

Abstract
Activation of AKT signaling by PTEN loss or PIK3CA mutations occurs frequently in human cancers, but targeting AKT has been difficult due to the mechanism-based toxicities of inhibitors that target the inactive conformation of AKT. Ipatasertib (GDC-0068) is a novel selective ATP-competitive small-molecule inhibitor of AKT that preferentially targets active phosphorylated AKT (pAKT) and is potent in cell lines with evidence of AKT activation. In this phase I study, ipatasertib was well tolerated; most adverse events were gastrointestinal and grade 1–2 in severity. The exposures of ipatasertib ≥200 mg daily in patients correlated with preclinical TGI90, and pharmacodynamic studies confirmed that multiple targets (i.e., PRAS40, GSK3β, and mTOR) were inhibited in paired on-treatment biopsies. Preliminary antitumor activity was observed; 16 of 52 patients (30%), with diverse solid tumors and who progressed on prior therapies, had radiographic stable disease, and many of their tumors had activation of AKT.
INTRODUCTION
AKT is one of the most frequently activated protein kinases in human cancers, and it plays a critical role in tumor growth, proliferation, survival, and resistance to anticancer therapy (1–5). AKT is the central node of the PI3K–AKT–mTOR signaling pathway and is encoded by three closely related genes: AKT1, AKT2, and AKT3. The AKT signaling pathway can be activated by multiple mechanisms in cancers, including loss of the tumor suppressor PTEN, activating mutations in AKT or in the p110α subunit of PI3K, PIK3CA, and/or increased receptor tyrosine kinase signaling. Taken together, these data provide a strong rationale for developing therapeutics to target the AKT pathway in human cancers.
AKT kinase function is activated following phosphorylation of two residues, threonine 308 (Thr308) and serine 473 (Ser473). Prior to phosphorylation, AKT assumes a pleckstrin homology (PH)–in conformation, which prevents access to and binding of ATP. Following phosphorylation, AKT adopts a catalytically active PH-out conformation, where the ATP-binding pocket is accessible. The mechanism of action of AKT inhibitors has focused on this conformational change, by being either allo steric (non–ATP-competitive), binding AKT in its inactive state, or ATP-competitive (active-site–directed), targeting the phosphorylated conformation (6). A phase I study with an allosteric inhibitor in patients with cancer reported that the most common adverse events (AE) were rash, fatigue, nausea, vomiting, diarrhea, and hyperglycemia (7). Given these toxicities, the preferential targeting of AKT by selective ATP-competitive AKT inhibitors may increase the therapeutic index by targeting tumors which presumably have more frequent AKT activation or a constitutively active form of AKT, while sparing normal cells that may have lower AKT activity.
Ipatasertib (GDC-0068; Genentech, Inc.) is a potent, novel, selective, ATP-competitive small-molecule inhibitor of all three isoforms of AKT (IC50 of 5–18 nmol/L; refs. 8, 9). Ipatasertib has a greater than 100-fold selectivity over other relevant kinases, including a greater than 600-fold selectivity over protein kinase A. Consistent with its mechanism, ipatasertib is potent both in preclinical cancer cell lines and in xenograft models with activation of AKT, including tumors with complete loss of PTEN (PTEN-null), decreased expression of PTEN, or mutations in PIK3CA (9). Because AKT is a central node in growth factor signaling, however, it was unclear whether AKT could be effectively targeted without causing serious toxicities in patients.
This phase I clinical study was designed to quantify the degree of AKT pathway suppression prior to observing dose-limiting toxicities (DLT) by measuring the level of pathway inhibition with doses of ipatasertib that are well tolerated by multiple methods, including reverse phase protein array (RPPA) from tumor biopsies, blood-based biomarkers, and fludeoxyglucose F18 ([18F]FDG)–positron emission tomography (PET) scans. The protocol-defined primary objectives of this phase I study were to evaluate the safety and tolerability of ipatasertib and to estimate the maximum tolerated dose (MTD). Additional objectives included characterization of ipatasertib pharmacokinetics (PK) and pharmacodynamics (PD) by (i) changes in the expression of AKT-related pathway biomarkers in paired tumor biopsies, (ii) changes in the levels of phosphorylated glycogen synthase kinase 3-beta (pGSK3β) in platelet-rich plasma (PRP), and (iii) tumor metabolic responses on PET scans.
RESULTS
Preclinical Studies
Preclinical studies suggested that high AKT activity predicts sensitivity to ipatasertib in cancer cells, especially in those with alterations in PTEN and/or PIK3CA (9). PTEN alterations are defined by PTEN mutations or protein loss (either complete loss or decreased expression) by Western blot analysis in cell lines or by IHC in patient-derived tumor xenografts. PIK3CA alterations are defined by mutations or amplifications. The efficacy of ipatasertib in cell-line models with or without alterations in PTEN or PIK3CA both in vitro (Supplementary Table S1) and in vivo in xenograft models (Supplementary Table S2) was evaluated. In vitro viability assays showed that cancer cell lines with PTEN loss or PIK3CA mutations are more likely to respond to ipatasertib, with lower IC50 values (mean 4.8 ± 0.56 μmol/L, median 2.2 μmol/L, n = 60) than those without alterations (mean 8.4 ± 0.48 μmol/L, median 10 μmol/L, n = 40, P < 0.0001; Fig. 1A). A significant difference is also observed when stratified using PTEN alterations alone, with lower IC50 values observed in cells with PTEN alterations (mean 3.8 ± 0.73 μmol/L, median 1.3 μmol/L, n = 33) than those without (mean 7.4 ± 0.46 μmol/L, median 10 μmol/L, n = 67; P < 0.0001). Similarly, when stratified with PIK3CA alone, lower IC50 values are also observed in cells with PIK3CA alterations (mean 5.0 ± 0.71 μmol/L, median 2.2 μmol/L, n = 40) than those without (mean 7.1 ± 0.5 μmol/L, median 10 μmol/L, n = 60; P < 0.05). Interestingly, several PIK3CA mutations that are present in the Single Nucleotide Polymorphism database (dbSNP) and have not been demonstrated to activate the PI3K/AKT pathway, such as I391M, did not associate with sensitivity to ipatasertib (Supplementary Table S1; ref. 9). In vivo xenografts implanted from cancer cell lines or patient-derived tumors also showed higher percentage tumor growth inhibition (%TGI) in models with alterations in PTEN or PIK3CA (mean %TGI 95 ± 11, median %TGI 97, n = 21) than those without (mean %TGI 38 ± 12, %TGI median 44, n = 6, P < 0.05; Fig. 1B). As an example, ipatasertib decreases the growth of a PTEN -null melanoma model (Fig. 1C) and a PIK3CA -mutant (H1047R) breast cancer model in a dose-dependent manner (Fig. 1D). As reported (9), ipatasertib is less effective in suppressing the growth of cell lines with RAS/RAF activation, as shown in a colon cancer xenograft with a KRASG13D mutation despite a PIK3CAH1047R mutation (Fig. 1E). Therefore, these preclinical studies suggest that patients with tumors having PTEN loss or PIK3CA mutations may respond to ipatasertib and, further, that the co-occurrence of mutations that activate the MAPK pathway may also serve as important biomarkers that can be used to further define a set of predictive biomarkers for ipatasertib activity.
Figure 1.
