

Abstract
Early successes in identifying and targeting individual oncogenic drivers, together with the increasing feasibility of sequencing tumor genomes, have brought forth the promise of genome-driven oncology care. As we expand the breadth and depth of genomic analyses, the biological and clinical complexity of its implementation will be unparalleled. Challenges include target credentialing and validation, implementing drug combinations, clinical trial designs, targeting tumor heterogeneity, and deploying technologies beyond DNA sequencing, among others. We review how contemporary approaches are tackling these challenges and will ultimately serve as an engine for biological discovery and increase our insight into cancer and its treatment.
Introduction
Cancer is a disease of the genome, and major strides have been made in our understanding and treatment of this heterogeneous collection of diseases, beginning with the initial identification of oncogenes and tumor suppressor genes to the development of the first generation of targeted therapies and culminating in the full annotation of the genomic landscape of the most common cancer types (Kandoth et al., 2013). Much of this progress can be traced to technological advances in sequencing, from capillary-based sequencing technologies to the modern massively parallel sequencing of today, collectively known as next-generation sequencing. These advances have enabled the routine genomic study of every tumor at the point of care and will redefine clinical management and translational research in transformative ways. Detailed genomic characterization of tumors is already driving the definition of a new taxonomy of human cancers that will, ultimately, complement current histology-based classifications (Hoadley et al., 2014). Routine genomic profiling will also improve prognostication of clinical outcomes, as has already been achieved with human epidermal growth factor receptor-2 (HER2) amplifications in breast cancer and mutations in FLT3, NPM1, KIT, and CEBPA in acute myelogenous leukemia. The farthest reaching consequence of routine tumor profiling, however, will be the identification of genetically driven tumor dependencies and vulnerabilities that will lead to the further development of precision therapies and combinatorial treatment approaches. In fact, as a preview of this concept, there are already a plethora of genomic alterations for which targeted therapies have been approved.
Although the promise of such progress is immense, there are many obstacles to broad implementation of genome-based cancer care. These challenges are both practical and scientific. Soon, all cancer patients will have the opportunity to obtain detailed genomic profiles of their tumors, but this is only the first and perhaps easiest step. How do we differentiate between therapeutically actionable alterations and biologically neutral passenger changes? How do we manage and prioritize the biologic credentialing of the large number of novel alterations now routinely identified through prospective tumor genomic-screening programs? How can we utilize genome-driven clinical trials to accelerate the biologic investigation of incompletely characterized alterations now that they are routinely being identified in patients receiving ongoing care? What strategies will be most effective in engendering prolonged response to targeted therapy and mitigating the consequences of tumor heterogeneity and acquired resistance? How do we ensure that our ever-expanding knowledge of the cancer genome and the therapeutic vulnerabilities encoded therein are shared among the biomedical community in a manner that maximizes further discovery? What depth and breadth of genomic characterization of each cancer type will be required, and how do we incorporate technologies in the clinic beyond DNA sequencing? How can we improve the efficiency of genomic hypotheses testing in the clinic, and how do we ensure we are learning the most we can from each treated patient? Finally, how do we target mutations that individually occur rarely but, in aggregate, affect a large proportion of the cancer population? Here, we review how contemporary approaches in precision oncology are beginning to address these key challenges and, in so doing, serve as an engine for biological discovery that will ultimately increase our insight into this complex set of diseases.
At the outset, we recognize that as with any new field of science and medicine, a diversity of views on the value of this approach is inevitable. The emerging field of precision medicine is no different, and some authoritative voices have raised appropriate concerns (Tannock and Hickman, 2016; Voest and Bernards, 2016). First, it has been pointed out that despite the immense complexity of the task at hand, there is a lack of much-needed collaboration among cancer institutions, and even in those situations in which tumor sequencing takes place, there is a low rate of patient participation in genomically matched trials. There is truth in this concern, and later on in this review, we will touch on some ongoing collaborative initiatives that are precisely aimed at addressing the current fragmentation of efforts and inefficiency in clinical trials participation. Another far more serious criticism questions whether this approach will work at all to begin with (Tannock and Hickman, 2016). In support of this view, one recently published randomized trial (the SHIVA study) found equivalent outcomes when patients with multiple tumor types were randomized to receive genomically matched versus conventional therapy (Le Tourneau et al., 2014). This study was designed to explore the off-label use of marketed drugs in a variety of unvalidated genomic alterations in multiple tumor types and provides good evidence of the inadequacy of legacy clinical trial paradigms for evaluating genome-driven medicine. The study was underpowered, the genomic alterations had not been validated as optimal targets, and the therapies used were not best in class but rather commercially available agents. For example, any alteration present in the PI3K/AKT/mTOR pathway led to treatment with the mTOR inhibitor everolimus despite the fact that there is strong evidence that PTEN deletions and PI3k/AKT mutations do not confer sensitivity to this agent. Our reaction, therefore, to this type of study is that the complexity of genomically driven oncology strongly argues that more narrowly focused studies that ask questions about specific genomic alterations or drugs rather than large randomized studies attempting to evaluate the entire approach will yield the most informative data.
In short, we believe that both the underlying science and early clinical successes of precision oncology support an optimistic viewpoint, and although we acknowledge the significant challenges that lay ahead, we have strived here to present a critically self-reflective but solutions-focused perspective on the field.
The Current and Emerging Clinical Landscape of Precision Oncology
Limited genomic data are already being used to guide diagnosis, inform prognosis, and support treatment decisions in a variety of cancers. The pioneering example of molecularly driven cancer medicine was the development and use of the kinase inhibitor imatinib for the treatment of chronic myelogenous leukemias (CML) that harbor the BCR-ABL1 balanced chromosomal translocation (Druker et al., 2006). For patients with this previously lethal form of leukemia, imatinib has dramatically improved their outcomes to the point that the survival of CML patients is now nearly identical to that of the general population (Bower et al., 2016). Similarly, the advent of HER2-targeted therapies for the treatment of women with newly diagnosed metastatic HER2-positive breast cancer has radically changed the outcome of what was until recently the most lethal form of breast cancer. A woman diagnosed with metastatic HER2-positive breast cancer can now anticipate a median survival of almost 5 years through the use of state-of-the-art dual-HER2 blockade compared to just 20 months prior to the advent of these therapies (Slamon et al., 2001; Swain et al., 2015). Moreover, the addition of HER2-directed therapy to routine chemotherapy for patients with early-stage HER2-positive breast cancer has improved cure rates by 35%–50% (Piccart-Gebhart et al., 2005).
From these early breakthrough therapies to today, cancer patients are benefiting from a diverse array of therapies targeting genomic somatic aberrations in their tumors, including amplifications, gain-of-function mutations, and structural rearrangements, as well as germline loss-of-function mutations in at least 11 different genes arising in 10 different cancer types (Table 1). These therapies have not only transformed the lives of many patients but also provided a powerful validation of the approach of precision cancer medicine. Individual genomic findings are also used to forgo therapies that are unlikely to result in clinical benefit, such as KRAS, NRAS, and BRAF mutations in colorectal cancers that would otherwise receive anti-epidermal growth factor (EGFR)-targeted therapies (De Roock et al., 2010). These approved targeted therapies, administered on the basis of genomic observations in patients, represent a new era in cancer therapy. In fact, genomic profiling in some form is now required for the appropriate clinical management of a variety of tumor types, including melanoma, gliomas, some sarcomas, as well as carcinomas of the lung, breast, thyroid, ovary, and colon.
Table 1.
Genes Used to Guide FDA-Approved Therapies
| Mutations Used to Select Targeted Therapy | |
|---|---|
| ABL1 | CML, ALL |
| EGFR | lung cancer |
| ALK | lung cancer |
| ROS1 | lung cancer |
| BRAF | melanoma |
| ERBB2 | breast and gastric cancer |
| KIT | gastrointestinal stromal tumor |
| PDGFRA | leukemia, MDS |
| PDGFRB | dermatofibrosarcoma protuberans |
| BRCA1 and BRCA2 (germline) | ovarian cancer |
| Mutations Used to Select against Targeted Therapy | |
| KRAS | colorectal cancer |
| NRAS | colorectal cancer |
| BRAF | colorectal cancer |
How then will ever-larger-scale prospective sequencing efforts further advance our field? We propose that emerging evidence indicates that a widespread implementation of prospective next-generation sequencing will be transformational at several levels. Large-scale sequencing studies have demonstrated that nearly all of the genomic alterations currently used to guide the selection of targeted therapy within specific disease contexts also occur across a variety of other cancer types, albeit typically at low frequencies (Chang et al., 2016; Kandoth et al., 2013). Targeting these clinically validated predictive biomarkers in a wider array of tumor types, therefore, is an opportunity to immediately extend the benefits of precision oncology to a broader population of patients and has been a major focus of recent clinical development efforts. Evidence is growing that this approach will work for at least some genomic alterations, but in a context-and tumor-dependent fashion. For instance, BRAF V600 mutations are found in approximately half of cutaneous melanomas, and the use of RAF and MEK inhibitors in these patients has been shown to improve survival, leading to the approval of four drugs targeting this pathway (Robert et al., 2015). However, nearly two-thirds of all BRAF V600 mutations occur in non-melanoma cancers, suggesting that this therapeutic strategy may be applicable to a much larger number of patients. Clinical trials have been conducted in tumor types in which the overall incidence of BRAF V600 mutations is sufficient to conduct disease-specific studies. These efforts have already identified clinical activity of these inhibitors in lung cancer, hairy cell leukemia, and thyroid cancer (Brose et al., 2016; Planchard et al., 2016; Tiacci et al., 2015).
Testing the efficacy of targeting BRAF V600 mutations in the broad array of cancer types that uncommonly harbor this alteration has been far more challenging and spurred the development of new clinical study paradigms. One increasingly important approach to treating patients with rare genomic variants has been the use of multiple-tumor-type, genomically selected “basket” studies. These studies revisit the tumor type as the traditional organizing principle of a clinical trial and instead group patients by the genomic alterations present in their tumors, thereby reflecting an increasingly accepted reclassification of human cancers not based on the organ of their origin but instead on the molecular abnormalities that drive their growth and progression (Hoadley et al., 2014). In this way, patients whose tumors harbor the qualifying genomic alterations are eligible for treatment regardless of cancer type. Tumor types anticipated to have a sufficient prevalence of the alteration(s) of interest are enrolled to their own “basket,” whereas all other patients are grouped in a remaining “all-comers” cohort. These studies recognize that response may be conditioned by the disease context in which the genomic alteration arises and therefore analyze efficacy independently for any tumor type that enrolls a sufficient number of patients. A variety of statistical designs have been utilized in these basket studies, and these approaches have been continuously iterated with each successive generation of studies (Cunanan et al., 2016). In doing so, basket trials have become a very efficient means of generating clinical efficacy datasets across a broad variety of tumor types treated with therapy targeting shared genomic alterations. The early results of these studies have already begun to change clinical practice. In the case of BRAF V600, we recently completed a basket study that is expected to lead to regulatory approval of the BRAF inhibitor vemurafenib in several additional tumor types, including a group of rare disorders collectively known as histiocytoses for which no active therapies were previously available (Hyman et al., 2015a). A similar basket study approach was undertaken to determine the efficacy of the poly-ADP-ribose polymerase (PARP) inhibitor olaparib in various solid tumors harboring loss-of-function germline BRCA1 and BRCA2 alterations. This study led directly to the first regulatory approval of a PARP inhibitor for women with germline BRCA1/2-associated ovarian cancer, as well as initial proof of efficacy in prostate and pancreas cancer (Kaufman et al., 2015).
Expanding the Druggable Genome
Expanding the use of therapy targeting previously validated genomic biomarkers in a larger set of tumor types is only the first step in broadening the utility of precision oncology. Maximizing the potential of genome-driven oncology will require understanding the clinical significance of a much broader set of potentially actionable alterations, both within specific tumor types and in a tumor-agnostic manner for targets where actionability is not conditioned by tumor lineage.
