for the 15q26.2q26.3 region and partial trisomy 15q26.
Discussion
Chromosomal translocations are a known cause of genetic disorders and can result in a syndromic condition, as seen in our 36-year-old male with intellectual disability, microcytic hypochromic anemia, seizures, low muscle tone, small rib cage/upper body cavity, scoliosis, and dysmorphic facial features. The patient’s duplicated 15q genetic material included the IGF1R gene; his deleted 16p region of genetic material included the SSTR5 and IGFALS genes. All 3 of these genes are known to be involved in growth. The IGF1R gene encodes the insulin-like growth factor 1 receptor required for normal response to the growth hormone, while SSTR5 encodes the somatostatin receptor 5. Autosomal dominant mutations in SSTR5 are associated with acromegaly that is resistant to the somatostatin analog treatment (OMIM 102200) [Ballarè et al., 2001]. The IGFALS gene encodes the insulin-like growth factor-binding protein, an acid-labile sub-unit with mutations associated with delayed growth and puberty (OMIM 601489). Our patient was 182 cm tall, putting him at the 70th percentile for height, but his marked S-shaped thoracic scoliosis and lumbar lordosis would impact his stature. Hence, our patient had a unique combination of a duplicated IGF1R gene from chromosome 15 and deleted SSTR5 and IGFALS genes affecting growth and stature from chromosome 16 along with the alpha hemoglobin genes. The duplicated IGF1R and deleted SSTR5 genes would have likely produced tall stature [Faivre et al., 2002]; however, the deleted IGFALS gene may have mediated this effect.
Our patient had a duplication of the CHSY1 gene; the loss is known to be associated with brachydactyly as well as autosomal recessive Temtamy preaxial brachydactyly syndrome (OMIM 605282). He had an increased total hand length of 20.9 cm (>97th percentile) and middle finger length of 8.3 cm (97th percentile). There is lack of current literature about the effects of a duplication of the CHSY1 gene on hand length, but a duplication may contribute to increased hand and middle finger size. A duplication of the IGF1R gene may also have contributed to hand size as well.
Our patient’s chromosome 16p13.3 deletion included the region involved with alpha-thalassemia mental retardation syndrome type 1 or alpha-thalassemia/mental retardation syndrome chromosome 16 related (ATR-16 syndrome) [Gibson et al., 2008]. The alpha-thalassemia mental retardation syndrome is due to a contiguous 16p13.3pter gene deletion and presents with variable clinical features that are nonspecific. The phenotype of this contiguous gene deletion is very similar to that of X-linked alpha thalassemia/mental retardation syndrome (ATR-X). The chromosome 16p13 deletion apparently accounts for our patient’s hypochromic microcytic anemia and probably contributes to his intellectual disability, low muscle tone and body habitus. Gibson et al. [2008] reported the following clinical features associated with a 16p13.3 deletion: low muscle tone, a high forehead, hypertelorism, a broad or prominent nasal bridge, and talipes equinovarus. Several of these features were seen in our patient. He also had hemoglobin genes deleted including HBZ, HBM, HBA2, HBA1, and HBQ1 involved in hemoglobin zeta, hemoglobin mu, Heinz body anemias, alpha thalassemia, hemoglobin H, alpha hemoglobin locus 2 and 1, and hemoglobin theta. However, hemoglobin electrophoresis was normal.
The CCDC78 gene was also deleted and associated with autosomal dominant centronuclear myopathy type 4 (OMIM 614807). Majczenko et al. [2012] described this syndrome as a myopathy with preserved ambulation, mild cognitive involvement, and no involvement of the facial, cardiac, or respiratory musculature. He had low muscle tone noted at birth.
Variants of the CACNA1H gene are associated with susceptibility to idiopathic generalized seizures (OMIM 611942). This gene is within the deleted region in our patient and may be associated with his seizures, which reportedly first occurred at 20 years of age. Heterozygous mutations of the IFT140 gene are also associated with short-rib thoracic dysplasia 9 with or without polydactyly (OMIM 266920). Our patient’s deletion included this gene, and he was observed to have a small rib cage during examination.