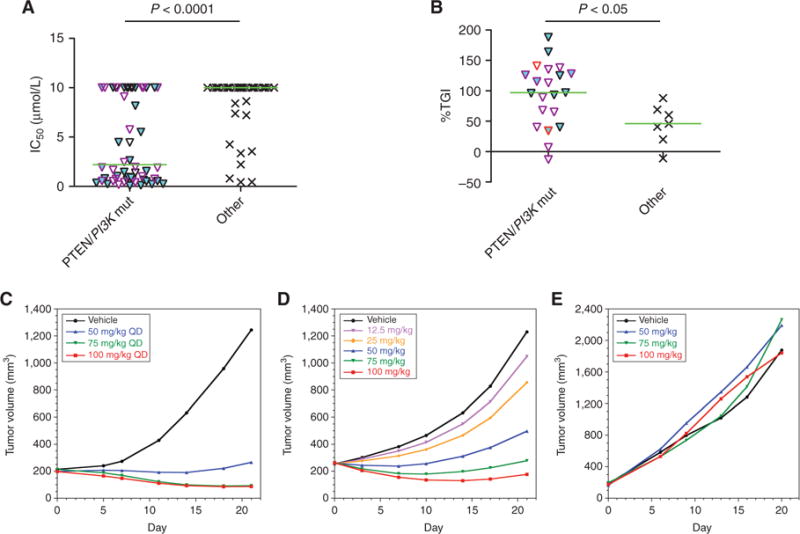
Preclinical cancer cell line and xenograft models with alterations in PTEN or PIK3CA are significantly more sensitive to ipatasertib both in vitro and in vivo. A, IC50 values of ipatasertib on cell viability in cancer cell lines with alterations in PTEN, including loss of expression, or in PIK3CA, including mutations, are plotted against those cell lines without known alterations in these genes. Cell lines with PTEN alterations are indicated with a cyan fill, whereas those with PIK3CA mutations that are found in the Catalogue of Somatic Mutations in Cancer (COSMIC) but not in the Single Nucleotide Polymorphism database (dbSNP) are indicated with a purple border. Cells with no PIK3CA mutations, or with PIK3CA mutations that are either not found in COSMIC or are present in the dbSNP, have black borders. Median values of each group are indicated as a green horizontal line, and P values are calculated by the Student t test between indicated groups. Ipatasertib treatment in cell lines with PTEN loss or PIK3CA mutations showed a significantly lower mean and median IC50 values (mean IC50 ±SEM 4.8 ± 0.56 μmol/L and median IC50 2.2 μmol/L) than those without known alterations (mean IC50 8.4 ± 0.48 μmol/L and median IC50 10 μmol/L). B, Cancer cell lines or patient-derived tumors were implanted into immunocompromised mice as xenograft models, and percentage tumor growth inhibition (%TGI) at day 21 after daily oral treatment with 100 mg/kg ipatasertib was calculated as described in Methods. The %TGI in xenograft models with alterations in PTEN (cyan fill) or PIK3CA (purple border for mutations, red border for amplifications) is significantly higher (mean %TGI ± SEM 95 ± 11% and median %TGI 97%) than in those models without alterations (mean %TGI 38 ± 12% and median %TGI 44%). C, Ipatasertib decreases the growth in vivo of the melanoma cell line 537Mel (PTEN -null) in a dose-dependent manner, as measured by mixed modeling that analyzes the repeated measurements of tumor volumes from the same animals over time. This method of analysis addresses both repeated measurements and modest dropout rates due to non–treatment-related discontinuation prior to study end. QD, once daily. D, Ipatasertib decreases the growth in vivo of the breast cancer cell line KPL4, which has a PIK3CAH1047R mutation. E, Ipatasertib does not suppress the growth of the colon cancer cell line HCT116, which has a KRASG13D mutation despite having a PIK3CAH1047R mutation.
Clinical Trial Patients
Fifty-two patients were enrolled in this three-stage study (April 2010 to May 2012): 30 patients in dose-escalation (stage 1) and 22 patients in the two dose-expansion cohorts of tumor-specific indications [n = 11 metastatic breast cancer (MBC), n = 5 metastatic castration-resistant prostate cancer (CRPC), stage 2] or all solid tumors (n = 6, stage 3). The median age was 57 years (range, 32–76 years), and the majority were female (n = 30, 58%; Table 1). Most patients had an Eastern Cooperative Oncology Group (ECOG) status of 1 (n = 32, 61%), and common cancer diagnoses were MBC (n = 16, 31%) or colorectal cancer (n = 14, 27%). The patients were extensively pretreated, with a median of 6 prior anticancer therapies (range, 1–17).
Table 1.
Patient characteristics at baseline
| All patients (n = 52)a | |
|---|---|
| Age, years | |
| Median | 57 |
| Range | 32–76 |
| Sex, n (%) | |
| Female | 30 (58) |
| Male | 22 (42) |
| ECOG PS, n (%) | |
| 0 | 20 (38) |
| 1 | 32 (62) |
| Primary tumor type, n (%) | |
| Breast | 16 (31) |
| Colorectal | 14 (27) |
| Prostate | 6 (12) |
| Chondrosarcoma | 2 (4) |
| Ovarian | 2 (4) |
| Other | 12 (22) |
| Prior systemic therapies | |
| Median | 6 |
| Range | 1–17 |
Abbreviations: n, number of patients; PS, performance status.
Fifty-two patients were enrolled in the study, but one enrolled patient was not treated with ipatasertib; therefore, a total of 51 patients were treated with ipatasertib.
PTEN status by IHC was obtained in the archival tumors from 37 of 52 patients (71%). The majority of tumors had decreased PTEN, as the H-score was <200 in 22 of 37 patients (59%), and 6 patients (16%) had tumors with complete PTEN loss (H-score 0). Forty-two of 52 patients (81%) had evaluable archival tumor samples for assessment of PIK3CA or AKT mutations. There were 6 PIK3CA mutations in the 42 samples (14%; H1047R mutation, n = 3; n = 1 for E545K, E542K, or R88Q), but only one AKT1 mutation (E17K; Supplementary Table S3).
As of February 2, 2015, all patients had discontinued treatment, primarily for disease progression (n = 42, 81%; Supplementary Table S4). The other reasons for discontinuation were patient withdrawal (n = 6, 12%), AEs (n = 2), physician decision (n = 1), and death due to disease progression (n = 1). Of the two patients who discontinued for AEs, one patient in the 600-mg cohort discontinued for grade 2 nausea and grade 1 vomiting, and the other patient in the 800-mg cohort discontinued for grade 3 asthenia, which was a DLT (discussed in the next section).
Determination of the MTD
Patients received ipatasertib orally (PO) once daily (QD) for 21 days continuously followed by 7 days off (21/7 dosing schedule) every 28 days at doses of 25 mg (n = 3), 50 mg (n = 3), 100 mg (n = 3), 200 mg (n = 3), 400 mg (n = 3), and 800 mg (n = 7). Two patients at the 800-mg dose experienced two DLTs (grade 3 asthenia, n = 1; and grade 3 nausea, n = 1). Both the grade 3 asthenia and the grade 3 nausea improved and/or resolved following holding of ipatasertib and initiation of supportive medications. Because of the DLTs, eight patients were then enrolled at 600 mg. No DLTs were observed, and no patients dose-reduced ipatasertib for the first cycle. Therefore, the MTD for ipatasertib was 600 mg PO QD on a 21/7 dosing schedule. Subsequent patients were enrolled in the expansion cohorts at the MTD of 600 mg.
Safety
Forty-seven of 51 treated patients (92%), across all doses, had at least one AE of any grade related to ipatasertib, as assessed by investigators (Supplementary Table S5). The most frequently reported grade ≥ 2 AEs related to ipatasertib were diarrhea (35%), nausea (27%), asthenia (25%), hyperglycemia (10%), decreased appetite (6%), rash (6%), and vomiting (6%; Table 2). The majority of AEs were grade 1–2 in severity and could be managed with supportive care and/or dose holds, such that patients could continue ipatasertib.
Table 2.
All grade ≥2 ipatasertib-related AEs in >5% of patients
| Number (%) of patients by ipatasertib daily dosea | ||||||||
|---|---|---|---|---|---|---|---|---|
| Adverse event | 25 mg (n = 3) |
50 mg (n = 3) |
100 mg (n = 3) |
200 mg (n = 3) |
400 mg (n = 3) |
600 mg (n = 29) |
800 mg (n = 7) |
All subjects (n = 51) |
| Any AEb,c | 0 | 2 (67) | 1 (33) | 1 (33) | 2 (67) | 21 (72) | 7 (100) | 34 (67) |
| Diarrhea | 0 | 0 | 0 | 0 | 0 | 14 (48) | 4 (57) | 18 (35) |
| Nausea | 0 | 0 | 0 | 0 | 1 (33) | 7 (24) | 6 (86) | 14 (27) |
| Asthenia | 0 | 2 (67) | 1 (33) | 1 (33) | 0 | 5 (17) | 4 (57) | 13 (25) |
| Hyperglycemia | 0 | 0 | 0 | 0 | 0 | 3 (10) | 2 (29) | 5 (10) |
| Decreased appetite | 0 | 0 | 0 | 0 | 0 | 2 (7) | 1 (14) | 3 (6) |
| Rash | 0 | 0 | 0 | 0 | 0 | 1 (3) | 2 (29) | 3 (6) |
| Vomiting | 0 | 0 | 0 | 0 | 0 | 1 (3) | 2 (29) | 3 (6) |
Abbreviation: n, number of patients.
Patients are categorized by initial dose cohort assignment, regardless of subsequent dose modification.
Grade 3 AEs that were assessed by the investigators as related to ipatasertib were diarrhea (n = 4 at 600 mg, 8%), asthenia (n = 3 at 600 mg and 800 mg, 6%), hypercholesterolemia (n = 1 at 100 mg, 2%), hyperglycemia (n = 1 at 600 mg, 2%), hypophosphatemia (n = 1 at 600 mg, 2%), nausea (n =1 at 800 mg, 2%), and toxic skin eruption (n = 1 at 600 mg, 2%).
Serious adverse events that were assessed by the investigators as related to ipatasertib were grade 3 asthenia (n = 1 at 800 mg, 2%), grade 3 diarrhea (n = 1 at 600 mg, 2%), grade 3 hyperglycemia (n = 1 at 600 mg, 2%), grade 3 toxic skin eruption (n = 1 at 600 mg, 2%), and grade 2 renal failure (n = 1 at 600 mg, 2%).