The successful targeting of BRAF and PARP in multiple tumor types suggests that targeting even rare genomic alterations independent of tumor type may be applicable to other driver genes as well. There are more than 30 promising genomic targets today (Table 2). This list is only a first iteration of a continuous process, and we anticipate the number of targets will continue to expand at a vibrant pace, although the final number of gene targets is difficult to predict. The cumulative work establishing these targets has already markedly increased the fraction of patients who are now believed to harbor at least one potentially actionable genomic alteration (Figure 1). Unlike the more common genomic alterations described above, however, many of these emerging targets are rare across all tumor types. An example of this phenomenon is the activating E17K mutation in AKT1, which leads to its pathologic localization to the plasma membrane, causing constitutive downstream signaling of the PI3K-AKT-mTOR axis (Carpten et al., 2007). AKT1 E17K is found in ~3% of cases of breast cancer and is even less common in endometrial, ovarian, cervical, lung, colorectal, and prostate cancers, as well as several additional tumor types (Bleeker et al., 2008). No single tumor type has a sufficient prevalence of AKT1 E17K to allow for a traditional disease-focused drug development strategy. Instead, and like BRAF before it, this AKT1 mutation is especially well suited for clinical hypothesis validation within a basket study. Indeed, AKT inhibitors have recently shown marked efficacy in AKT1 E17K-mutation-positive solid tumors in a basket study, and clinical development is now progressing toward regulatory approval (Hyman et al., 2015b). Similarly, a basket study of the pan-HER kinase inhibitor neratinib in solid tumors harboring activating mutations in ERBB2 and ERBB3, which also occur infrequently across a large number of cancer types, has reported promising preliminary results (Bose et al., 2013; Hyman et al., 2016).
Table 2.
Genes with Clinical Evidence Supporting Them as Targets for Drug Development
| Gene | Alteration(s) |
|---|---|
| RET | M918, fusions |
| MET | exon 14 splice, amplifications |
| AKT1 | E17K |
| ERBB2 | activating missense mutations |
| FGFR1/2/3 | fusions, amplifications, activating missense mutations |
| FLT3 | ITD, D835 |
| IDH1 | R132 |
| IDH2 | R140 |
| MAP2K1 (MEK1) | activating missense mutations |
| MTOR | activating missense mutations |
| BRAF | non-V600-activating mutations, fusions |
| NTRK1/2/3 | fusions |
| NRG1 | fusions |
| PIK3CA | activating missense mutations |
| Homologous recombination deficiencies (BRCA1, BRCA2, PALB2, RAD50, ATM, RAD51, RAD51B/C/D, FANCA, CHEK1/2) | inactivating alterations |
| ARAF | S214 |
| EGFR | rare activating missense mutations, insertions |
| TSC1/2 | inactivating alterations |
| SMARCA4 | inactivating alterations |
| SMARRB1 | inactivating alterations |
Figure 1. Druggable Alterations in Oncology Today and in the Near Future.
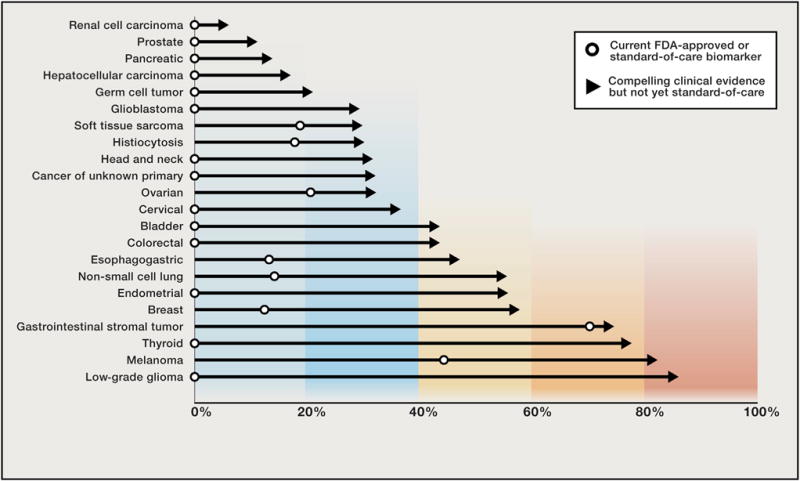
The percent of patients by cancer type harboring a biomarker that, at present, guides the use of either FDA-approved or standard-of-care therapies (open circles) compared to the fraction of patients in the same tumor type harboring a genomic alteration with compelling clinical evidence that it predicts response to a drug but neither the genomic biomarker nor the drug are standard-of-care yet in that indication represented by an arrow.
However, unlike AKT1, where the vast majority of activating mutations occur at a single allele, activating mutations in ERBB2 are distributed throughout the extracellular, juxtamembrane, and kinase domains of the gene encoding for the transmembrane HER2 tyrosine kinase receptor. Thus far, more than 10 recurrent missense mutations, in addition to various insertions and even rarer in-frame fusions, have been described. Moreover, the pattern of ERBB2 mutations varies by tumor type (Chang et al., 2016). Consequently, responses to HER2-targeted therapy in ERBB2-mutant tumors may be dependent, in part, on potential differences in function of each mutation, as well as each variant’s sensitivity to pharmacological inhibition with a specific inhibitor. Indeed, the unique biochemical properties of individual genomic variants within the same gene have been a recurrent observation across oncogenes such as BRAF and KRAS, as well as tumor suppressors such as TP53 (Halevy et al., 1990; Karnoub and Weinberg, 2008; Yao et al., 2015). These genomic nuances add significant complexity to the interpretation of genomic data and clinical decision making. Furthermore, as with the basket studies mentioned above, responses can be conditioned by the tissue of origin, and this must therefore be taken into account. Given the multitude of factors that can condition response to therapy, larger studies that enroll sufficient numbers of patients with different mutant alleles and tumor types are necessary to reach definitive conclusions regarding such intricacies.
Additional strategies exist to expand the druggable genome beyond the targeting of individual gain-of-function missense variants. There are increasing efforts to target a growing number of kinase fusions in various cancer types. As first exemplified by BCR-ABL1 in leukemia, targeting structural rearrangements resulting in constitutively activated kinases can lead to dramatic efficacy and has become a renewed focus of precision oncology. Drugs targeting kinase fusions involving ALK, ROS1, ABL1, PDGFRA, and PDGFRB have already transformed the care of patients with lung cancer, leukemia, and sarcoma. The increasing adoption of DNA-and especially RNA-sequencing technologies capable of detecting dozens of known and novel kinase fusions have demonstrated that these genomic events are more common and implicate a larger number of kinases and tumor types than previously recognized (Stransky et al., 2014). As a result, ongoing studies are already evaluating targeted therapy in additional sets of fusions involving RET, FGFR1, FGFR2, FGFR3, NRG1, BRAF, RAF1, NTRK1, NTRK2, NTRK3, and PRKCA across a diversity of cancer types (Drilon et al., 2016b; Tabernero et al., 2015). In fact, preliminary data from studies targeting these novel kinase fusions suggest that the rate and depth of response as well as the diversity of tumor histologies that will be sensitive may be greater than for many of the current clinically credentialed missense mutations. As mentioned, we have been particularly struck by the greater magnitude of responses observed when targeting many of these “fusion gene targets” and speculate that these difficult-to-acquire genomic alterations may indicate a higher level of oncogenic dependency on these complex lesions. For instance, fusions involving the neurotrophic receptor tyrosine kinases (NTRK1, 2, and 3) occur in approximately 0.5% of diverse solid tumors and hematological malignancies, for which NTRK inhibitors have demonstrated nearly uniform response (D.S. Hong et al., 2016; AACR Annual Meeting abstract; Vaishnavi et al., 2015). As a consequence of these impressive early clinical results, the US Food and Drug Administration recently granted Breakthrough Therapy Designation to one of these agents for the treatment of any solid tumor harboring an NTRK-fusion transcript, regardless of cancer lineage (Loxo, 2016). We are, therefore, finally on the verge of seeing a genomically targeted therapy approved and used based solely on the presence of a genomic alteration, regardless of the tumor type in which it arises. Similar data have emerged from studies of selective fibroblast growth factor receptor (FGFR) inhibitors in FGFR2/3 fusion-positive cancers, including cholangiocarcinomas, bladder cancers, and gliomas (Tabernero et al., 2015). Although these results targeting kinase fusions are exciting for the field, they raise a key challenge. Many of these gene fusions are also present, and sometimes enriched, in pediatric cancers for which optimal available therapies are lacking. Historically, targeted drug development in pediatrics has lagged beyond that for adults, and pediatric patients have become a sadly underserved population when it comes to precision medicine. To begin to address this deficit, we have recently had success progressively lowering the minimum permitted age in several genomically driven studies, a change prompted by the dramatic responses observed in adults and the enthusiastic support of health regulators.
Navigating Biological Complexity in Precision Oncology
Credentialing Therapeutically Actionable Mutations
The shift toward larger panel and whole-exome sequencing has led to the identification of increasing numbers of somatic mutations in potentially actionable cancer genes, the vast majority of which lack biological or clinical validation. This knowledge gap significantly impairs our ability to fully utilize data generated by prospective profiling to guide patient care and to implement comprehensive precision-oncology programs. To begin unraveling the biological complexity of both common and rare alleles, we must create a systematic framework to catalog genomic alterations and characterize their frequency within and across cancer types. Large-scale consortia efforts such as The Cancer Genome Atlas have provided a vital first step but have predominantly studied primary untreated tumors, and of the 33 unique cancer types profiled to date, only 9 qualify as rare tumors (http://cancergenome.nih.gov/). On the other hand, prospective clinical sequencing initiatives reflect a greater diversity and distribution of cancer types seen in patients with advanced disease, the group most in need of new individualized treatment strategies (Hyman et al., 2015c). Moreover, many of these samples are collected after the tumors have been exposed to prior therapies and therefore possess mutations that only arise upon selective therapeutic pressure, such as activating mutations in ESR1 (the gene encoding for the estrogen receptor [ER]) in patients with ER-positive advanced breast cancers that have progressed after anti-estrogen therapies (Toy et al., 2013).
Assembling large representative databases of clinically sequenced cancers is only the first step toward saturating the discovery, and eventual clinical validation, of actionable variants. The next step is the development of a mutational taxonomy that classifies each aberration on the basis of its abnormal function and druggability. To this end, computational frameworks have been developed for analyzing large-scale sequencing data in order to identify individual positions and genes recurrently mutated, both within individual tumor types and across cancers, more often than expected in the absence of selection (Chang et al., 2016; Lawrence et al., 2014). Such statistically principled approaches that operationalize different facets of how mutations, both driver and passenger, accrue in cancer genomes will become increasingly necessary as the community aggregates larger datasets where repeated observations of even passenger alterations are expected. These early efforts have begun to bear fruit, identifying previously uncharacterized and potentially druggable variants, and have the potential to significantly expand the proportion of patients that may benefit from precision-oncology approaches. For instance, a recent analysis of PIK3CA hotspots in more than 11,000 sequenced tumors identified not only the well-characterized helical and kinase domain mutations E545 and H1047 but also 16 additional statistically significant recurrent alterations (Chang et al., 2016). These additional hotspots accounted for 32% of all the PIK3CA mutations observed in the overall cohort, potentially significantly expanding the number of patients eligible for inhibitors targeting this pathway. It remains unknown when, as a community, we will saturate the detection of rare recurrent mutations in PIK3CA and other clinically actionable genes such as ERBB2, EGFR, and MET. Such efforts will also help the basic and translational cancer research community to prioritize biologic investigation or novel variants and, in doing so, facilitate new target discovery.