There were multiple other genes within our patient’s distal partial trisomy 15q26, including NR2F2, involved with heart development [Al Turki et al., 2014]; SPATA8, associated with spermatogenesis associated protein [Nie and Xiang, 2006]; SYNM, which may play a role in muscle integrity [Mizuno et al., 2001]; MEF2A, associated with autosomal dominant coronary artery disease (OMIM 608320); ADAMTS17, associated with autosomal recessive Weill-Marchesani-like syndrome (OMIM 613195); CERS3, associated with autosomal recessive congenital ichthyosis (OMIM 615023); LINS, encoding a regulator of the WNT pathway [Akawi et al., 2013]; ASB7 gene, which is predicted to be involved in protein degradation [Kile et al., 2002]; ALDH1A3, associated with autosomal recessive isolated microphthalmia (OMIM 615113); LRRK1, which undergoes intramolecular autophosphorylation [Korr et al., 2006]; VIMP, possibly involved in insulin resistance [Gao et al., 2003]; SNRPA1, required for cell division [Kittler et al., 2004], and TM2D3, which contains several features of a G protein-coupled receptor. There were multiple other genes within our patient’s distal partial monosomy 16p13.3, including AXIN1, associated with autosomal dominant hepatocellular carcinoma (OMIM 114550) and caudal duplication anomaly (OMIM 607864); LMF1, associated with autosomal recessive combined lipase deficiency (OMIM 246650); GNPTG, associated with autosomal recessive mucolipidosis III (OMIM 252605); CLCN7, associated with autosomal dominant and recessive osteopetrosis (OMIM 166600, OMIM 611490); HAGH, associated with autosomal dominant glyoxylase II deficiency [Valentine et al., 1970], and NME3 which may be involved in hematopoiesis [Venturelli et al., 1995]. These additional genes related to the partial trisomy 15q26 and partial monosomy 16p13.3 may or may not be related to our patient’s phenotype. As many genes are involved in our patient’s unbalanced translocation, we are unable to determine what role these genes may play in his phenotype. We are also unable to determine any potential interaction between genes which may also play a role in our patient’s phenotype.
Conclusion
We found this clinical case worthy of reporting, as of this date, there are no reports regarding a derivative chromosome 16 due to an unbalanced translocation involving the distal chromosome 15q. Here, we present the unique phenotype represented by this derivative chromosome 16 affecting several important genes for not only growth and physical development and cognition, but also for hematological disorders and hemoblobinopathies. The authors would encourage the report of other similarly affected individuals to further characterize the clinical outcomes and presentations due to distal partial trisomy 15q and partial monosomy 16p13.3.
References
- Akawi NA, Al-Jasmi F, Al-Shamsi AM, Ali BR, Al-Gazali L. LINS, a modulator of the WNT signaling pathway, is involved in human cognition. Orphanet J Rare Dis. 2013;8:87. doi: 10.1186/1750-1172-8-87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al Turki S, Manickaraj AK, Mercer CL, Gerety SS, Hitz MP, et al. Rare variants in NR2F2 cause congenital heart defects in humans. Am J Hum Genet. 2014;94:574–585. doi: 10.1016/j.ajhg.2014.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballarè E, Persani L, Lania AG, Filopanti M, Giammona E, et al. Mutation of somatostatin receptor type 5 in an acromegalic patient resistant to somatostatin analog treatment. J Clin Endocrinol Metab. 2001;86:3809–3814. doi: 10.1210/jcem.86.8.7787. [DOI] [PubMed] [Google Scholar]
- Blennow E, Telenius K, de Vos D, Larsson C, Henriksson P, et al. Tetrasomy 15q: two marker chromosomes with no detectable alpha-satellite DNA. Am J Hum Genet. 1994;54:877–883. [PMC free article] [PubMed] [Google Scholar]
- Chen CP, Lin YH, Au HK, Su YN, Hsu CY, et al. Chromosome 15q overgrowth syndrome: prenatal diagnosis, molecular cytogenetic characterization, and perinatal findings in a fetus with dup(15)(q26.2q26.3) Taiwan J Obstet Gynecol. 2011;50:359–365. doi: 10.1016/j.tjog.2011.07.004. [DOI] [PubMed] [Google Scholar]