Grade 3 AEs that were assessed by the investigators as related to ipatasertib occurred at doses ≥ 600 mg: diarrhea (n = 4, 600 mg), asthenia (n = 1, 600 mg and n = 2, 800 mg), hypercholesterolemia (n = 1, 100 mg), hyperglycemia (n = 1, 600 mg), hypophosphatemia (n = 1, 600 mg), nausea (n = 1, 800 mg), and toxic skin eruption (n = 1, 600 mg; Supplementary Table S6). Most grade 3 AEs were managed with supportive care. The grade 3 hypercholesterolemia reversed following a cholesterol-lowering medication (simvastatin), and the patient continued ipatasertib. The grade 3 toxic skin eruption resolved following holding of ipatasertib. There were no grade 4 AEs or deaths related to ipatasertib. Five serious adverse events (SAE) related to ipatasertib were seen, including grade 3 asthenia (n = 1, 800 mg), grade 3 diarrhea (n = 1, 600 mg), grade 3 hyperglycemia (n = 1, 600 mg), grade 3 toxic skin eruption (n = 1, 600 mg), and grade 2 renal failure (n = 1, 600 mg). Two of 51 patients (4%) discontinued for ipatasertib-related AEs (grade 3 asthenia; grade 2 nausea and grade 1 vomiting). Eight patients (16%) experienced AEs that led to dose reduction (most frequently due to diarrhea or asthenia, in dose cohorts of 600 mg and 800 mg) and remained on study until disease progression. No cases of colitis or pneumonitis were reported.
Pharmacokinetics
The exposures seen in patients were compared with those observed at the efficacious doses from preclinical models to determine if the same efficacious exposures could be achieved in patients (Supplementary Fig. S1). At steady state, ipatasertib as low as 200 mg (3.52 μmol/L*hour) resulted in over 90% TGI (TGI90) in preclinical xenograft models with PTEN -null status, including LNCaP prostate cancer, U87MG glioma, and HGC-27 gastric cancer (9) cell lines. Therefore, the clinical PK data demonstrated that potentially efficacious exposures could be achieved in patients.
PK data from 30 patients treated with ipatasertib during dose escalation (25–800 mg) showed that ipatasertib was rapidly absorbed, with median time to peak concentration (tmax) ranging from 0.5 to 3 hours. Mean terminal half-life ranged from 31.9 to 53 hours at doses >100 mg (Table 3); at 600 mg, mean half-life was 45.8 hours (range, 36.7–55.0 hours). Mean accumulation ratio (AUC0–24,ss/AUC0–24, day 1) of ipatasertib at doses of 200 mg to 800 mg ranged from 1.80 to 2.44 (Table 3). A metabolite of ipatasertib, G-037720, was also evaluated in this study. Based on preclinical studies, metabolite G-037720 was expected to be a main metabolite for ipatasertib. Median tmax for this metabolite ranged from 0.5 to 3 hours. G-037720 had a mean half-life of 28.8 to 49.4 hours at ipatasertib ≥ 100 mg, which is similar to ipatasertib, indicating that this metabolite is formation-rate limited. At the MTD of 600 mg, the mean metabolite ratio (metabolite to parent ratio; AUC0–24, ss, metabolite/AUC0–24, ss, parent) was 0.398 (44.5% coefficient of variation).
Table 3.
Mean PK of ipatasertib following a single dose (day 1) and multiple doses at steady state (day 15)
| Day 1 | Day 15 | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Analyte | Dose | tmax (h) | Cmax (ng/mL) | AUC0–24 (ng • h/mL) | t1/2 (h) | tmax (h) | Cmax (ng/mL) | AUC0–24,ss (ng • h/mL) | Accumulation ratio |
| Ipatasertib | 25 mg (n = 3) |
0.50 (0.5–3.08) |
8.93 (112%) |
31.8 (93.7%) |
4.46 (2.75–7.66) |
0.5 (0.50–3.07) |
8.92 (111%) |
88.3 (77.8%) |
2.78 (15.7%) |
| 50 mg (n = 3) |
2.92 (2.00–2.98) |
13.3 (46.0%) |
70.3 (41.1%) |
10.1 (8.08–15.3) |
1.00 (0.50–3.00) |
33.9 (68.7%) |
171 (11.0%) |
2.43 (32.1%) |
|
| 100 mg (n = 3/2)a |
2.97 (1.33–3.00) |
59.8 (21.4%) |
260 (16.6%) |
31.9 (29.9–36.2) |
0.500–0.67 | 76.2, 53.0 | 568, 239 | 2.57,0.927 | |
| 200 mg (n = 3) |
3.00 (1.00–3.00) |
130 (4.45%) |
726 (13.2%) |
39.1 (27.8–66.9) |
1.00 (0.50–2.00) |
183 (18.2%) |
1770 (14.6%) |
2.44 (28.1%) |
|
| 400 mg (n = 3) |
1.00 (0.50–1.00) |
506 (25.3%) |
1651 (15.1%) |
53.0 (45.2–64.3) |
1.00 (0.50–1.50) |
838 (50.2%) |
3210 (22.3%) |
1.95 (36.5%) |
|
| 600 mg (n = 7) |
0.58 (0.50–4.03) |
488 (41.4%) |
2670 (39.4%) |
45.8 (36.7–55.0) |
0.83 (0.50–2.12) |
748 (36.7%) |
4450 (43.8%) |
1.80 (73.6%) |
|
| 800 mg (n = 7/5)b |
2.00 (0.50–4.00) |
479 (79.9%) |
3220 (75.0%) |
43.8 (34.4–66.6) |
3.00 (0.50–5.00) |
879 (74.5%) |
6670 (66.1%) |
2.22 (20.0%) |
|
NOTE: All parameters are reported as geometric mean (%CV) except tmax, which is reported as median (range) and t1/2, which is reported as geometric mean (range). In the case of n = 2, individual values are reported.
Abbreviations: Accumulation ratio, AUC0–24,ss (day 15)/AUC0–24 (day 1); AUC0–24,ss, area under concentration–time curve from time 0 to 24 hours at steady state; AUC0–24, area under concentration–time curve from time 0 to 24 hours at single dose; Cmax, highest observed plasma concentration; CV, coefficient of variation; t1/2, terminal half-life; tmax, time to maximum concentration
Consists of 3 patients for day 1 and 2 patients for day 15.
Consists of 7 patients for day 1 and 5 patients for day 15.
An analysis of ipatasertib dose/exposure (AUC0–24,ss) to the grades of the four most common AEs related to ipatasertib (diarrhea, nausea, fatigue/asthenia, and rash) shows that the severity of these AEs increased relative to the dose and exposure of ipatasertib (Supplementary Fig. S2A). At an ipatasertib AUC ≤4000 ng*h/mL (corresponding to ipatasertib ≤400 mg), AEs were infrequent. Any observed AEs were mostly asthenia and nausea (grade 1–2). At an ipatasertib AUC ≥ 6,000 ng*h/mL (corresponding to doses ≥ 600 mg), the most common AEs increased in severity of grade, and rash became more prevalent. All grade 3 AEs occurred at ipatasertib ≥600 mg.
Time to the first onset of diarrhea, nausea, fatigue, and rash occurred during cycle 1 (Supplementary Fig. S2B). Nausea and diarrhea tended to occur within the first week, whereas fatigue and rash occurred within the first two weeks. Prolonged treatment with ipatasertib did not generally lead to the first onset of these AEs, as diarrhea or rash did not occur in patients after the first month, and only two patients developed fatigue or nausea after two months. Time to onset for the worst grade of these most common AEs also occurred early in cycle 1 (Supplementary Fig. S2C). Onset of the first grade 1–2 nausea or the first grade 1 diarrhea occurred within 7 days, whereas higher grades of nausea and diarrhea occurred within 14 days. The worst grade of fatigue (grade 3) and rash (grade 2) occurred slightly later at 7 to 14 days. Chronic dosing of ipatasertib did not lead to the higher grade AEs, as no grade 3 nausea or grade 2 rash was seen after cycle 1, and only 3 patients developed grade 2–3 fatigue or diarrhea after cycle 2. Therefore, these most common AEs were often manageable in cycle 1, and most resolved, which allowed patients to resume ipatasertib.
Pharmacodynamics
The PD of ipatasertib in patients were assessed via changes in the following AKT pathway markers: phosphorylated GSK3β (pGSK3β) in PRP; glucose, insulin, and c-peptide levels in serial blood samples; tumor metabolic responses on [18F]FDG-PET scans; and downstream AKT pathway components, including pPRAS40, pGSK3β, pmTOR, p4EBP1, and pS6, in on-treatment tumor biopsies versus pretreatment biopsies via RPPA.