Novel allele prioritization is necessary, but not sufficient. The mutational diversity in already therapeutically actionable genes is profound and, in many cases, may reflect differences in phenotype that translate into unique pharmacologic dependencies that in silico approaches alone cannot capture reliably. For example, BRAF V600 is among the most common somatic mutations in cancer and results in a constitutively active oncoprotein that signals as a RAF monomer (Davies et al., 2002). Consequently, BRAF V600 is sensitive to RAF inhibitors that preferentially bind activated RAF monomers (Bollag et al., 2010). Conversely, less frequent but recurrent BRAF variants including K601, L597, and G469 similarly lead to constitutive BRAF activation but signal primarily as homodimers and are insensitive to pharmacologic inhibition with first-generation RAF inhibitors (Poulikakos et al., 2011; Yao et al., 2015). Still other BRAF mutations such as V599 and G465 impair BRAF kinase activity but appear to activate MEK through RAS-dependent mechanisms (Wan et al., 2004). Finally, the impact of many other BRAF mutations, including many occurring outside of the kinase domain, on RAF/RAS signaling are still uncharacterized. Computational approaches alone cannot yet reliably predict the class to which a previously uncharacterized BRAF variant belongs and therefore cannot be used to guide selection of pharmacologic therapy. Moreover, purely genomic approaches do not account for signaling pathway cross-talk and feedback mechanisms that cloud simple genotype-drug response relationships but are responsible for clinically important observations such as the lack of response of KRAS mutant tumors to MEK inhibitors.
How then do we begin to capture at scale these nuances among an enormous number of as yet to be tested alleles? Interrogating each of these experimentally is time prohibitive. Conversely, techniques that facilitate rapid biochemical characterization of large numbers of individual genomic variants simultaneously have been developed to address this need (Kim et al., 2016; Kitzman et al., 2015). Utilizing recent advances in synthetic biology, these massively parallel functional genomic approaches use high-throughput methodologies to characterize the preliminary function of nearly all possible missense mutations within target genes or pathways of interest. Work is now underway using these methods to build large catalogs of the biochemical properties of thousands of individual missense mutations. This approach is exemplified by a recent report on the phenotypic characterization of a comprehensive set of MAPK1 (ERK2) missense mutants (Brenan et al., 2016). Employing saturation mutagenesis using a DNA synthesis-based approach (mutagenesis by integrated TilEs; Melnikov et al., 2014), these investigators were able to screen 6,810 of 6,821 possible (99.84%) MAPK1 missense mutants for gain and loss of function, as well as drug sensitivity. This screen confirmed that previously identified recurrently mutated alleles such as MAPK1 E322 were gain of function but, importantly, also identified other candidate functional alleles that are not yet known to be recurrent in existing tumor-sequencing databases. Soon, these technologies will mature such that mutations can be studied in high throughput in vivo and test a broader panel of molecular and biochemical phenotypes. This high-throughput strategy for allele prioritization can then be followed by traditional functional genetic studies of greater depth using a variety of modern molecular biology techniques, including CRISPR gene editing.
Although it is clear that a combination of multiple approaches are necessary to scale up the generation of essential preclinical data of a large number of candidate alleles, those efforts are primarily focused on exploring individual alleles in isolation. However, this is not typically how these alleles arise in patient tumors. Indeed, one of the open questions in precision oncology is how best to intervene therapeutically in patients whose tumors present with multiple actionable mutations. To complicate things further, the tumor microenvironment may also dictate how a particular patient will respond to therapy targeting a specific genetic alteration. For example, in colon cancers, but curiously not in other tumor types, inhibiting BRAF V600 is partially reversed by abundant EGF in the microenvironment, which activates a bypass pathway (Prahallad et al., 2012). Ultimately, this dependency framework will be necessary to guide rational drug combinations, as well as to prioritize targeting of one of potentially multiple alterations in genomically complex tumors.
A New Model for Accelerated Clinical Testing
The scale and potential biological complexity of still uncharacterized genomic variants may not be addressable through traditional clinical testing paradigms. The absence of detailed biochemical profiling of each genomic alteration is also a principal challenge to physicians trying to incorporate somatic mutational data into novel treatments for patients with advanced cancers and in need of such therapies. Biomarker development has historically proceeded from basic target discovery to biologic validation, culminating in clinical testing. Although this process has supported significant advancements, the time necessary to validate each target in this manner is impractical for many of the aforementioned reasons. In a reversal of fortunes, the clinical validation of a target is now frequently occurring prior to its full biological and functional characterization in the laboratory. We have begun to demonstrate that genome-driven clinical trials designed to evaluate laboratory-derived hypotheses can also be used to biologically credential new targets that have not undergone extensive biologic investigation, thus accelerating drug development (Figure 2). In this model, clinical studies can become platforms for exploring the functional consequences of novel genomic alterations detected in the same patient populations. Such studies can inform laboratory studies that, in turn, refine our understanding of the role these genomic aberrations play in disease pathogenesis, drug response, and resistance. Although unconventional in approach, many of the patients currently having their tumors genomically profiled are heavily pretreated and have no remaining standard therapeutic options. What level of evidence supporting the actionability of a potential genomic target should be required in this setting? A proof-of-principle of this approach was recently undertaken in a heavily pretreated patient with breast cancer with an ERBB2 L869R mutant tumor treated on a basket study with an ERBB2 tyrosine kinase inhibitor (neratinib) for solid tumors harboring ERBB2 mutations (ClinicalTrials.gov, NCT01953926). At the time, this patient’s tumor was identified as harboring an ERBB2 mutation; this particular variant was not known to be recurrent and had not been functionally characterized. However, it was noted that the ERBB2 L869 allele is paralogous to EGFR L861, a known activating EGFR mutation that is sensitive to EGFR tyrosine kinase inhibitors (Yang et al., 2015). Moreover, computational and three-dimensional modeling of ERBB2, EGFR, and the paralogous residue mutations predicted a similar constitutively activating signaling phenotype. On the basis of these findings, as well as the absence of standard therapeutic options for the patient, she was enrolled onto the study and ultimately achieved a partial response lasting for more than 1 year (Hyman et al., 2016). In addition to the significant benefit afforded this patient, this response simultaneously provides valuable insight into the biologic function of the novel L869R allele.
Figure 2. Approaches to Novel Target Validation.
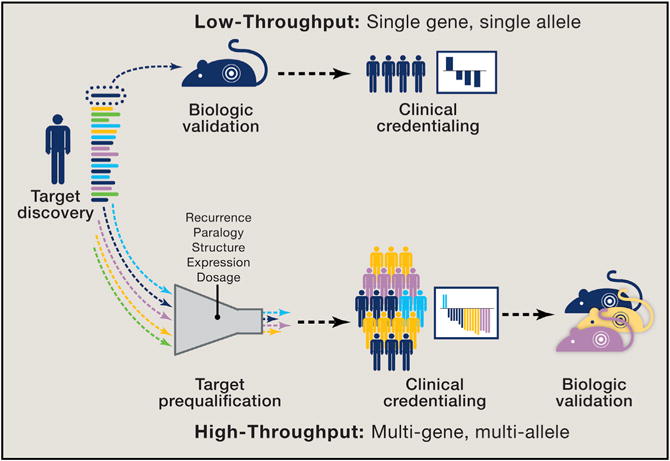
Discovery of a novel target is traditionally followed by biologic validation before proceeding with clinical credentialing. This “monogenic” low-throughput process involves evaluation of one genomic alteration at a time. Next-generation sequencing at the point of care now routinely identifies novel genomic alterations in patients who are in need of new treatment strategies and cannot wait for initial biologic validation. In a “polygenic” high-throughput model, novel genomic alterations observed in patients undergo an initial prequalification using a computational framework that considers allelic recurrence, paralogy, structure, expression, and gene dosage to evaluate the likelihood of clinical relevance. Patients whose tumors harbor qualifying alterations that are identified as potentially activating can then be enrolled in genome-driven clinical trials and the responses observed, potentially providing initial clinical credentialing before biologic characterization. In this model, clinical studies become platforms for exploring the functional consequences of novel genomic alterations detected in the same patient populations.
In the absence of comprehensive biochemical profiling of all possible mutations in commonly mutated oncogenes, it is essential that we establish principled approaches for analyzing novel variants in this way to determine whether and how they should be acted upon. Under this model, computational frameworks that combine analysis of statistically significant recurrence, sequence paralogy, and protein structure can be useful if applied judicially and when there are no more-definitive biologic data. Stakeholders, including physicians and their patients, must then decide whether the predictions generated by these multifaceted analyses, which cannot replace biologic validation, provide sufficient preliminary evidence to justify acting on them. This assessment must necessarily take into account the alternative treatment options available, to the extent any exist, as a greater degree of speculation may be appropriate in patients with no alternative evidence-based options. The goal here is to direct these patients to clinical studies that will collect and report treatment outcome in a way that contributes to the overall knowledge base while at the same time offering a real possibility of clinical benefit. Responses in this setting are essential clinical evidence that, even when anecdotal, can prompt extensive biological exploration of a given allele using traditional molecular biology techniques, studies that might also be performed in parallel to clinical testing via a co-clinical trial approach. Although these responses can be extremely informative, the absence of response cannot similarly be taken as conclusive evidence of the absence of biologic function and, therefore, should not a priori stifle further biologic evaluation of novel alleles.
The approach of enrolling patients with candidate but incompletely characterized genomic drivers can be paired with other study best practices, including generation of advanced models such as organoids and patient-derived xenografts, that can be used to further explore basic biology (when paired to the clinical outcome) from the patient who contributed the sample. Moreover, these patient-derived models permit us to study not only the initial response phenotype but also the adaptive signaling responses that underlie these outcomes.
Managing Tumor Heterogeneity and Acquired Resistance
Tumors and the potentially therapeutically actionable mutations that drive them evolve constantly from an ancestral cell and as a function of therapy, resulting in both temporal and spatial genomic heterogeneity (Gerlinger et al., 2012). Next-generation sequencing has transformed our understanding of intratumoral heterogeneity, and this phenomenon has been implicated in both the development of acquired resistance and lesion-specific differential response to targeted therapy (Diaz et al., 2012; Misale et al., 2012; Russo et al., 2016). Despite this, the degree to which this tumor heterogeneity will ultimately impact the utility of genome-driven oncology remains unclear.
From a therapeutic perspective, the early truncal mutations that are essential oncogenic drivers are typically shared by all sites of disease, even in patients with advanced and heavily pretreated cancers, a fact that may mitigate some of the potential consequences of intratumoral heterogeneity. As an example, BRAF V600 mutations first arise in dysplastic nevi before they progress to malignant melanoma and remain as a key tumoral dependency despite the marked genomic heterogeneity characteristic of melanomas as a result of UV-induced DNA damage. Nevertheless, therapy targeting even a truncal mutation will still alter the cellular milieu as the incumbent oncogene-addicted clone is depleted, facilitating a permissive environment for the outgrowth of cellular populations that were previously less fit. Hence, under the pressure of selective targeted therapy, these initial subclonal genomic mediators of acquired resistance often become the dominant clone, as exemplified by the emergence of EGFR T790M in EGFR mutant non-small cell lung cancer patients treated with first-generation inhibitors (Hata et al., 2016). Even in instances where more complex polyclonal resistance emerges (different co-existing cell populations driven by genetically distinct mechanisms of resistance), these alterations often converge on specific genes or pathways, suggesting that even these scenarios could be managed with drugs or drug combinations that target this evolutionary bottleneck (Juric et al., 2015).
The eventual development of acquired resistance has, therefore, been a near universal observation with targeted cancer therapy. Although the individual mechanisms underlying this are varied, two are common–target reactivation due to secondary genomic alterations or activation of upstream effectors and pathway reactivation mediated by activation of downstream or bypass effectors. The recognition that resistance to targeted therapies is often mediated by secondary mutation of the drug target has already led to the development of drugs that maintain efficacy despite these tumor adaptations. Resistance to imatinib in BCR-ABL1 fusion-positive CML is driven almost entirely by secondary mutations in the drug-binding site, ATP-binding pocket, catalytic domain, and activation loop (Milojkovic and Apperley, 2009). Identification of these mechanisms has led to the development and subsequent approval of even more potent BCR-ABL1 tyrosine kinase inhibitors that maintain activity in the setting of various secondary mutations (Cortes et al., 2012; Saglio et al., 2010). Similarly, second-and third-generation inhibitors targeting EGFR mutant and ALK fusion-positive non-small cell lung cancer have been successfully developed to manage on-target resistance (Jänne et al., 2015; Shaw et al., 2014,2016).