- Christofolini DM, Piazzon FB, Evo C, Mafra FA, Cosenza SR, et al. Complex small supernumerary marker chromosome with a 15p/16p duplication: clinical implications. Mol Cytogenet. 2014;7:29. doi: 10.1186/1755-8166-7-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faivre L, Gosset P, Cormier-Daire V, Odent S, Amiel J, et al. Overgrowth and trisomy 15q26.1-qter including the IGF1 receptor gene: report of two families and review of the literature. Eur J Hum Genet. 2002;10:699–706. doi: 10.1038/sj.ejhg.5200879. [DOI] [PubMed] [Google Scholar]
- Gao Y, Walder K, Sunderland T, Kantham L, Feng HC, et al. Elevation in Tanis expression alters glucose metabolism and insulin sensitivity in H4IIE cells. Diabetes. 2003;52:929–934. doi: 10.2337/diabetes.52.4.929. [DOI] [PubMed] [Google Scholar]
- Gibson WT, Harvard C, Qiao Y, Somerville MJ, Lewis ME, Rajcan-Separovic E. Phenotype-gentoype characterization of alpha-thalassemia mental retardation syndrome due to isolated monosomy of 16p13.3. Am J Med Genet A. 2008;146A:225–232. doi: 10.1002/ajmg.a.32056. [DOI] [PubMed] [Google Scholar]
- Jones KL. Duplication 15q syndrome. In: Jones KL, editor. Smith’s Recognizable Patterns of Human Malformation. 6. Elsevier Saunders; Philadelphia: 2006. pp. 58–59. [Google Scholar]
- Kile BT, Schulman BA, Alexander WS, Nicola NA, Martin HME, Hilton DJ. The SOCS box: a tale of destruction and degradation. Trends Biochem Sci. 2002;27:235–241. doi: 10.1016/s0968-0004(02)02085-6. [DOI] [PubMed] [Google Scholar]
- Kittler R, Putz G, Pelletier L, Poser I, Heninger AK, et al. An endoribonuclease-prepared siR-NA screen in human cells identifies genes essential for cell division. Nature. 2004;432:1036–1040. doi: 10.1038/nature03159. [DOI] [PubMed] [Google Scholar]
- Korr D, Toschi L, Donner P, Pohlenz HD, Kreft B, Weiss B. LRRK1 protein kinase activity is stimulated upon binding of GTP to its Roc domain. Cell Signal. 2006;18:910–920. doi: 10.1016/j.cellsig.2005.08.015. [DOI] [PubMed] [Google Scholar]
- Majczenko K, Davidson AE, Camelo-Piragua S, Agrawal PB, Manfready RA, et al. Dominant mutation of CCDC78 in a unique congenital myopathy with prominent internal nuclei and atypical cores. Am J Hum Genet. 2012;91:365–371. doi: 10.1016/j.ajhg.2012.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizuno Y, Thompson TG, Guyon JR, Lidov HGW, Brosius M, et al. Desmuslin, an inter-mediate filament protein that interacts with alpha-dystrobrevin and desmin. Proc Natl Acad Sci USA. 2001;98:6156–6161. doi: 10.1073/pnas.111153298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson M, Quinonez S, Ackley T, Lyer RK, Innis JW. Multiple congenital anomalies and developmental delay in a boy associated with a de novo 16p13.3 deletion. Am J Hum Genet A. 2010;155A:612–617. doi: 10.1002/ajmg.a.33808. [DOI] [PubMed] [Google Scholar]
- Nie D, Xiang Y. Molecular cloning and characterization of a novel human testis-specific gene by use of digital differential display. J Genet. 2006;85:57–62. doi: 10.1007/BF02728971. [DOI] [PubMed] [Google Scholar]
- Rowe AG, Abrams L, Qu Y, Chen E, Cotter PD. Tetrasomy 15q25 → qter: cytogenetic and molecular characterization of an analphoid supernumerary marker chromosome. Am J Med Genet. 2000;93:393–398. [PubMed] [Google Scholar]
- Su PH, Chen JY, Chen SJ. Siblings with deletion 22q13.3 and trisomy 15q26 inherited from a maternally balanced translocation. Pediatr Neonatol. 2010;52:287–289. doi: 10.1016/j.pedneo.2011.06.008. [DOI] [PubMed] [Google Scholar]
- Valentine WN, Paglia DE, Neerhout RC, Konrad PN. Erythrocyte glyoxalase II deficiency with coincidental hereditary elliptocytosis. Blood. 1970;36:797–808. [PubMed] [Google Scholar]
- Venturelli D, Martinez R, Melotti P, Casella I, Peschle C, et al. Overexpression of DR-nm23, a protein encoded by a member of the nm23 gene family, inhibits granulocyte differentiation and induces apoptosis in 32Dcl3 myeloid cells. Proc Natl Acad Sci USA. 1995;92:7435–7439. doi: 10.1073/pnas.92.16.7435. [DOI] [PMC free article] [PubMed] [Google Scholar]