Blood samples were obtained from patients at scheduled time points after ipatasertib on days 1 and 15 of cycle 1 and were evaluated for pGSK3β and total GSK3β in PRP. Levels of pGSK3β decreased over time in PRP samples, compared with GSK3β, and both a dose- and concentration-dependent pharmacologic response in relationship to ipatasertib occurred on days 1 (Fig. 2, top) and 15 (Fig. 2, bottom). pGSK3β reached a nadir at 2 hours and remained suppressed at 24 hours. At steady state of ipatasertib, checked at day 15 in cycle 1, pGSK3β had a sustained decrease. Overall, an analysis of exposure relative to pGSK3β inhibition shows ≥80% inhibition at ipatasertib ≥400 mg, and even at doses <100 mg, there is still >50% inhibition (Supplementary Fig. S3).
Figure 2.
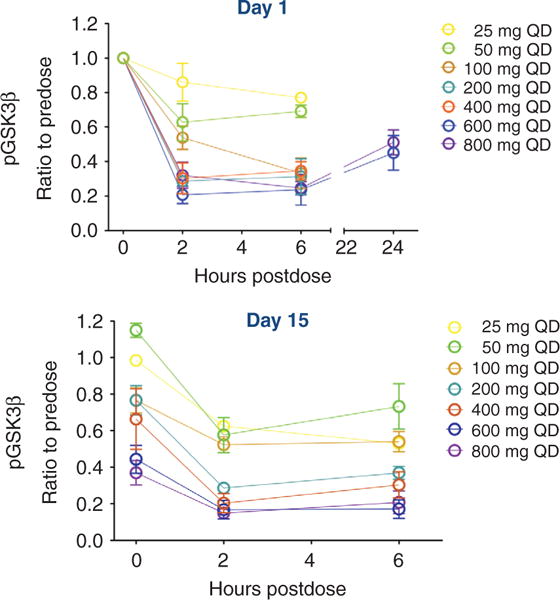
Ipatasertib shows evidence of PD inhibition of AKT signaling in patients as evaluated by the suppression of pGSK3β in PRP. The ratio of pGSK3β to total GSK3β was evaluated in PRP from patients in all the dosing cohorts (25–800 mg) on day 1 (top) and on day 15 (bottom) of cycle 1 and was graphed as a function of time (in hours) following a single dose of ipatasertib. In all patients, pGSK3β decreased in a time-and dose-dependent manner, reaching a nadir at 2 hours after dose and remaining suppressed at 24 hours after dose. Inhibition of pGSK3β was also sustained at day 15 of dosing (bottom).
AKT also plays a key role in glucose homeostasis and mediates the metabolic effects of insulin downstream of the insulin receptor. Blood samples from patients on days 1 and 15 of cycle 1 were evaluated for changes in the levels of insulin, glucose, and c-peptide. Almost all patients dosed with ipatasertib ≥200 mg had transient nonfasting (post-meal) asymptomatic hyperglycemia (Supplementary Fig. S4A). At day 15, there was a mild dose-dependent glucose elevation, which began at 100 mg. Increases of glucose by 50 mg/dL were considered physiologic based on patients dosed in the expansion cohort with midazolam as a substrate (data not shown); therefore, increases by >50 mg/dL were supraphysiologic. Beginning at 600 mg, higher glucoses can be observed, which increased further at 800 mg. Reciprocal increases in insulin were also seen, beginning at 100 mg (Supplementary Fig. S4B). Higher elevations in insulin, up to 500-fold, were seen with ipatasertib ≥600 mg.
Although glucose and insulin levels increased following ipatasertib, the elevations were transient, and patients were asymptomatic; the levels generally returned to baseline within 6 hours. A representative plot of glucose and insulin over time in one patient dosed with ipatasertib 600 mg is shown (Supplementary Fig. S4C, green line). For all patients in stage 1, ipatasertib for two cycles did not significantly elevate baseline glucoses (Supplementary Fig. S4D). Glycated hemoglobin A1c (HbA1c) obtained at the end of study did not change significantly when compared with baseline at screening, including patients at 800 mg (Supplementary Table S7). HbA1c remained within the normal range independent of the dose or duration of ipatasertib, even in patients treated for a year (data not shown).
Because blood-based PK and PD assessments decreased AKT signaling by approximately 50% and confirmed that the TGI90 exposures from preclinical models can be achieved at doses ≥200 mg (from Supplementary Fig. S1), pretreatment and on-treatment tumor biopsies were then collected from consenting patients to evaluate the effect of ipatasertib on multiple AKT pathway effectors, as measured by RPPA. Consistent with the mechanism of action of targeting pAKT, the binding of ATP-competitive ipatasertib stabilized the pAKT in a conformation in which the phosphorylated residues are inaccessible to phosphatases (6), ipatasertib caused a dose-dependent accumulation of pAKT (Thr308 and Ser473) in the biopsies (Fig. 3A). As a result of ipatasertib binding and locking the pAKT in the nonfunctional state, the expression of multiple targets (i.e., pPRAS40, p4EBP1, pS6, and pmTOR) was downregulated in a dose-dependent manner, beginning at 100 mg (Fig. 3A and B). In addition, RPPA analysis in the tumor biopsies showed upregulation of pERK and pHER3, two targets outside the AKT pathway (Fig. 3A and C), suggesting that a compensatory feedback mechanism may be activated following ipatasertib.
Figure 3.
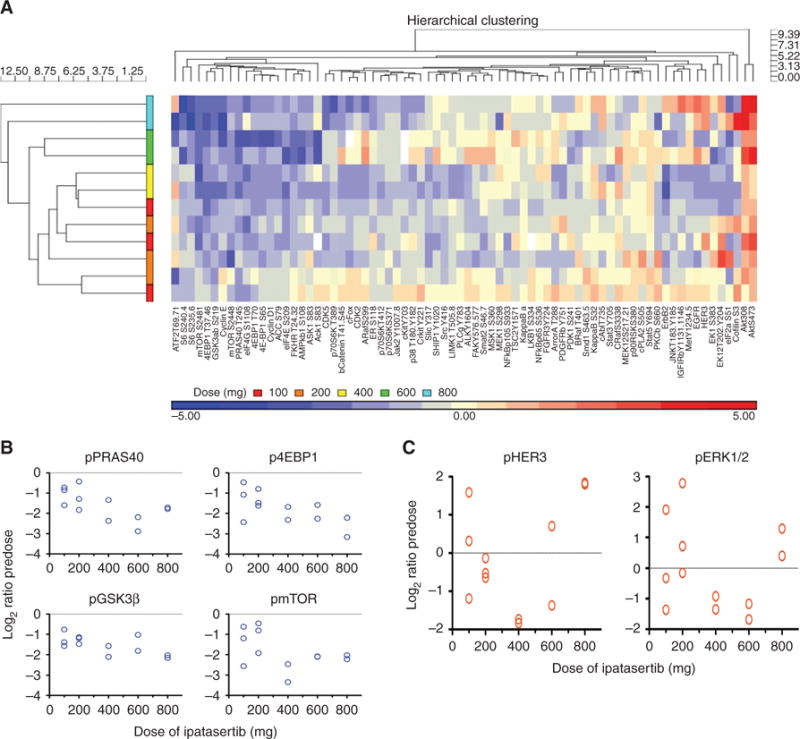
Downregulation of AKT pathway targets by ipatasertib in on-treatment tumor biopsies compared with pretreatment biopsies by RPPA analysis. A, On-treatment tumor biopsies from consenting patients were evaluated by RPPA for multiple AKT pathway targets, including pAKT, pPRAS40, p4EBP1, pS6, and pERK. Protein expression of the target from the on-treatment biopsy was compared with the pretreatment biopsy to determine if ipatasertib caused upregulation (marked in red, up to 5-fold increase as shown in the legend along the bottom), downregulation (blue, up to 5-fold), or no significant changes (yellow). Each horizontal row shows a patient, ranked by increasing doses of ipatasertib from 100 mg (bottom) to 800 mg (top). The targets were organized by hierarchical clustering of expression levels. Inhibition of AKT with ipatasertib resulted in a dose-dependent accumulation of pAKT (Thr308 and Ser473, highlighted in red, right side), consistent with its mechanism of action. Consequently, ipatasertib downregulated multiple targets of AKT, including pPRAS40, p4EBP1, pS6, and pmTOR (highlighted in blue, left side). Downregulation of these targets occurred in a dose-dependent manner, as a greater degree of target inhibition occurred with ipatasertib ≥400 mg. A compensatory feedback mechanism following ipatasertib also occurred, as upregulation of pERK and pHER3 (highlighted in red, right side) was observed with ipatasertib as low as 100 mg. B, As an example, ipatasertib downregulated four targets of AKT: pPRAS40, pGSK3β, p4EBP1, and pmTOR, in the on-treatment biopsy compared with the pretreatment biopsy (expressed as log 2 ratio with each circle representing a patient). This inhibition can be observed at doses of ipatasertib as low as 100 mg, and the degree of inhibition increases in a dose-dependent manner. C, As an example, AKT inhibition by ipatasertib modulated two signaling pathways, pHER3 and pERK, in the on-treatment biopsy compared with the pretreatment biopsy. These modulations are directionally heterogeneous across the doses; upregulation can be observed at doses of ipatasertib as low as 100 mg.