Acquired resistance can also be driven by activation of bypass mechanisms via genomic alterations affecting upstream and downstream effectors and through pathway-independent mechanisms whose identification has uncovered biochemically important facets of key cancer genes and pathways. For example, the increased expression of the androgen receptor in castrate-resistant metastatic prostate cancers ultimately led to the discovery of their continued dependence on androgen signaling despite apparent resistance to androgen deprivation and first-generation antagonists. This insight ultimately led to the successful development of second-generation androgen receptor inhibitors (Chen et al., 2004; Scher et al., 2012). Similarly, identification of new mutations in the ligand-binding domain of ESR1 in patients with ER-positive breast cancer treated with certain classes of antiestrogens led to the discovery that these mutations induce ligand-independent, ER-dependent gene transcription (Toy et al., 2013). This, in turn, has led to the development of selective ER degraders (Lai et al., 2015). Recently, deletions of JAK2 and B2M were implicated in resistance to programmed death 1 (PD-1) checkpoint blockade in melanoma, alluding to the critical role these effectors may play in patients with regard to tumor-antigen presentation and immune surveillance (Zaretsky et al., 2016).
Undoubtedly, acquired resistance will impact every facet of precision oncology. This is not an indictment of the therapeutic approach but will require us to develop strategies to affect more durable responses in our patients. The identification of many of the aforementioned mechanisms of resistance has revealed novel biochemical and signaling phenotypes and demonstrated first principles of how multiple signaling pathways interact via cross-talk and feedback. As a result, drug combinations, when applied in a biologically rational and synergistic manner, can delay the onset of resistance (Baselga et al., 2012; Turner et al., 2015). On the other hand, although preclinical modeling may nominate potential mechanisms of resistance, few of these mechanisms may manifest clinically in patients. This further emphasizes the importance of prospectively identifying the genetic alterations acquired with, or selected by, treatment in the clinic and in real time to focus biologic investigation. In this manner, acquired resistance is teaching us as much about underlying biological dependencies as is the identification of sensitizing lesions.
Data and Knowledge Sharing
Accomplishing our goals will necessitate data and knowledge sharing by the clinical and biomedical community. Indeed, the mutational heterogeneity and complexity of human cancers is greater than can be reflected by the data any single center or commercial lab can generate. Recognizing the critical importance of collaboration, early on our center developed an institution-wide genomics biospecimen protocol (ClinicalTrials.gov, NCT01775072) that includes a framework for genomic data sharing that can raise important privacy concerns (Hyman et al., 2015c). The use of similar genomic biospecimen protocols has become a common feature of academic centers pursuing precision-oncology programs, and we view this as critical to the success of the field. Maximizing the utility of now federated genomic datasets will require not only consent for data sharing but also bioinformatic approaches to harmonizing variants generated by disparate sequencing platforms, each with their own unique tissue requirements, performance characteristics, and variant calling pipelines. To address this need, the American Association of Cancer Research (AACR) has launched Project GENIE (Genomics, Evidence, Neoplasia, Information, Exchange), which is developing a regulatory-grade registry that aggregates and links clinical sequencing data from tens of thousands of cancer patients treated at multiple international academic medical centers. Unlike commercial laboratories that have limited clinical annotation and no follow-up on the samples they receive, institutional datasets have longitudinal follow-ups that when aggregated are uniquely powered to answer previously unaddressed questions. For instance, EGFR inhibitors are currently approved for the treatment of non-small cell lung cancers with the most common mutational hotspots, including L858R and exon 19 deletions. Little is known, however, about less common EGFR variants (such as mutations in exon 18) and how they might similarly guide evidence-based prescribing practices for these widely available agents. Efforts to address this knowledge gap have been unable to aggregate sufficiently large numbers of patients. Project GENIE and related efforts are well suited to answer these outstanding questions and immediately expand the proportion of patients who receive and benefit from precision oncology. Other complementary public and private efforts, including the NIH Genomic Data Commons (Grossman et al., 2016), the Global Alliance for Genomics and Health (Siu et al., 2016), and Molecular Evidence Development Consortium (http://med-c.org/), are aggregating such genomic data to evaluate clinical utility. In fact, data sharing was a cornerstone recommendation of the Blue Ribbon Panel Report issued as part of Vice President Biden’s Cancer Moonshot initiative.
Sharing data is only one piece of this puzzle. How do we similarly curate, standardize, and aggregate knowledge about the biological and clinical relevance of individual genomic alterations to guide principled clinical decision making in prospectively sequenced patients? The medical genetics community has long recognized the value of high-quality interpretations of individual genomic variants, which are relied upon to make definitive recommendations to patients involving complex issues such as cancer screening, risk-reducing surgery, and reproduction. To facilitate this decision making, this community has created a number of resources, most prominently ClinVar (https://www. ncbi.nlm.nih.gov/clinvar/) (Landrum et al., 2016). To similarly systematically curate somatic variants, a number of clinical knowledge bases have been created, including OncoKB (oncokb.org), MyCancerGenome (mycancergenome.org), CIViC (civic.genome.wustl.edu), Cancer Genome Interpreter (CGI, cancergenomeinterpreter.org), CANDL (candl.osu.edu), and the Personalized Cancer Medicine Knowledge Base (https://pct.mdanderson.org/). Like ClinVar, these knowledge bases have established levels of actionability and support community contribution with expert curation. Centralization, standardization, and timely updates of these critical data resources are necessary and could be catalyzed by a single national or international effort so that a single principled recommendation per variant can be democratized to any treating oncologist.
Next-Generation Sequencing in the Clinic
An essential pillar of precision oncology is ensuring that we identify, at the point of care and in every cancer patient, the genomic alterations on which the growth and progression of their diseases depend. A practical and technical challenge to implementing this paradigm has been the limited quantity and quality of tumor material typically available for testing. To establish an initial cancer diagnosis, patients often undergo only a small tumor core biopsy or fine-needle aspiration. A portion of this material is consumed for standard diagnostic evaluation. Moreover, nearly all surgical and biopsy specimens are fixed in formalin and may be stored for years in suboptimal conditions before they are utilized for profiling. Biopsies and surgical specimens are frequently mixed with stromal tissue, further limiting tumor content. These limitations in tissue quality and quantity make obtaining comprehensive whole-genome sequencing, whole-transcriptome sequencing, and phosphoproteomics impractical or even impossible for many cancer patients.
Despite these limitations, DNA enrichment and sequencing technologies have matured to the point where they can now generate reliable results on individual tumors within clinically meaningful time frames using small amounts of paraffin-embedded tumor tissue. These approaches have varied from small gene panels that sequence only recurrently mutated “hotspot” positions (Meric-Bernstam et al., 2015) to targeted gene panels typically sequencing the entire coding regions of 50–500 genes (Hyman et al., 2015c) to whole-exome and -genome sequencing. Smaller hotspot-based panels, which typically utilize amplicon-based sequencing, miss many of the less common but potentially actionable alterations that in aggregate constitute the majority of genomic variants in cancer and, in addition, are not well suited for detecting copy-number alterations and structural rearrangements. Conversely, larger targeted panels, which typically utilize hybridization enrichment, can sequence the entire coding region of targeted genes, are capable of detecting copy-number alterations, as well as select structural rearrangements, and cover most, if not all, of the genomic targets for which there are currently drugs in clinical development. Importantly, these hybrid capture platforms perform well with small tumor specimens and provide sufficient sensitivity for the detection of actionable alterations in samples with low tumor purity. In addition, these panels can be easily expanded to include new genomic content as the actionable genome expands. Consequently, targeted gene panels using this technology have been favored by many of the high-volume academic institutions and commercial laboratories pioneering the use of this testing (Cheng et al., 2015; Frampton et al., 2013). Although broader whole-exome sequencing provides the opportunity to detect recurrent alterations in genes not previously implicated in cancer, and is therefore useful for discovery, it requires more input DNA, typically offers lower coverage, and often has less sensitivity for structural rearrangements due to limited sequencing of intronic regions (Beltran et al., 2015). Whole-genome sequencing similarly offers lower coverage and also remains cost prohibitive and therefore impractical for most patients. In the near term, these different approaches will continue to represent tradeoffs in terms of sequencing breadth, depth, and practicality of overall adoption. In weighing these tradeoffs, we believe that the rarity of promising genomic alterations currently dictates that clinical genomic profiling strategies be optimized to screen more patients using sufficiently broad targeted gene panels rather than fewer patients with even more comprehensive assays. Although even further from clinical implementation, single-cell DNA and RNA sequencing are also actively being investigated as a means of providing unprecedented resolution into the heterogeneity of each tumor, as well as its microenvironment (Tirosh et al., 2016)
The increased sensitivity afforded by targeted next-generation sequencing has substantially improved our ability to identify known actionable alterations in patients and has also led to the discovery of new potentially druggable alterations. One recent example is MET splice site mutations, which promote MET exon 14 skipping that results in augmented MET kinase activity through loss of ubiquitin-mediated degradation and appear sensitive to MET inhibitors such as crizotinib (A. Drilon et al., 2016a; ASCO Annual Meeting abstract; Peschard et al., 2001). These heterogeneous alterations, which are present in ~3% of lung adenocarcinomas and rarely in other cancer types, can span 200 bases and are not easily detected by hotspot-based sequencing approaches (Frampton et al., 2015).
Technical advances are now accelerating the development of genomic profiling strategies suitable for sequencing tumor-derived cell free DNA (cfDNA) from plasma with high sensitivity and specificity. These assays combine deep targeted hybrid capture sequencing with the use of unique molecular identifiers that allow for the differentiation of sequencing artifacts from very low-allele-frequency mutations (Kivioja et al., 2011; Lanman et al., 2015). The potential advantages of cfDNA, also known as a “liquid biopsy,” compared to tumor sequencing are many. Tumor biopsies are complex, invasive, and expensive, which precludes their widespread implementation and repeated use in patients. Conversely, cfDNA only requires a blood draw and can be repeated as frequently as clinically indicated with little risk to the patient. Furthermore, cfDNA sequencing may better capture the genomic heterogeneity of patient disease by detecting mutations that are both shared and private to individual tumor sites. cfDNA sequencing can also be used to monitor response to targeted therapy. In fact, real-time analysis of cfDNA may determine response to therapy within days of treatment initiation, as opposed to weeks with conventional imaging studies. As the sensitivity and specificity of cfDNA profiling improves, as does our ability to interpret the presence and biological significance of rare mutations in circulation, we envision the power of cfDNA being utilized as a screening tool to detect early-stage cancers, when the likelihood of cure would be far higher.
The Reach of Precision Oncology into the Germline: The Case for Integrated Testing
The growing compendium of germline polymorphisms established by large-scale projects such as the EXaC consortium (http://exac.broadinstitute.org/) has improved the ability to distinguish between somatic and germline variants in tumor-only prospective sequencing. Despite this progress, these databases are under-represented by individuals of non-European ancestry and do not entirely address private germline variations that are unlikely to be sufficiently captured without orders of magnitude of additional germline data, if ever. As a result, clinical tumor sequencing, where inaccurate variant classification could potentially lead to improper treatment selection, is increasingly utilizing patient-matched normal controls with the primary objective of distinguishing between germline and somatic variants. However, matched germline sequencing also facilitates simultaneous diagnosis of cancer predisposition syndromes that are themselves therapeutically actionable. Early evidence from large-scale clinical sequencing initiatives suggests a higher rate of pathologic germline variants in both adult and pediatric cancer patients than would be predicted from conventional germline screening guidelines based on personal and family history (Schrader et al., 2016; Zhang et al., 2015). The utility of detecting germline mutations has been recently highlighted in prostate cancer, for which putative loss-of-function germline mutations in BRCA2, ATM, CHEK2, BRCA1, and PALB2 are found in nearly 12% of patients, a rate higher than expected (Pritchard et al., 2016). Importantly, these genomic alterations are all potentially targetable with PARP inhibitors, as well as other inhibitors of DNA-damage repair or DNA-damaging agents. Similarly, defects in mismatch repair through germline, somatic, and epigenetic mechanisms may be targetable with immune checkpoint blockade and are readily detectable through tumor and matched germline sequencing (Le et al., 2015). These findings demonstrate that combined tumor and matched germline sequencing not only improves the detection of somatic mutations but also simplifies comprehensive testing for cancer patients while further expanding the scope of actionable alterations. Moreover, although variants of unknown significance plague the clinical interpretation of most germline alleles, combined analysis with the corresponding tumor that reveals somatic loss of heterozygosity can implicate a germline variant of uncertain significance in the absence of functional data and perhaps inform therapy in advanced patients who otherwise lack treatment options. Therefore, such integrative analysis may provide insights that individual testing cannot capture.