To evaluate PD across all metastatic disease in a cohort of patients with MBC, [18F]FDG-PET scans were conducted at baseline and days 22 to 28 of cycles 1 and 2. Ten patients with MBC with PI3K/AKT activation in archival tumors based on site information were dosed with ipatasertib 600 mg, and 8 of 9 (89%) had a target lesion metabolic PET response by modified European Organisation for Research and Treatment of Cancer (EORTC) criteria (10), defined by SUVmax decreases ≥20% in cycle 1, after being on ipatasertib for two weeks (Fig. 4A). One patient did not have a metabolic response but had discontinued ipatasertib 10 days prior to the scan in cycle 1 and thus was excluded from the analysis. Based on central pathology testing, most patients with target lesion metabolic PET responses also had archival tumors with PI3K/AKT activation, including at least one of the following: loss of PTEN (n = 1, H-score 0), decreased PTEN (n = 5; H-scores 20, 100, 115, 165, and 200), PIK3CA mutations (n = 3, H1047R, E545K, or E542K), and AKT1 mutation (n = 1, E17K). One patient with MBC with an AKT1E17K mutation had a complete metabolic PET response (Fig. 4B) as described previously (11).
Figure 4.
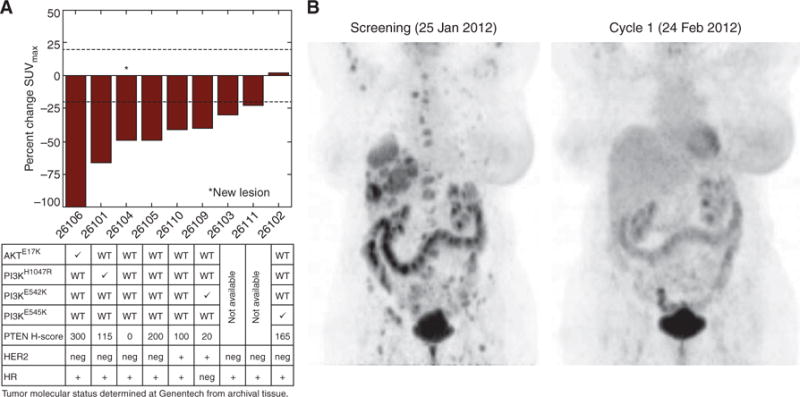
Patients with MBC with tumors having PTEN loss and/or PIK3CA/AKT mutations showed target lesion metabolic responses by [18 F]FDG-PET following ipatasertib. A, Nine patients with MBC, with archival tumors having activation of the PI3K/AKT pathway as determined by the investigator, were dosed with ipatasertib 600 mg and had [18F]FDG-PET scans done at screening and in cycle 1 after two weeks. The maximum percentage change in SUV between the on-treatment and pretreatment PET scans (SUVmax) in target lesions was then graphed for each patient, ranked by order of response. Eight of the nine evaluable patients (89%) had a metabolic PET response, as defined by an SUVmax decrease ≥20% in the target lesions. One patient (ID# 26102) did not have a PET response, but this patient also stopped the study drug in cycle 1 at the time of the PET scan. Most of the patients with metabolic PET responses also had archival tumors with activation of the PI3K/AKT pathway, including decreased PTEN expression (n = 6), PIK3CA mutations (n = 3), and an AKT1 mutation (n = 1), as determined centrally. WT, wild-type. B, One patient with MBC (HER2-negative) harboring an AKT1E17K mutation in the archival tumor sample exhibited a complete metabolic response at cycles 1 and 2. All FDG-avid target lesions identified at baseline were indistinguishable from background on the on-treatment PET scans (% SUVmax decrease set to 100%).
Antitumor Activity
Across all dosing cohorts for ipatasertib (25–800 mg), 16 of 47 patients (34%) who had post-treatment tumor assessments had a best overall response of stable disease (SD) or incomplete response (IR) as assessed by the investigators by Response Evaluation Criteria in Solid Tumors (RECIST) criteria, version 1.0; and six patients (8.5%) had investigator-assessed progression-free survival greater than 6 months (which is 18% of patients in the 400 mg and 600 mg cohorts who had SD/IR >24 weeks; Supplementary Table S8). At the MTD of 600 mg for ipatasertib, 11 of 25 patients (44%) had a best overall response of SD/IR. SD/IR was observed in patients diagnosed with a wide range of cancers, including colon, ovarian, prostate, breast (both HER2-positive and HER2-negative), and lung cancers, and chondrosarcoma. Most of these patients were heavily pretreated, with a median number of 4 prior lines of therapy (including adjuvant and/or metastatic treatment).
Activation of AKT signaling by complete loss of PTEN (H-score of 0) or by mutations of PIK3CA or AKT in the archival tumors also tended to correlate with stable radiographic criteria for patients treated with ipatasertib. Of the 52 patients in the study, the PIK3CA and/or AKT mutation status of the archival tumors was known for 42 patients (81%), as the others did not have available samples (Supplementary Table S3). Of the 41 patients who had post-treatment tumor assessments and known tumor molecular status, radiographic SD as best response was observed in 6 of the 9 patients (67%) who had complete loss of PTEN, PIK3CA, or AKT mutations in their tumor, whereas 3 of the 9 patients (33%) had progressive disease as best response. For patients without alterations, 10 of the 32 patients (31%) had SD, and 22 of 32 (69%) had progressive disease as best response. Radiographic SD was not observed in patients who had known KRAS mutations (G12V/D/A or Q61L, n = 7). The PTEN status from archival tumors was known for 11 of 16 patients (69%) who had a best response of SD/IR, and the PIK3CA and AKT1 mutation status was known for 13 of the 16 (81%) patients. Molecular analysis revealed that 10 of 16 patients (63%) who had a best response of SD/IR had tumors with alterations in the PI3K/AKT pathway, including complete PTEN loss (n = 3, 30%), decreased PTEN with H-score between 1 and 200 (median H-score 132, n = 6, 60%), PIK3CA mutation (H1047R, n = 2, 20%), or AKT1 mutation (E17K, n = 1, 10%). For patients who did not have SD/IR tumor responses, the PTEN status from archival tumors was known for 23 patients and the PIK3CA and AKT1 mutation status was known for 25 patients: complete PTEN loss (n = 1, 4%), decreased PTEN (median H-score 140, n = 13, 56%), PIK3CA mutations (H1047R or E545K, n = 2, 8%), AKT1 mutation (0%), and KRAS mutations (G12V/D/A or Q61L, n = 7, 28%; Supplementary Table S8). Decreased PTEN expression in archival samples was not mutually exclusive from PIK3CA and/or AKT mutations and was also not limited to specific tumors.
Time on study in relationship to activation of AKT signaling was then examined. The mean time on study for 12 of 42 patients (29%) with evaluable archival samples who were considered to have AKT diagnostic-positive tumors (Dx+, defined by complete loss of PTEN, and PIK3CA or AKT1 mutations), was 185 days (standard deviation 292 days; median 75; range, 21–1,072). In contrast, the mean time on study for the 30 patients who did not have AKT diagnostic-positive tumors was 71 days (standard deviation 49 days; median 63; range, 23–245). The presence of KRAS or NRAS mutations, however, correlated with decreased time on study. Nine patients had KRAS or NRAS mutations, and their mean time on study was 51 days (standard deviation 21 days; median 57; range, 23–84) compared with a mean time on study of 116 days (standard deviation 193 days; median 64; range, 21–1,072) for the patients without such mutations.
Antitumor activity of ipatasertib was observed in particular in two patients. In the first example, a 57-year-old male patient with metastatic CRPC with bone metastases, who had progressed on bicalutamide after 600 days prior to study entry, remained on study with ipatasertib 600 mg for 1,072 days. His prostate-specific antigen (PSA) doubling time improved from 1.62 months (prior to study) to 14.66 months while on study. This patient also tolerated ipatasertib 600 mg, and, of note, his archival prostate cancer had complete loss of PTEN. In the second example, a 68-year-old female patient with MBC (HER2-negative), who had progressed on chemotherapy (nonpegylated liposomal doxorubicin, cyclophosphamide, and most recently capecitabine for 90 days prior to study), had a complete metabolic PET response in cycle 1 (Fig. 4B; ref. 11). Her CA15-3 tumor marker declined by more than 50%, and she remained on study for 235 days. This patient’s archival tumor had an AKT1E17K mutation.
DISCUSSION
AKT is a central node in the signaling cascade of multiple receptor tyrosine kinases in both cancer cells and normal cells, so it was unclear at the onset of this study whether AKT could be effectively targeted without significant toxicities. A prior phase I study of an allosteric AKT inhibitor was limited by multiple toxicities, predominantly rash (7). In contrast, this first-in-human study reports that an ATP-competitive AKT inhibitor, ipatasertib, was generally well tolerated. In addition to tolerability at relatively high exposures of ipatasertib up to and including the MTD of 600 mg, patients exhibited multiple lines of evidence for AKT pathway knockdown at ipatasertib ≥100 mg. Collectively, these data demonstrate that AKT can be inhibited safely in patients with an ATP-competitive inhibitor.
The degree of PD inhibition in the AKT pathway in blood-and tumor-based biomarkers following ipatasertib was quantified. Blood-based PD biomarkers (including pGSK3β, glucose, insulin, and c-peptide) showed PD knockdown with ipatasertib, starting even at a lower dose of 100 mg. Furthermore, the clinical exposure of ipatasertib at 100 mg was equivalent to TGI 60 in the most sensitive preclinical models. The results of pretreatment and on-treatment biopsies also confirmed that ipatasertib, beginning at 100 mg, downregulated multiple AKT effectors, including pPRAS40, pGSK3β, pS6, and p4EBP. In addition, RPPA data showed evidence of feedback upregulation of compensatory pathways (HER3 and ERK) in response to ipatasertib (12). These data represent the first characterization in clinical samples that an ATP-competitive AKT inhibitor can cause broad AKT pathway inhibition, which may result in feedback upregulation.
In terms of safety, the predominant ipatasertib-related AEs were diarrhea, nausea, asthenia/fatigue, and rash. Evaluation of ipatasertib exposure/dose with severity of the most common related AEs confirmed that ipatasertib from 100 mg to 400 mg was well tolerated, as most AEs were grade 1–2 in severity with a low prevalence of grade 3. Most AEs occurred early in cycle 1 and could be managed or resolved with supportive care, such that many patients continued ipatasertib. Overall, ipatasertib was not associated with life-threatening toxicities, such as pneumonitis or cytopenias, which have been observed with other inhibitors of the AKT/PI3K/mTOR pathway. The incidence of clinically significant hyperglycemia or rash is low with ipatasertib compared with other PI3K (13) or allosteric AKT inhibitors (7). The reduced toxicity may be attributed to the ability of ipatasertib to selectively inhibit the active signaling conformation of AKT, which may be more prevalent in cancer cells, while sparing normal cells that may not be dependent on constitutive AKT activation. Alternatively, incomplete target inhibition may also have impact on efficacy in association with reduced toxicity. Nevertheless, the safety and activity of ipatasertib may translate into an increased therapeutic window when compared with the safety profile of other AKT inhibitors (nonselective for activated states) at doses with similar antitumor activity. Importantly, the efficacy in the diagnostic-positive patients as defined by PTEN loss and/or PIK3CA/AKT mutations may be enhanced without increased toxicity. A higher percentage of patients in the study stage 3 who are either Dx+ MBC or CRPC (∼60% patients had PTEN loss) had SD and prolonged time on study.
Although treatment with ipatasertib was associated with an approximately >80% inhibition of multiple AKT targets resulting in [18F]FDG-PET metabolic responses (including a complete metabolic response), no radiographic RECIST responses were observed, as the best response was prolonged SD. Patients who had SD on ipatasertib also tended to have activation of AKT signaling in their archival tumors. Patients with AKT- or PIK3CA -mutant tumors (n = 7) also tended to stay on study longer with ipatasertib when compared with those patients without mutations (n = 33), with median time on study of 84 versus 63 days, respectively. Interpreting time on study in patients with and without AKT pathway alterations and RAS mutations is challenging in such a heterogeneous population. Certainly, the enrichment of PIK3CA mutations in breast cancer (typically ER + cancers, which tend to have an indolent natural history) and the enrichment of RAS mutations in colon cancer could skew the results. Collectively, 28 patients had tumors with PI3K/AKT pathway alterations, including complete PTEN loss, decreased PTEN (H-score 1–200), and PIK3CA or AKT mutations; however, none of these patients had radiographic RECIST responses. Lack of substantial single-agent RECIST radiographic activity has also been reported for other in-class inhibitors, including AKT (7, 14) or pan-PI3K (15, 16). In contrast, isoform-selective PI3K inhibitors, including PI3K-alpha-selective in solid tumors or PI3K-delta-selective in hematologic malignancies, have shown single-agent RECIST responses, even at low doses (13, 17).
There may be four possible hypotheses for why ipatasertib did not result in significant objective tumor shrinkage. First, this phase I study enrolled patients with diagnostic-positive and diagnostic-negative tumors, and an insufficient number of patients with diagnostic-positive tumors may have enrolled to adequately assess a RECIST response rate. Although about 80% of patients had evaluable archival samples, only 6 tumors had PIK3CA mutations and only one tumor had an AKT1 mutation. Second, mutational activation of the PI3K/AKT pathway may not cause classic “oncogene addiction,” compared with other activating mutations. Third, more substantial AKT pathway inhibition (for example, >80% pathway knockdown) may be required for RECIST radiographic responses. Finally, even though ipatasertib inhibited AKT signaling, pathway feedback and cross-talk may have limited any responses. Such feedback mechanisms have also been hypothesized to have limited the antitumor activity of an allosteric AKT inhibitor (7), and our preclinical studies have shown that enhanced HER3 and ERK stimulation via feedback limits the in vitro activity of PI3K/AKT inhibitors (12).
To circumvent this compensatory feedback, we hypothesize that an appropriate combination strategy with ipatasertib and hormonal therapy, chemotherapy, or targeted agents will need to be explored. The identification of key feedback pathways in this study has led to rational future combination approaches that are currently being evaluated in phase Ib/II studies with ipatasertib. For example, upregulation of the RAS pathway following ipatasertib suggests that coadministration of ipata-sertib with inhibitors of the RAS/MEK pathway may be more effective. In conclusion, because of its favorable safety profile and therapeutic index, ipatasertib may be a good combination partner with multiple anticancer agents, and ipatasertib is currently being evaluated in phase Ib/II studies in metastatic prostate, gastric, and breast cancers.
METHODS
Preclinical Studies
Cell viability assays were performed as described (9). Plates (384-well) were seeded with 2,000 cells/well/54 μL and incubated at 37°C under 5% CO2 for approximately 16 hours. Ipatasertib was diluted in dimethyl sulfoxide and added 6 μL/well in quadruplicate. After incubation for 4 days, viable cells were estimated using CellTiter-Glo (Promega), and total luminescence was measured on a Wallac Multilabel Reader (PerkinElmer). The concentration of drug resulting in IC50 was calculated from a 4-parameter curve analysis (XLfit, IDBS) and was determined from a minimum of 3 experiments. For cell lines that failed to achieve an IC50, the highest tested concentration (10 μmol/L) was used.
In vivo efficacy was evaluated in tumor cell line and patient-derived xenograft models as described (9). All in vivo studies were reviewed and approved by Genentech’s Institutional Animal Care and Use Committee (IACUC). Cells or tumor fragments were implanted subcutaneously into the flanks of female nude (nu/nu) or SCID/beige mice (Charles River Laboratories or Taconic), and the tumors were monitored until they reached mean volumes of 150 to 350 mm3. Ipatasertib was formulated in 0.5% methylcellulose/0.2% Tween-80 (MCT) and administered daily, via oral gavage. Tumor volumes were determined using digital calipers (Fred V. Fowler Company) using the formula (L × W × W)/2. To appropriately analyze the repeated measurements of tumor volumes from the same animals, a mixed-modeling approach was used. Percentage tumor growth inhibition was calculated as a percentage of AUC for the dose group per day in relation to vehicle, such that %TGI = 100 × [1 − (AUCtreatment/d)/(AUCvehicle/d)]. Tumor sizes and body weights were recorded twice weekly. Mice with tumor volumes ≥2,000 mm3 or losses in body weight ≥20% were euthanized per IACUC guidelines.
Clinical Trial Design and Conduct
This open-label phase I clinical study used a 3 + 3 dose-escalation design to evaluate increasing oral doses of ipatasertib and occurred at two centers in Spain (Vall d’Hebron, Barcelona, and University of Valencia) from November 2009 to January 2015 (Trial registration ID: NCT 01090960). The study was conducted in accordance with good clinical practice guidelines, applicable laws, and regulations, and in accordance with Declaration of Helsinki. Regulatory authorities and institutional review boards of each site approved the study, and all subjects provided written informed consent prior to enrollment. The trial was designed by the academic authors in collaboration with the sponsor, Genentech, Inc., and trial medications were provided by the sponsor.