A Future for Genome-Driven Oncology
We have described a framework that can begin to successfully navigate the scientific challenges and broaden the scope and utility of genome-driven oncology. Equally important will be optimizing the way we evaluate the resulting genomic hypotheses in the clinic. Maximizing progress will require us to improve every step in the precision medicine ecosystem, beginning with how we sequence patients, identifying the best targets from this testing, notifying stakeholders, improving access to relevant clinical studies, and finally, making sure these studies are appropriately designed to advance the field.
The Hallmarks of a Precision-Oncology Study: Learning More from Each Patient
Clinical studies evaluating a genomic-driven hypothesis should be designed to learn from each case in an unprecedented way. To capture maximal information from each enrolled patient, precision-oncology studies should include, when possible, not only traditional clinical response evaluation but also the systematic analysis of patient-derived biospecimens and even potential clinical strategies to overcome adaptive or acquired resistance in real time (Figure 3). We propose that the hallmarks of a modern precision-oncology study include four primary scientific objectives: identification of the target, confirmation of target inhibition, biologic credentialing of the target, and description of the mechanisms underlying acquired resistance. Collection and analysis of biospecimens should be organized around, and driven by, these key objectives. For example, pretreatment tumor and liquid biopsies should be used to confirm the presence of the target and define the broader genomic context in which it arises. Confirmation of target engagement and early adaptive responses to target inhibition can be evaluated by tumor biopsies obtained shortly after the initiation of treatment, when early compensatory feedback mechanisms that may modify treatment efficacy can be observed, potentially nominating rational combinatorial strategies. Target engagement can also be assessed through functional imaging studies, such as 18F-fluoroestradiol positron emission tomography (PET), or indirectly via analysis of circulating cfDNA, exosomes, and tumor cells. Biologic target validation can be greatly facilitated through generation of patient-derived xenografts or organoids at the time of tumor biopsy. As described earlier, the identification of mechanisms that result in de novo and acquired resistance in patients is critical and often requires comparative analysis of pre-and post-treatment tumor using a variety of experimental approaches, including DNA and RNA sequencing, phosphoprotein analysis, and immune profiling. Interrogating tumor-derived cfDNA from plasma can also identify the emergence of resistance at an early stage and identify clonal evolution as a response to therapy that may facilitate the addition of other agents or lead to a switch to alternative targeted agents entirely in order to prevent progression of disease (Dawson et al., 2013; Thress et al., 2015). This approach can provide rapid clinical confirmation of underlying resistance mechanisms nominated by preclinical models even when treating small numbers of patients. The study of circulating tumor cells can be considered when key study-related biological questions require intact cells, and it can be visualized as a “live liquid biopsy” where cells can be interrogated in an unprecedented fashion (Yu et al., 2013). Similarly, tumor-derived exosomes appear to contain not only DNA but also RNA, microRNA, proteins, and lipids from cancer cells and provide an opportunity for the non-invasive, multidimensional profiling of cancer (Hoshino et al., 2015). The viability of this approach has been facilitated by techniques that selectively enrich for tumor-derived exosomes (Melo et al., 2015). Finally, for those patients who ultimately succumb to their disease, rapid autopsy programs can provide invaluable insights as well as sustaining models to further accelerate drug development (Juric et al., 2015).
Figure 3. The Hallmarks of a Precision-Oncology Study.
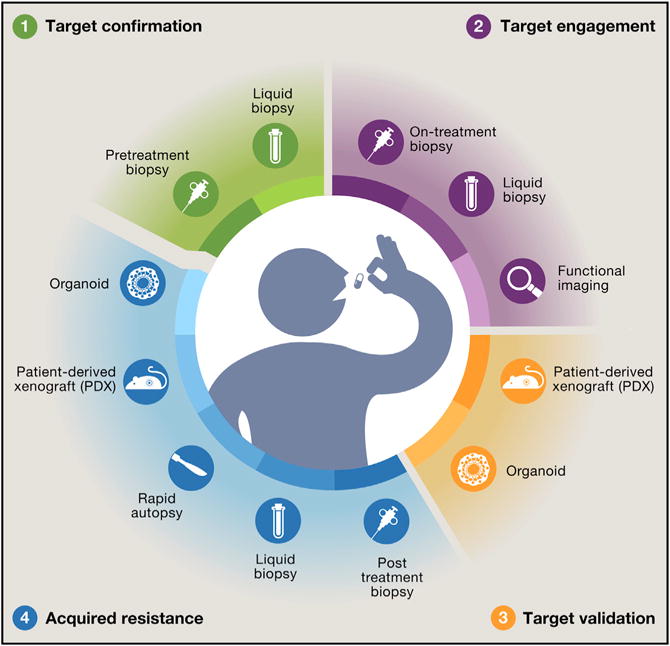
Shown are multiple facets of a modern oncology trial that not only refines a biomarker hypothesis in a scientifically principled manner but also can serve as an engine to drive new scientific discoveries. The hallmarks of a modern precision-oncology study include four primary scientific objectives: identification of the target, confirmation of target inhibition, biologic credentialing of the target, and description of the mechanisms underlying acquired resistance. Collection and analysis of biospecimens should be organized around, and driven by, these key objectives.
Unexplored Facets of Sensitizing Biomarkers
Optimizing genome-driven oncology will likely demand that we move beyond simple classifications of biomarkers as being present or absent in a given patient. Additional facets of a genomic alteration, including absolute copy number, clonality, and zygosity, may condition its function or modify response to targeted therapy. Advanced analytical techniques, originally developed to generate precise estimates of subclonal heterogeneity, allele-specific absolute copy number, and zygosity from whole-exome sequencing, are now being scaled down to targeted sequencing data to address this unmet need (Carter et al., 2012; Shen and Seshan, 2016).
An important and still unresolved clinical question is the degree to which subclonal heterogeneity of a sensitizing genomic biomarker affects the likelihood of response. With modern analytic techniques, it is now possible to catalog the clonality of the individual genomic variants within a single sequenced site and incorporate evolutionary inference to identify the order in which they were acquired within “molecular” time. These analyses will provide unique insights into how tumors evolve and permit an additional dimension of genotype-to-response-phenotype correlation. Similarly, copy-number estimates made by most clinically implemented next-generation sequencing bioinformatic pipelines do not currently correct for tumor purity or provide estimates of allele-specific absolute copy number. An increasing body of evidence, however, suggests that high absolute copy number of gene amplifications, including MET and FGFR2, are necessary to condition response (D.R. Camidge et al., 2014; ASCO Annual Meeting abstract; Pearson et al., 2016). As a result, standardized reporting of allele-specific absolute copy number will enhance the optimal genomic selection of patients when targeting oncogene amplifications.
Understanding the zygosity of genomic alterations involving tumor-suppressor genes may also be important for targeted therapies that exploit synthetic lethality. For instance, PARP inhibitors have shown efficacy in treating homologous recombination-deficient (HRD) tumors with deleterious germline BRCA1/2 mutations. One clinically important question is whether PARP inhibitors may also be effective in patients whose tumors harbor homologous recombination deficiency driven by somatic mutations. Preliminary data suggest that somatic alterations in key HRD genes may require biallelic inactivation to sensitize tumors to PARP inhibitors (Mateo et al., 2015). Computational techniques capable of determining whether somatic truncating variants in HRD genes are accompanied by loss of heterozygosity of the remaining wild-type allele may therefore be necessary to optimally select patients for these therapies. Similarly, recent data demonstrate that biallelic loss of SMARCA4 (BRG1) and SMARCB1 (INI1) in rhabdoid carcinomas and epithelioid sarcomas, respectively, may be targetable with EZH2 inhibitors (Kim et al., 2015). Efforts are currently underway to extend this therapeutic strategy to a wider range of epigenetic modifiers, including BAP1 and ARID1A (Bitler et al., 2015; LaFave et al., 2015).
DNA-based signatures are also emerging as an important tool with which to select targeted therapy. For example, several DNA-based gene signatures have been developed to detect the genomic instability associated with homologous recombination deficiency caused by inactivation of BRCA1, BRCA2, and potentially a variety of other related genes (Telli et al., 2016). Demonstrating the potential value of this approach, a recent phase III study in ovarian cancer utilizing a DNA-based homologous recombination signature demonstrated that signature-positive patients treated with the PARP inhibitor niraparib versus placebo had a near tripling of their progression-free survival (12.9 versus 3.8 months) compared to signature-negative patients (Mirza et al., 2016). These homologous recombination-deficiency signatures, also sometimes referred to as “BRCA-ness” or “genome scar” assays, also demonstrate how the field will begin to extend precision-oncology approaches into tumor types, such as prostate and pancreatic cancer, that otherwise do not harbor frequent actionable genomic variants.
Moving beyond DNA
Although DNA sequencing provides a wealth of information, genomic alterations are only one of several important biologic drivers of cancer. As a consequence, it is understood that DNA sequencing will not be sufficient to optimally select patients for all classes of targeted therapy. In the laboratory, high-throughput technologies, including RNA sequencing, genomewide DNA methylation profiling, microRNA profiling, and phosphoprotein arrays, have been extensively used to further improve our understanding of the biologic dependencies of cancer. Of these various technologies, the one that is closest to the clinic is RNA sequencing, also sometimes referred to as transcriptome sequencing. The incremental value of RNA sequencing in addition to existing DNA sequencing is two-fold. First, when compared to targeted or whole-exome DNA sequencing, RNA sequencing is better suited to detecting structural rearrangements resulting in fusion gene products. Along these lines, we have recently instituted in our center a “cancer of unknown driver” initiative whereby cancers for which prospective targeted DNA sequencing fails to identify a genomic driver are reflexed to targeted RNA sequencing with the goal of identifying cryptogenic kinase fusions. This approach has already identified novel and potentially actionable kinase fusions involving NTRK1/2/3, FGFR2/3, BRAF, NRG1, RET, ERBB2, and AKT1. Similar approaches have been adopted by some high-volume commercial laboratories in cancer types such as sarcomas and hematologic malignancies in which the diversity of potential gene fusions cannot be efficiency interrogated by targeted DNA-based methods (He et al., 2016). In addition to enhanced detecting sensitivity for structural rearrangements in the genome, RNA sequencing offers the ability to measure the transcription of both mutant and wild-type proteins. In breast cancer, earlier approaches to measuring gene-expression profiling are already being used to provide a more refined prognosis than that based on standard clinical risk factors alone and, in doing so, guide treatment decisions (Cardoso et al., 2016). The understanding of the transcriptome at the point of care in each patient will provide an invaluable additional dimension of information, and we believe it is the next frontier for precision oncology. Pilot programs have already begun evaluating the feasibility of incorporating broader RNA sequencing into prospective precision-medicine programs (Roychowdhury et al., 2011). The preliminary results have been encouraging but are limited by the quantity and type of tumor material required, as well as challenges associated with scaling this approach to larger patient volumes. Over time, we expect these technical, scientific, and financial obstacles to be solved, and we must begin to consider now how to begin to incorporate these technologies into the clinic.