Eligibility Criteria
Eligible subjects were adult patients ≥18 years of age with incurable, locally advanced or metastatic solid malignancies that had progressed or failed to respond to at least one prior regimen or for which standard therapy either does not exist or has proven ineffective or intolerable. There was no limit to the number or types of prior treatments. Subjects had to have evaluable or measurable disease per RECIST version 1.0, although patients with prostate cancer with nonmeasurable disease were eligible if they had two rising PSA levels that met PSA Working Group criteria for progression (18), and patients with ovarian cancer with nonmeasurable disease were eligible if they had two rising cancer antigen-125 (CA-125) tumor marker levels. All patients had to have ECOG performance status of 0 or 1 at screening and had to have acceptable renal, hepatic, and bone marrow function based on screening studies within 14 days prior to dosing. Patients were not eligible if they had undergone anticancer treatment within 4 weeks prior to study, including chemotherapy, hormonal therapy (except hormone replacement or gonadotropin-release hormone agonists for prostate cancer), immunotherapy, biologic therapy, radiation therapy (except palliative radiation to bony metastases), or herbal therapy as cancer therapy. Additional exclusion criteria included known untreated malignancies of the brain, HIV infection, history of type 1 or 2 diabetes requiring insulin (patients with diabetes on a stable dose of oral diabetes medications were allowed to enroll), grade ≥2 hypercholesterolemia or hypertriglyceridemia, or other comorbidities that may render the patient at high risk from complications.
Treatments
This study enrolled patients in three stages: stage 1 consisted of all patients in dose escalation, stage 2 was expansion cohort of patients with metastatic CRPC or diagnostic-positive MBC as defined by PTEN loss and/or PIK3CA/AKT mutations, and stage 3 consisted of a second expansion cohort of all solid tumors. After a 28-day screening period, patients received ipatasertib orally once daily for 21 consecutive days, followed by 7 days off (21/7 dosing schedule), during a 28-day cycle. In stage 1, patients were given a dose of ipatasertib on day 1, followed by PK measurements 7 days later. Ipatasertib dosing then resumed on day 8 with daily oral dosing on a 21/7 dosing schedule; thus, the first cycle for DLT assessment was 35 days, whereas all remaining cycles were 28 days. The starting dose of ipatasertib in stage 1 was 25 mg, and the dose was doubled for each cohort, unless a safety signal triggered a modified Fibonacci design or slower dose escalations.
At least three evaluable subjects were enrolled in each cohort. DLTs were evaluated during the first cycle in stage 1 (days 1 to 35), with severity assessed according to NCI Common Terminology Criteria for Adverse Events version 3.0, and included the following AEs assessed by investigators as related to ipatasertib: Grade ≥3 nonhematologic, nonhepatic organ system, nonmetabolic (hyperglycemia and hyperlipidemia) toxicity excluding alopecia and grade 3 diarrhea, nausea, or vomiting that responds to standard-of-care therapy; grade ≥4 thrombocytopenia; grade ≥4 neutropenia lasting >5 days or accompanied by fever; fasting grade ≥4 hyperglycemia or two episodes of fasting grade 3 hyperglycemia within 7 days despite an oral antidiabetic medication for ≥2 weeks; drug interruption of three or more doses for drug-related hyperglycemia during the DLT window; grade ≥4 fasting hypercholesterolemia or hypertriglyceridemia for ≥2 weeks despite a lipid-lowering agent; and grade ≥3 serum bilirubin or hepatic transaminases or values < 5 times the upper limit of normal due to known liver metastases. The MTD was defined as the highest dose at which fewer than one-third of patients in a cohort (e.g., <2 of 6) experienced a DLT. Dose expansions then occurred at the MTD for all patients in stages 2 and 3.
Safety assessments were conducted at screening, weekly during cycle 1 (on days 1, 8, 15, 22, and 29), every 2 weeks during cycles 2 through 6 (days 1 and 15), and every cycle beyond cycle 7 (day 1). Assessments included monitoring and recording AEs and SAEs and measuring hematology, clinical chemistry, and urinalysis variables. All measurable disease was assessed at screening, at the end of cycle 2, and at every even-numbered cycle thereafter.
PK and PD Assessments
Plasma levels for ipatasertib and its metabolite (G-037720) were measured from blood samples collected at predose and at 0.5, 1, 2, 3, 4, 6, and 24 hours after dose on days 1 and 15 of cycle 1. Blood samples were centrifuged, and plasma aliquots were stored at −20°C. The concentration of ipatasertib and its metabolite was analyzed in plasma using validated LC/MS-MS, and PK parameters were determined using a noncompartmental method (WinNonlin 5.2.1; Pharsight).
Archival tumor samples were requested from all patients. PTEN status was assessed centrally at Genentech by an IHC assay as described (12). Briefly, samples were fixed and stained with hematoxylin and eosin and were then stained with a primary rabbit monoclonal anti-PTEN antibody (Cell Signaling Technology, Cat # 9559). The absence or presence of PTEN was then assessed by IHC, when compared with expression in surrounding stroma, and the value was recorded as an H-score ranging from 0 (complete loss) to 400 (full expression). The PIK3CA or AKT mutation status of the archival tumor was assessed centrally via the TaqMan genotyping assay, as described (19).
Tumor biopsies in consenting patients were conducted at baseline and cycle 1 (days 15–21) for PD assessments after two criteria were met: (i) PK data confirmed that greater than the requisite TGI60 exposures from preclinical models were achieved, and (ii) ≥50% change in a blood-based PD biomarker for AKT was achieved. At ipatasertib ≥100 mg, three consenting patients per cohort had tumor biopsies, and no complications were observed. Needle core biopsies were snap-frozen and evaluated by RPPA for AKT pathway effectors, including pAKT, pGSK3β, pPRAS40, pmTOR, p4EBP1, pS6, and pERK (12).
Blood samples were obtained at scheduled time points after ipatasertib on days 1 and 15 for an analysis of pGSK3β using an electrochemiluminescence assay (Luminex xMPA; Luminex Corporation). The pGSK3β level was normalized to total GSK3β protein in each PRP sample, and fraction inhibition of pGSK3β was measured relative to day 1 baseline (12). The levels of glucose and insulin were also evaluated in blood samples.
Whole-body [18F]FDG-PET scans were performed at baseline and days 22 to 28 of cycles 1 and 2 for 10 patients with MBC treated with 600 mg ipatasertib in stage 2. Modified from EORTC guidelines (10), an on-treatment PET metabolic response was defined as average percentage decrease in maximum standardized uptake value (SUVmax) in target lesions ≥20% relative to pretreatment.
Evaluation of Tumor Response
Tumor response was assessed radiographically by RECIST version 1.0 criteria following every 2 cycles of treatment (or approximately every 2 months). Patients who were deriving clinical benefit were allowed to continue on study if they had not developed a DLT, unacceptable toxicity, or disease progression.
Statistical Analysis
All 52 patients were included in the analyses. PK evaluations used actual sampling times and doses, and plasma concentration–time data were analyzed using noncompartmental methods. PD assessments were conducted, with increases calculated for each marker relative to baseline. Radiographic responses were categorized per RECIST version 1.0 as complete response, partial response, stable disease, or progressive disease (20). Measurable baseline lesions were defined as lesions that could be measured in at least one dimension with a longest diameter ≥20 mm or ≥10 mm with a spiral computed tomography scan.
Supplementary Material
SIGNIFICANCE.
Potent inhibition of AKT signaling with ipatasertib was associated with a tolerable safety profile and meaningful disease control in a subgroup of patients. Targeting pAKT with an ATP-competitive inhibitor provides a greater therapeutic window than allosteric inhibitors. Further investigation with ipatasertib is ongoing in phase II studies.
Acknowledgments
The authors thank Hartmut Koeppen for assistance with the IHC analysis, Vikram Malhi and Yuzhing Deng for assistance with the clinical pharmacology analysis, and Marie Wagle for assistance with the biomarker analysis.
Grant Support
This study was funded by Genentech, Inc. (ClinicalTrials.gov trial registration ID: NCT01090960).
Footnotes
Note: Supplementary data for this article are available at Cancer Discovery Online (http://cancerdiscovery.aacrjournals.org/).
Disclosure of Potential Conflicts of Interest
N. Budha has ownership interest (including patents) in Roche. J. Zhu has ownership interest in Roche. W.Y. Chan is a stock holder of Genentech/Roche. Y. Yan has ownership interest (including patents) in Roche/Genentech. K. Lin has ownership interest (including patents) in Roche. J. Tabernero is a consultant/advisory board member for Amgen, Bayer, Boehringer Ingelheim, Celgene, Chugai, Genentech, Lilly, MSD, Merck Serono, Novartis, Pfizer, Roche, Sanofi, Symphogen, Taiho, and Takeda. No potential conflicts of interest were disclosed by the other authors.
One of the Editors-in-Chief is an author on this article. In keeping with the AACR’s editorial policy, the peer review of this submission was managed by a senior member of Cancer Discovery’s editorial team; a member of the AACR Publications Committee rendered the final decision concerning acceptability.