Expanding Accessibility and Enrollment to Precision-Oncology Studies
The timely and efficient execution of precision-oncology studies, like those we detail here, represents a true engineering challenge on top of an already inefficient clinical-trials model. In fact, less than 5% of cancer patients are enrolled into clinical trials. Therefore, lack of execution represents a real threat that needs to be addressed. The components of this engineering problem include routine tumor sequencing as a central component of cancer care, the input and annotation of genomic findings into the medical records, and both patient and physician education. Furthermore, robust methods for connecting patients with rare actionable alterations to clinical trials targeting these genomic variants are needed. Within our own institution, we have developed a system that automatically identifies, tracks, and recruits patients with qualifying genomic alterations to the appropriate genome-driven study (Eubank et al., 2016). This system has allowed treating physicians to rely on domain experts to interpret the actionability of individual genomic alterations and identify relevant and immediately available treatment opportunities for their patients. The broader field urgently needs a similar centralized genomic “clearing house” wherein the results of clinical genomic sequencing can be shared and matched in real time against the qualifying genomic alterations being targeted by individual studies nationwide. Unlike current static study registries such as ClinicalTrials.gov, such a system would ensure a privacy-compliant two-way exchange between patients and study sponsors who could indicate the specific genomic variants of interest and provide a path to enrollment for qualifying patients. Several groups, including the Global Alliance for Genomics and Health, are working to develop such a resource (Siu et al., 2016).
Facilitating the identification of highly relevant and immediately available genomically matched studies is only the first step; patients must be able to readily access these studies. Geographical considerations remain an important barrier to study access. The current generation of precision-oncology studies are beginning to address this problem in a number of ways, from supporting patient travel to study centers to bringing the study to the patient once they are identified. These efforts have significantly improved access to genomically matched therapy studies in the community, where the majority of cancer patients continue to be treated. Access to genomically matched therapy has also been improved by the consolidation of precision-oncology trials into larger protocols that have alternatively been referred to as “master,” “umbrella,” or “molecular allocation” studies. These protocols generally offer multiple therapeutic options matched to the patient’s individual tumor genome. Several of these studies, such as the iSpy2 (NCT01042379), Lung-MAP (NCT02154490), ALCHEMIST (NCT02194738), and BATTLE-2 (NCT01248247) trials, have explored genomically defined subtypes of specific cancers. Master protocols can also offer treatment across a variety of tumor types and, in this way, essentially become a collection of individual basket studies; these include NCI-MATCH (NCT02465060), MyPathway (NCT02091141), and the American Society of Clinical Oncology (ASCO) Targeted Agent and Profiling Utilization Registry (TAPUR) (NCT02693535). The most ambitious effort to date is the NCI-MATCH study, which is anticipated to have more than 30 unique treatment arms assigned primarily on the basis of genomic selection criteria. Unlike many related efforts, NCI-MATCH incorporates centralized high-multiplexed tumor genomic screening, with the plan to biopsy and sequence 6,000 patients. This feature has made NCI-MATCH particularly attractive to community oncologists who might otherwise not have access to high-quality tumor genetic screening.
Conclusion
This is a transformative time for cancer therapy. The feasibility of establishing the detailed molecular portraits of individual cancers, even at the point of care, is no longer the primary obstacle to progress. Similarly, new highly potent and selective purpose-built inhibitors are being developed at a time when our understanding of actionable mutations in cancer genomes is improving steadily, resulting in continuous erosion of the market share of what has been called the undruggable genome. What is most lacking, therefore, is the knowledge of how best to use these new and powerful tools that are already available to us. In short, we have an engineering problem. The strategies we propose here establish a framework to begin to address this critical knowledge and implementation gap. We cannot achieve the progress needed with conventional approaches. Rapid progress demands a new degree of collaboration and information exchange between basic and translational laboratory scientists and clinical investigators acting as equal partners. Evaluating the genomic hypotheses that emerge from this collaboration will require us to improve the efficiency with which we sequence patients, annotate results, identify accountable alterations, notify involved parties, enroll into relevant studies, and finally, interpret the outcome and iterate as necessary. If we do so, we will finally have the tools to make truly transformative insights into the basic biology of cancer and its treatment.
Acknowledgments
We thank Alison M. Schram and Maurizio Scaltriti for their critical reading of this manuscript, Jianjiong Gao and Nikolaus Schultz for clinical annotation of sequenced specimens, and Scott Johnson for assistance with medical graphics. The authors acknowledge support from the Prostate Cancer Foundation, the Sontag Foundation, the Josie Robertson Foundation, the Breast Cancer Research Foundation, the Geoffrey Beene Cancer Research Center, the Marie-Josée and Henry R. Kravis Center for Molecular Oncology, Cycle for Survival, and National Institutes of Health awards R01 CA190642-01A1 (J.B.), P50 CA092629 (B.S.T.), R01 CA207244 (D.M.H. and B.S.T.), U54 OD202355 (B.S.T.), and P30 CA008748. J.B. is a member of the scientific advisory board of GRAIL.
References
- Baselga J, Campone M, Piccart M, Burris HA, 3rd, Rugo HS, Sahmoud T, Noguchi S, Gnant M, Pritchard KI, Lebrun F, et al. Everolimus in postmenopausal hormone-receptor-positive advanced breast cancer. N Engl J Med. 2012;366:520–529. doi: 10.1056/NEJMoa1109653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beltran H, Eng K, Mosquera JM, Sigaras A, Romanel A, Rennert H, Kossai M, Pauli C, Faltas B, Fontugne J, et al. Whole-exome sequencing of metastatic cancer and biomarkers of treatment response. JAMA Oncol. 2015;1:466–474. doi: 10.1001/jamaoncol.2015.1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bitler BG, Aird KM, Garipov A, Li H, Amatangelo M, Kossenkov AV, Schultz DC, Liu Q, Shih IeM, Conejo-Garcia JR, et al. Synthetic lethality by targeting EZH2 methyltransferase activity in ARIDIA-mutated cancers. Nat Med. 2015;21:231–238. doi: 10.1038/nm.3799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bleeker FE, Felicioni L, Buttitta F, Lamba S, Cardone L, Rodolfo M, Scarpa A, Leenstra S, Frattini M, Barbareschi M, et al. AKT1(E17K) in human solid tumours. Oncogene. 2008;27:5648–5650. doi: 10.1038/onc.2008.170. [DOI] [PubMed] [Google Scholar]
- Bollag G, Hirth P, Tsai J, Zhang J, Ibrahim PN, Cho H, Spevak W, Zhang C, Zhang Y, Habets G, et al. Clinical efficacy of a RAF inhibitor needs broad target blockade in BRAF-mutant melanoma. Nature. 2010;467:596–599. doi: 10.1038/nature09454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bose R, Kavuri SM, Searleman AC, Shen W, Shen D, Koboldt DC, Monsey J, Goel N, Aronson AB, Li S, et al. Activating HER2 mutations in HER2 gene amplification negative breast cancer. Cancer Discov. 2013;3:224–237. doi: 10.1158/2159-8290.CD-12-0349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bower H, Björkholm M, Dickman PW, Hoglund M, Lambert PC, Andersson TM. Life expectancy of patients with chronic myeloid leukemia approaches the life expectancy of the general population. J Clin Oncol. 2016;34:2851–2857. doi: 10.1200/JCO.2015.66.2866. [DOI] [PubMed] [Google Scholar]
- Brenan L, Andreev A, Cohen O, Pantel S, Kamburov A, Cacchiarelli D, Persky NS, Zhu C, Bagul M, Goetz EM, et al. Phenotypic characterization of a comprehensive set of MAPK1/ERK2 missense mutants. Cell Rep. 2016;17:1171–1183. doi: 10.1016/j.celrep.2016.09.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brose MS, Cabanillas ME, Cohen EE, Wirth LJ, Riehl T, Yue H, Sherman SI, Sherman EJ. Vemurafenib in patients with BRAF(V600E)-positive metastatic or unresectable papillary thyroid cancer refractory to radioactive iodine: a non-randomised, multicentre, open-label, phase 2 trial. Lancet Oncol. 2016;17:1272–1282. doi: 10.1016/S1470-2045(16)30166-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cardoso F, van’t Veer LJ, Bogaerts J, Slaets L, Viale G, Delaloge S, Pierga JY, Brain E, Causeret S, DeLorenzi M, et al. MINDACT Investigators 70-gene signature as an aid to treatment decisions in early-stage breast cancer. N Engl J Med. 2016;375:717–729. doi: 10.1056/NEJMoa1602253. [DOI] [PubMed] [Google Scholar]
- Carpten JD, Faber AL, Horn C, Donoho GP, Briggs SL, Robbins CM, Hostetter G, Boguslawski S, Moses TY, Savage S, et al. Atransforming mutation in the pleckstrin homology domain of AKT1 in cancer. Nature. 2007;448:439–444. doi: 10.1038/nature05933. [DOI] [PubMed] [Google Scholar]
- Carter SL, Cibulskis K, Helman E, McKenna A, Shen H, Zack T, Laird PW, Onofrio RC, Winckler W, Weir BA, et al. Absolute quantification of somatic DNA alterations in human cancer. Nat Biotechnol. 2012;30:413–421. doi: 10.1038/nbt.2203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang MT, Asthana S, Gao SP, Lee BH, Chapman JS, Kandoth C, Gao J, Socci ND, Solit DB, Olshen AB, et al. Identifying recurrent mutations in cancerrevealswidespread lineagediversityand mutational specificity. Nat Biotechnol. 2016;34:155–163. doi: 10.1038/nbt.3391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen CD, Welsbie DS, Tran C, Baek SH, Chen R, Vessella R, Rosenfeld MG, Sawyers CL. Moleculardeterminants of resistance to antiandrogen therapy. Nat Med. 2004;10:33–39. doi: 10.1038/nm972. [DOI] [PubMed] [Google Scholar]
- Cheng DT, Mitchell TN, Zehir A, Shah RH, Benayed R, Syed A, Chandramohan R, Liu ZY, Won HH, Scott SN, et al. Memorial Sloan Kettering-Integrated Mutation Profiling of Actionable Cancer Targets (MSK-IMPACT): A hybridization capture-based next-generation sequencing clinical assayforsolid tumor molecular oncology. J Mol Diagn. 2015;17:251–264. doi: 10.1016/j.jmoldx.2014.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cortes JE, Kantarjian H, Shah NP, Bixby D, Mauro MJ, Flinn I, O’Hare T, Hu S, Narasimhan NI, Rivera VM, et al. Ponatinib in refractory Philadelphia chromosome-positive leukemias. N Engl J Med. 2012;367:2075–2088. doi: 10.1056/NEJMoa1205127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunanan KM, Gonen M, Shen R, Hyman DM, Riely GJ, Begg CB, Iasonos A. Basket trials in oncology: a trade-off between complexity and efficiency. J Clin Oncol. 2016;35:271–273. doi: 10.1200/JCO.2016.69.9751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies H, Bignell GR, Cox C, Stephens P, Edkins S, Clegg S, Teague J, Woffendin H, Garnett MJ, Bottomley W, et al. Mutations of the BRAF gene in human cancer. Nature. 2002;417:949–954. doi: 10.1038/nature00766. [DOI] [PubMed] [Google Scholar]