Disclaimer
The authors were fully responsible for all content and editorial decisions, were involved at all stage of manuscript development, and have approved the final version.
Authors’ Contributions
Conception and design: C. Saura, D. Roda, R. Morales-Barrera, L. Musib, K. Lin, Y. Yan, P. Patel, J. Baselga, J. Tabernero
Development of methodology: C. Saura, D. Roda, Y. Yan, P. Patel, J. Baselga, J. Tabernero
Acquisition of data (provided animals, acquired and managed patients, provided facilities, etc.): C. Saura, D. Roda, S. Roselló, M. Oliveira, T. Macarulla, J.A. Pérez-Fidalgo, R. Morales-Barrera, M. Nannini, Y. Yan, J. Tabernero, A. Cervantes
Analysis and interpretation of data (e.g., statistical analysis, biostatistics, computational analysis): C. Saura, D. Roda, L. Musib, N. Budha, J. Zhu, M. Nannini, W.Y. Chan, S.M. Sanabria Bohórquez, R.D. Meng, K. Lin, Y. Yan, P. Patel, J. Tabernero
Writing, review, and/or revision of the manuscript: C. Saura, D. Roda, S. Roselló, M. Oliveira, T. Macarulla, J.A. Pérez-Fidalgo, R. Morales-Barrera, L. Musib, J. Zhu, M. Nannini, W.Y. Chan, R.D. Meng, K. Lin, Y. Yan, P. Patel, J. Tabernero, A. Cervantes
Administrative, technical, or material support (i.e., reporting or organizing data, constructing databases): D. Roda, R. Morales-Barrera, L. Musib, W.Y. Chan, R.D. Meng, J. Tabernero
Study supervision: C. Saura, D. Roda, T. Macarulla, W.Y. Chan, R.D. Meng, P. Patel, J. Baselga
Other (co-investigator; interventional radiology procedures; performing serial biopsies): J.M. Sanchis-García
References
- 1.Bellacosa A, de Feo D, Godwin AK, Bell DW, Cheng JQ, Altomare DA, et al. Molecular alterations of the AKT2 oncogene in ovarian and breast carcinomas. Int J Cancer. 1995;64:280–5. doi: 10.1002/ijc.2910640412. [DOI] [PubMed] [Google Scholar]
- 2.Manning BD, Cantley LC. AKT/PKB signaling: navigating downstream. Cell. 2007;129:1261–74. doi: 10.1016/j.cell.2007.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jiang BH, Liu LZ. AKT signaling in regulating angiogenesis. Curr Cancer Drug Targets. 2008;8:19–26. doi: 10.2174/156800908783497122. [DOI] [PubMed] [Google Scholar]
- 4.Tokunaga E, Oki E, Egashira A, Sadanaga N, Morita M, Kakeji Y, et al. Deregulation of the Akt pathway in human cancer. Curr Cancer Drug Targets. 2008;8:27–36. doi: 10.2174/156800908783497140. [DOI] [PubMed] [Google Scholar]
- 5.Robey RB, Hay N. Is Akt the “Warburg kinase”?-Akt-energy metabolism interactions and oncogenesis. Semin Cancer Biol. 2009;19:25–31. doi: 10.1016/j.semcancer.2008.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lin K, Lin J, Wu WI, Ballard J, Lee BB, Gloor SL, et al. An ATP-site on-off switch that restricts phosphatase accessibility of Akt. Sci Signal. 2012;5:ra37. doi: 10.1126/scisignal.2002618. [DOI] [PubMed] [Google Scholar]
- 7.Yap TA, Yan L, Patnaik A, Fearen I, Olmos D, Papadopoulos K, et al. First-in-man clinical trial of the oral pan-AKT inhibitor MK-2206 in patients with advanced solid tumors. J Clin Oncol. 2011;29:4688–95. doi: 10.1200/JCO.2011.35.5263. [DOI] [PubMed] [Google Scholar]
- 8.Blake JF, Xu R, Bencsik JR, Xiao D, Kallan NC, Schlachter S, et al. Discovery and preclinical pharmacology of a selective ATP-competitive Akt inhibitor (GDC-0068) for the treatment of human tumors. J Med Chem. 2012;55:8110–27. doi: 10.1021/jm301024w. [DOI] [PubMed] [Google Scholar]
- 9.Lin J, Sampath D, Nannini MA, Lee BB, Degtyarev M, Oeh J, et al. Targeting activated Akt with GDC-0068, a novel selective Akt inhibitor that is efficacious in multiple tumor models. Clin Cancer Res. 2013;19:1760–72. doi: 10.1158/1078-0432.CCR-12-3072. [DOI] [PubMed] [Google Scholar]
- 10.Young H, Baum R, Cremerius U, Herholz K, Hoekstra O, Lammertsma AA, et al. Measurement of clinical and subclinical tumour response using [18F]fluorodeoxyglucose and positron emission tomography: review and 1999 EORTC recommendations. European Organization for Research and Treatment of Cancer (EORTC) PET Study Group. Eur J Cancer. 1999;35:1773–82. doi: 10.1016/s0959-8049(99)00229-4. [DOI] [PubMed] [Google Scholar]
- 11.De Mattos-Arruda L, Weigelt B, Cortes J, Won HH, Ng CK, Nuciforo P, et al. Capturing intra-tumor genetic heterogeneity by de novo mutation profiling of circulating cell-free tumor DNA: a proof-of-principle. Ann Oncol. 2014;25:1729–35. doi: 10.1093/annonc/mdu239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yan Y, Serra V, Prudkin L, Scaltriti M, Murli S, Rodriquez O, et al. Evaluation and clinical analyses of downstream targets of the Akt inhibitor GDC-0068. Clin Cancer Res. 2013;19:6976–86. doi: 10.1158/1078-0432.CCR-13-0978. [DOI] [PubMed] [Google Scholar]
- 13.Dienstmann R, Rodon J, Serra V, Tabernero J. Picking the point of inhibition: a comparative review of PI3K/AKT/mTOR pathway inhibitors. Mol Cancer Ther. 2014;13:1021–31. doi: 10.1158/1535-7163.MCT-13-0639. [DOI] [PubMed] [Google Scholar]
- 14.Yap TA, Yan L, Patnaik A, Tunariu N, Biondo A, Fearen I, et al. Interrogating two schedules of the AKT inhibitor MK-2206 in patients with advanced solid tumors incorporating novel pharmacodynamics and functional imaging biomarkers. Clin Cancer Res. 2014;20:5672–85. doi: 10.1158/1078-0432.CCR-14-0868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rodon J, Braña I, Siu LL, De Jonge MJ, Hormji N, Mills D, et al. Phase I dose-escalation and –expansion study of buparlisib (BKM120), an oral pan-Class I PI3K inhibitor, in patients with advanced solid tumors. Invest New Drugs. 2014;32:670–81. doi: 10.1007/s10637-014-0082-9. [DOI] [PubMed] [Google Scholar]
- 16.Sarker D, Ang JE, Baird R, Kristeleit R, Shah K, Moreno V, et al. First-in-human phase I study of pictilisib (GDC-0941), a potent pan-class I phosphatidylinositol-3-kinase (PI3K) inhibitor, in patients with advanced solid tumors. Clin Cancer Res. 2015;21:77–86. doi: 10.1158/1078-0432.CCR-14-0947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gopal AK, Kahl BS, de Vos S, Wagner-Johnston ND, Schuster SJ, Jurczak WJ, et al. PI3K-delta inhibition by idelalisib in patients with relapsed indolent lymphoma. N Engl J Med. 2014;13:1008–18. doi: 10.1056/NEJMoa1314583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bubley GJ, Carducci M, Dahut W, Dawson N, Daliani D, Eisenberger M, et al. Eligibility and response guidelines for phase II clinical trials in androgen-independent prostate cancer: recommendations from the Prostate-Specific Antigen Working Group. J Clin Oncol. 1999;17:3461–7. doi: 10.1200/JCO.1999.17.11.3461. [DOI] [PubMed] [Google Scholar]
- 19.Punnoose EA, Atwal S, Liu W, Raja R, Fine BM, Hughes BG, et al. Evaluation of circulating tumor cells and circulating tumor DNA in non-small cell lung cancer: association with clinical endpoints in a phase II trial of pertuzumab and erlotinib. Clin Cancer Res. 2012;18:2391–401. doi: 10.1158/1078-0432.CCR-11-3148. [DOI] [PubMed] [Google Scholar]
- 20.Therasse P, Arbuck SG, Eisenhauer EA, Wanders J, Kaplan RS, Rubinstein L, et al. New guidelines to evaluate the response to treatment in solid tumors. European Organization for Research and Treatment of Cancer, National Cancer Institute of the United States, National Cancer Institute of Canada. J Natl Cancer Inst. 2000;92:205–16. doi: 10.1093/jnci/92.3.205. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.