- Dawson SJ, Tsui DW, Murtaza M, Biggs H, Rueda OM, Chin SF, Dunning MJ, Gale D, Forshew T, Mahler-Araujo B, et al. Analysis ofcirculating tumor DNAto monitor metastatic breast cancer. N Engl J Med. 2013;368:1199–1209. doi: 10.1056/NEJMoa1213261. [DOI] [PubMed] [Google Scholar]
- De Roock W, Claes B, Bernasconi D, De Schutter J, Biesmans B, Fountzilas G, Kalogeras KT, Kotoula V, Papamichael D, Laurent-Puig P, et al. Effects of KRAS, BRAF, NRAS, and PIK3CA mutations on the efficacy of cetuximab plus chemotherapy in chemotherapy-refractory metastatic colorectal cancer: a retrospective consortium analysis. Lancet Oncol. 2010;11:753–762. doi: 10.1016/S1470-2045(10)70130-3. [DOI] [PubMed] [Google Scholar]
- Diaz LA, Jr, Williams RT, Wu J, Kinde I, Hecht JR, Berlin J, Allen B, Bozic I, Reiter JG, Nowak MA, et al. The molecularevolution of acquired resistance to targeted EGFR blockade in colorectal cancers. Nature. 2012;486:537–540. doi: 10.1038/nature11219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drilon A, Rekhtman N, Arcila M, Wang L, Ni A, Albano M, Van Voorthuysen M, Somwar R, Smith RS, Montecalvo J, et al. Cabozantinib in patients with advanced RET-rearranged non-small-cell lung cancer: an open-label, single-centre, phase 2, single-arm trial. Lancet Oncol. 2016b;17:1653–1660. doi: 10.1016/S1470-2045(16)30562-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Druker BJ, Guilhot F, O’Brien SG, Gathmann I, Kantarjian H, Gattermann N, Deininger MW, Silver RT, Goldman JM, Stone RM, et al. IRIS Investigators Five-year follow-up of patients receiving imatinib for chronic myeloid leukemia. N Engl J Med. 2006;355:2408–2417. doi: 10.1056/NEJMoa062867. [DOI] [PubMed] [Google Scholar]
- Eubank MH, Hyman DM, Kanakamedala AD, Gardos SM, Wills JM, Stetson PD. Automated eligibility screening and monitoring for genotype-driven precision oncology trials. J Am Med Inform Assoc. 2016;23:777–781. doi: 10.1093/jamia/ocw020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frampton GM, Fichtenholtz A, Otto GA, Wang K, Downing SR, He J, Schnall-Levin M, White J, Sanford EM, An P, et al. Development and validation of a clinical cancer genomic profiling test based on massively parallel DNA sequencing. Nat Biotechnol. 2013;31:1023–1031. doi: 10.1038/nbt.2696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frampton GM, Ali SM, Rosenzweig M, Chmielecki J, Lu X, Bauer TM, Akimov M, Bufill JA, Lee C, Jentz D, et al. Activation of MET via diverse exon 14 splicing alterationsoccurs in multipletumortypes and confers clinical sensitivity to MET inhibitors. Cancer Discov. 2015;5:850–859. doi: 10.1158/2159-8290.CD-15-0285. [DOI] [PubMed] [Google Scholar]
- Gerlinger M, Rowan AJ, Horswell S, Larkin J, Endesfelder D, Gronroos E, Martinez P, Matthews N, Stewart A, Tarpey P, et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N Engl J Med. 2012;366:883–892. doi: 10.1056/NEJMoa1113205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grossman RL, Heath AP, Ferretti V, Varmus HE, Lowy DR, Kibbe WA, Staudt LM. Toward a shared vision for cancer genomic data. N Engl J Med. 2016;375:1109–1112. doi: 10.1056/NEJMp1607591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halevy O, Michalovitz D, Oren M. Different tumor-derived p53 mutants exhibit distinct biological activities. Science. 1990;250:113–116. doi: 10.1126/science.2218501. [DOI] [PubMed] [Google Scholar]
- Hata AN, Niederst MJ, Archibald HL, Gomez-Caraballo M, Siddiqui FM, Mulvey HE, Maruvka YE, Ji F, Bhang HE, Krishnamurthy Radhakrishna V, et al. Tumor cells can follow distinct evolutionary paths to become resistant to epidermal growth factor receptor inhibition. Nat Med. 2016;22:262–269. doi: 10.1038/nm.4040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He J, Abdel-Wahab O, Nahas MK, Wang K, Rampal RK, Intlekofer AM, Patel J, Krivstov A, Frampton GM, Young LE, et al. Integrated genomic DNA/RNA profiling of hematologic malignancies in the clinical setting. Blood. 2016;127:3004–3014. doi: 10.1182/blood-2015-08-664649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoadley KA, Yau C, Wolf DM, Cherniack AD, Tamborero D, Ng S, Leiserson MD, Niu B, McLellan MD, Uzunangelov V, et al. Multiplatform analysis of 12 cancer types reveals molecular classification within and across tissues of origin. Cell. 2014;158:929–944. doi: 10.1016/j.cell.2014.06.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoshino A, Costa-Silva B, Shen TL, Rodrigues G, Hashimoto A, Tesic Mark M, Molina H, Kohsaka S, Di Giannatale A, Ceder S, et al. Tumour exosome integrins determine organotropic metastasis. Nature. 2015;527:329–335. doi: 10.1038/nature15756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyman DM, Puzanov I, Subbiah V, Faris JE, Chau I, Blay JY, Wolf J, Raje NS, Diamond EL, Hollebecque A, et al. Vemurafenib in multiple nonmelanoma cancers with BRAF V600 mutations. N Engl J Med. 2015a;373:726–736. doi: 10.1056/NEJMoa1502309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyman DM, Smyth L, Bedard PL, Oza A, Dean E, Armstrong A, Lima J, Bando H, Kabos P, Perez-Fidalgo JA, et al. AZD5363, a catalytic pan-Akt inhibitor, in Akt1 E17K mutation positive advanced solid tumors. Mol Cancer Ther. 2015b;14 Abstract B109. [Google Scholar]
- Hyman DM, Solit DB, Arcila ME, Cheng DT, Sabbatini P, Baselga J, Berger MF, Ladanyi M. Precision medicine at Memorial Sloan Kettering Cancer Center: clinical next-generation sequencing enabling next-generation targeted therapy trials. Drug Discov Today. 2015c;20:1422–1428. doi: 10.1016/j.drudis.2015.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyman DM, Piha-Paul S, Rodon J, Saura C, Puzanov I, Shapiro G, Loi S, Joensuu H, Hanrahan A, Modi S, et al. Neratinib for ERBB2 mutant, HER2 non-amplified, metastatic breast cancer: Preliminary analysis from a multicenter, open-label, multi-histology phase II basket trial. Cancer Res. 2016;76 Abstract PD5–05. [Google Scholar]
- Jänne PA, Yang JC, Kim DW, Planchard D, Ohe Y, Ramalingam SS, Ahn MJ, Kim SW, Su WC, Horn L, et al. AZD9291 in EGFR inhibitor-resistant non-small-cell lung cancer. N Engl J Med. 2015;372:1689–1699. doi: 10.1056/NEJMoa1411817. [DOI] [PubMed] [Google Scholar]
- Juric D, Castel P, Griffith M, Griffith OL, Won HH, Ellis H, Ebbesen SH, Ainscough BJ, Ramu A, Iyer G, et al. Convergent loss of PTEN leads to clinical resistance to a PI(3)Kα inhibitor. Nature. 2015;518:240–244. doi: 10.1038/nature13948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kandoth C, McLellan MD, Vandin F, Ye K, Niu B, Lu C, Xie M, Zhang Q, McMichael JF, Wyczalkowski MA, et al. Mutational landscape and significance across 12 major cancer types. Nature. 2013;502:333–339. doi: 10.1038/nature12634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karnoub AE, Weinberg RA. Ras oncogenes: split personalities. Nat Rev Mol Cell Biol. 2008;9:517–531. doi: 10.1038/nrm2438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaufman B, Shapira-Frommer R, Schmutzler RK, Audeh MW, Friedlander M, Balmaña J, Mitchell G, Fried G, Stemmer SM, Hubert A, et al. Olaparib monotherapy in patients with advanced cancer and a germline BRCA1/2 mutation. J Clin Oncol. 2015;33:244–250. doi: 10.1200/JCO.2014.56.2728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim E, Ilic N, Shrestha Y, Zou L, Kamburov A, Zhu C, Yang X, Lubonja R, Tran N, Nguyen C, et al. Systematic functional interrogation of rare cancer variants identifies oncogenic alleles. Cancer Discov. 2016;6:714–726. doi: 10.1158/2159-8290.CD-16-0160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim KH, Kim W, Howard TP, Vazquez F, Tsherniak A, Wu JN, Wang W, Haswell JR, Walensky LD, Hahn WC, et al. SWI/SNF-mutant cancers depend on catalytic and non-catalytic activity of EZH2. Nat Med. 2015;21:1491–1496. doi: 10.1038/nm.3968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitzman JO, Starita LM, Lo RS, Fields S, Shendure J. Massively parallel single-amino-acid mutagenesis. Nat Methods. 2015;12:203–206. 4, 206. doi: 10.1038/nmeth.3223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kivioja T, Vähärautio A, Karlsson K, Bonke M, Enge M, Linnarsson S, Taipale J. Counting absolute numbers of molecules using unique molecular identifiers. Nat Methods. 2011;9:72–74. doi: 10.1038/nmeth.1778. [DOI] [PubMed] [Google Scholar]
- LaFave LM, Béguelin W, Koche R, Teater M, Spitzer B, Chramiec A, Papalexi E, Keller MD, Hricik T, Konstantinoff K, et al. Loss of BAP1 function leads to EZH2-dependent transformation. Nat Med. 2015;21:1344–1349. doi: 10.1038/nm.3947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai A, Kahraman M, Govek S, Nagasawa J, Bonnefous C, Julien J, Douglas K, Sensintaffar J, Lu N, Lee KJ, et al. Identification of GDC-0810 (ARN-810), an orally bioavailable selective estrogen receptor degrader (SERD) that demonstrates robust activity in tamoxifen-resistant breast cancer xenografts. J Med Chem. 2015;58:4888–4904. doi: 10.1021/acs.jmedchem.5b00054. [DOI] [PubMed] [Google Scholar]
- Landrum MJ, Lee JM, Benson M, Brown G, Chao C, Chitipiralla S, Gu B, Hart J, Hoffman D, Hoover J, et al. ClinVar: public archive of interpretations of clinically relevant variants. Nucleic Acids Res. 2016;44(D1):D862–D868. doi: 10.1093/nar/gkv1222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanman RB, Mortimer SA, Zill OA, Sebisanovic D, Lopez R, Blau S, Collisson EA, Divers SG, Hoon DS, Kopetz ES, et al. Analytical and clinical validation of a digital sequencing panel for quantitative, highly accurate evaluation of cell-free circulating tumor DNA. PLoS ONE. 2015;10:e0140712. doi: 10.1371/journal.pone.0140712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrence MS, Stojanov P, Mermel CH, Robinson JT, Garraway LA, Golub TR, Meyerson M, Gabriel SB, Lander ES, Getz G. Discovery and saturation analysis of cancer genes across 21 tumour types. Nature. 2014;505:495–501. doi: 10.1038/nature12912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le DT, Uram JN, Wang H, Bartlett BR, Kemberling H, Eyring AD, Skora AD, Luber BS, Azad NS, Laheru D, et al. PD-1 blockade in tumors with mismatch-repair deficiency. N Engl J Med. 2015;372:2509–2520. doi: 10.1056/NEJMoa1500596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Tourneau C, Paoletti X, Servant N, Bièche I, Gentien D, Rio Frio T, Vincent-Salomon A, Servois V, Romejon J, Mariani O, et al. Randomised proof-of-concept phase II trial comparing targeted therapy based on tumour molecular profiling vs conventional therapy in patients with refractory cancer: results of the feasibility part of the SHIVA trial. Br J Cancer. 2014;111:17–24. doi: 10.1038/bjc.2014.211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loxo. Press release: Loxo Oncology receives breakthrough therapy designation from US Food and Drug Administration for LOXO-101. 2016 https://ir.loxooncology.com/press-releases/loxo-oncology-receives-breakthrough-therapy-designation-from-u.s.-food-and-drug-administration-for-loxo-101.
- Mateo J, Carreira S, Sandhu S, Miranda S, Mossop H, Perez-Lopez R, Nava Rodrigues D, Robinson D, Omlin A, Tunariu N, et al. DNA-repair defects and olaparib in metastatic prostate cancer. N Engl J Med. 2015;373:1697–1708. doi: 10.1056/NEJMoa1506859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melnikov A, Rogov P, Wang L, Gnirke A, Mikkelsen TS. Comprehensive mutational scanning of a kinase in vivo reveals substrate-dependent fitness landscapes. Nucleic Acids Res. 2014;42:e112. doi: 10.1093/nar/gku511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melo SA, Luecke LB, Kahlert C, Fernandez AF, Gammon ST, Kaye J, LeBleu VS, Mittendorf EA, Weitz J, Rahbari N, et al. Glypican-1 identifies cancer exosomes and detects early pancreatic cancer. Nature. 2015;523:177–182. doi: 10.1038/nature14581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meric-Bernstam F, Brusco L, Shaw K, Horombe C, Kopetz S, Davies MA, Routbort M, Piha-Paul SA, Janku F, Ueno N, et al. Feasibility of large-scale genomic testing to facilitate enrollment onto genomically matched clinical trials. J Clin Oncol. 2015;33:2753–2762. doi: 10.1200/JCO.2014.60.4165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milojkovic D, Apperley J. Mechanisms of resistance to imatinib and second-generation tyrosine inhibitors in chronic myeloid leukemia. Clin Cancer Res. 2009;15:7519–7527. doi: 10.1158/1078-0432.CCR-09-1068. [DOI] [PubMed] [Google Scholar]
- Mirza MR, Monk BJ, Herrstedt J, Oza AM, Mahner S, Redondo A, Fabbro M, Ledermann JA, Lorusso D, Vergote I, et al. Niraparib maintenance therapy in platinum-sensitive, recurrent ovarian cancer. N Engl J Med. 2016;375:2154–2164. doi: 10.1056/NEJMoa1611310. [DOI] [PubMed] [Google Scholar]
- Misale S, Yaeger R, Hobor S, Scala E, Janakiraman M, Liska D, Valtorta E, Schiavo R, Buscarino M, Siravegna G, et al. Emergence of KRAS mutations and acquired resistance to anti-EGFR therapy in colorectal cancer. Nature. 2012;486:532–536. doi: 10.1038/nature11156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearson A, Smyth E, Babina IS, Herrera-Abreu MT, Tarazona N, Peckitt C, Kilgour E, Smith NR, Geh C, Rooney C, et al. High-level clonal FGFR amplification and response to FGFR inhibition in a translational clinical trial. Cancer Discov. 2016;6:838–851. doi: 10.1158/2159-8290.CD-15-1246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peschard P, Fournier TM, Lamorte L, Naujokas MA, Band H, Langdon WY, Park M. Mutation of the c-Cbl TKB domain binding site on the Met receptor tyrosine kinase converts it into a transforming protein. Mol Cell. 2001;8:995–1004. doi: 10.1016/s1097-2765(01)00378-1. [DOI] [PubMed] [Google Scholar]
- Piccart-Gebhart MJ, Procter M, Leyland-Jones B, Goldhirsch A, Untch M, Smith I, Gianni L, Baselga J, Bell R, Jackisch C, et al. Trastuzumab after adjuvant chemotherapy in HER2-positive breast cancer. N Engl J Med. 2005;353:1659–1672. doi: 10.1056/NEJMoa052306. [DOI] [PubMed] [Google Scholar]
- Planchard D, Kim TM, Mazieres J, Quoix E, Riely G, Barlesi F, Souquet PJ, Smit EF, Groen HJ, Kelly RJ, et al. Dabrafenib in patients with BRAF(V600E)-positive advanced non-small-cell lung cancer: a single-arm, multicentre, open-label, phase 2 trial. Lancet Oncol. 2016;17:642–650. doi: 10.1016/S1470-2045(16)00077-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poulikakos PI, Persaud Y, Janakiraman M, Kong X, Ng C, Moriceau G, Shi H, Atefi M, Titz B, Gabay MT, et al. RAF inhibitor resistance is mediated by dimerization of aberrantly spliced BRAF(V600E) Nature. 2011;480:387–390. doi: 10.1038/nature10662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prahallad A, Sun C, Huang S, Di Nicolantonio F, Salazar R, Zecchin D, Beijersbergen RL, Bardelli A, Bernards R. Unresponsiveness of colon cancer to BRAF(V600E) inhibition through feedback activation of EGFR. Nature. 2012;483:100–103. doi: 10.1038/nature10868. [DOI] [PubMed] [Google Scholar]
- Pritchard CC, Mateo J, Walsh MF, De Sarkar N, Abida W, Beltran H, Garofalo A, Gulati R, Carreira S, Eeles R, et al. Inherited DNA-repair gene mutations in men with metastatic prostate cancer. N Engl J Med. 2016;375:443–53. doi: 10.1056/NEJMoa1603144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robert C, Karaszewska B, Schachter J, Rutkowski P, Mackiewicz A, Stroiakovski D, Lichinitser M, Dummer R, Grange F, Mortier L, et al. Improved overall survival in melanoma with combined dabrafenib and trametinib. N Engl J Med. 2015;372:30–39. doi: 10.1056/NEJMoa1412690. [DOI] [PubMed] [Google Scholar]
- Roychowdhury S, Iyer MK, Robinson DR, Lonigro RJ, Wu YM, Cao X, Kalyana-Sundaram S, Sam L, Balbin OA, Quist MJ, et al. Personalized oncology through integrative high-throughput sequencing: a pilot study. Sci Transl Med. 2011;3:111ra121. doi: 10.1126/scitranslmed.3003161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russo M, Siravegna G, Blaszkowsky LS, Corti G, Crisafulli G, Ahronian LG, Mussolin B, Kwak EL, Buscarino M, Lazzari L, et al. Tumor heterogeneity and lesion-specific response to targeted therapy in colorectal cancer. Cancer Discov. 2016;6:147–153. doi: 10.1158/2159-8290.CD-15-1283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saglio G, Kim DW, Issaragrisil S, le Coutre P, Etienne G, Lobo C, Pasquini R, Clark RE, Hochhaus A, Hughes TP, et al. Nilotinib versus imatinib for newly diagnosed chronic myeloid leukemia. N Engl J Med. 2010;362:2251–2259. doi: 10.1056/NEJMoa0912614. [DOI] [PubMed] [Google Scholar]
- Scher HI, Fizazi K, Saad F, Taplin ME, Sternberg CN, Miller K, de Wit R, Mulders P, Chi KN, Shore ND, et al. Increased survival with enzalutamide in prostate cancer after chemotherapy. N Engl J Med. 2012;367:1187–1197. doi: 10.1056/NEJMoa1207506. [DOI] [PubMed] [Google Scholar]
- Schrader KA, Cheng DT, Joseph V, Prasad M, Walsh M, Zehir A, Ni A, Thomas T, Benayed R, Ashraf A, et al. Germline variants in targeted tumor sequencing using matched normal DNA. JAMA Oncol. 2016;2:104–111. doi: 10.1001/jamaoncol.2015.5208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw AT, Kim DW, Mehra R, Tan DS, Felip E, Chow LQ, Camidge DR, Vansteenkiste J, Sharma S, De Pas T, et al. Ceritinib in ALK-rearranged non-small-cell lung cancer. N Engl J Med. 2014;370:1189–1197. doi: 10.1056/NEJMoa1311107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw AT, Friboulet L, Leshchiner I, Gainor JF, Bergqvist S, Brooun A, Burke BJ, Deng YL, Liu W, Dardaei L, et al. Resensitization to crizotinib by the lorlatinib ALK resistance mutation L1198F. N Engl J Med. 2016;374:54–61. doi: 10.1056/NEJMoa1508887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen R, Seshan VE. FACETS: allele-specific copy number and clonal heterogeneity analysis tool for high-throughput DNA sequencing. Nucleic Acids Res. 2016;44:e131. doi: 10.1093/nar/gkw520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siu LL, Lawler M, Haussier D, Knoppers BM, Lewin J, Vis DJ, Liao RG, Andre F, Banks I, Barrett JC, et al. Facilitating a culture of responsible and effective sharing of cancer genome data. Nat Med. 2016;22:464–471. doi: 10.1038/nm.4089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slamon DJ, Leyland-Jones B, Shak S, Fuchs H, Paton V, Bajamonde A, Fleming T, Eiermann W, Wolter J, Pegram M, et al. Use of chemotherapy plus a monoclonal antibody against HER2 for metastatic breast cancer that overexpresses HER2. N Engl J Med. 2001;344:783–792. doi: 10.1056/NEJM200103153441101. [DOI] [PubMed] [Google Scholar]
- Stransky N, Cerami E, Schalm S, Kim JL, Lengauer C. The landscape of kinase fusions in cancer. Nat Commun. 2014;5:4846. doi: 10.1038/ncomms5846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swain SM, Baselga J, Kim SB, Ro J, Semiglazov V, Campone M, Ciruelos E, Ferrero JM, Schneeweiss A, Heeson S, et al. Pertuzumab, trastuzumab, and docetaxel in HER2-positive metastatic breast cancer. N Engl J Med. 2015;372:724–734. doi: 10.1056/NEJMoa1413513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabernero J, Bahleda R, Dienstmann R, Infante JR, Mita A, Italiano A, Calvo E, Moreno V, Adamo B, Gazzah A, et al. Phase I dose-escalation study of JNJ-42756493, an oral pan-fibroblast growth factor receptor inhibitor, in patients with advanced solid tumors. J Clin Oncol. 2015;33:3401–3408. doi: 10.1200/JCO.2014.60.7341. [DOI] [PubMed] [Google Scholar]
- Tannock IF, Hickman JA. Limits to personalized cancer medicine. N Engl J Med. 2016;375:1289–1294. doi: 10.1056/NEJMsb1607705. [DOI] [PubMed] [Google Scholar]
- Telli ML, Timms KM, Reid J, Hennessy B, Mills GB, Jensen KC, Szallasi Z, Barry WT, Winer EP, Tung NM, et al. Homologous recombination deficiency (HRD) score predicts response to platinum-containing neoadjuvant chemotherapy in patients with triple-negative breast cancer. Clin Cancer Res. 2016;22:3764–3773. doi: 10.1158/1078-0432.CCR-15-2477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thress KS, Paweletz CP, Felip E, Cho BC, Stetson D, Dougherty B, Lai Z, Markovets A, Vivancos A, Kuang Y, et al. Acquired EGFR C797S mutation mediates resistance to AZD9291 in non-small cell lung cancer harboring EGFR T790M. Nat Med. 2015;21:560–562. doi: 10.1038/nm.3854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiacci E, Park JH, De Carolis L, Chung SS, Broccoli A, Scott S, Zaja F, Devlin S, Pulsoni A, Chung YR, et al. Targeting mutant BRAF in relapsed or refractory hairy-cell leukemia. N Engl J Med. 2015;373:1733–1747. doi: 10.1056/NEJMoa1506583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tirosh I, Izar B, Prakadan SM, Wadsworth MH, 2nd, Treacy D, Trombetta JJ, Rotem A, Rodman C, Lian C, Murphy G, et al. Dissecting the multicellular ecosystem of metastatic melanoma by single-cell RNA-seq. Science. 2016;352:189–196. doi: 10.1126/science.aad0501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toy W, Shen Y, Won H, Green B, Sakr RA, Will M, Li Z, Gala K, Fanning S, King TA, et al. ESR1 ligand-binding domain mutations in hormone-resistant breast cancer. Nat Genet. 2013;45:1439–1445. doi: 10.1038/ng.2822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner NC, Ro J, André F, Loi S, Verma S, Iwata H, Harbeck N, Loibl S, Huang Bartlett C, Zhang K, et al. Palbociclib in hormone-receptor-positive advanced breast cancer. N Engl J Med. 2015;373:209–219. doi: 10.1056/NEJMoa1505270. [DOI] [PubMed] [Google Scholar]
- Vaishnavi A, Le AT, Doebele RC. TRKing down an old oncogene in a new era of targeted therapy. Cancer Discov. 2015;5:25–34. doi: 10.1158/2159-8290.CD-14-0765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voest EE, Bernards R. DNA-guided precision medicine for cancer: a case of irrational exuberance? Cancer Discov. 2016;6:130–132. doi: 10.1158/2159-8290.CD-15-1321. [DOI] [PubMed] [Google Scholar]
- Wan PT, Garnett MJ, Roe SM, Lee S, Niculescu-Duvaz D, Good VM, Jones CM, Marshall CJ, Springer CJ, Barford D, Marais R, Cancer Genome Project Mechanism of activation of the RAF-ERK signaling pathway by oncogenic mutations of B-RAF. Cell. 2004;116:855–867. doi: 10.1016/s0092-8674(04)00215-6. [DOI] [PubMed] [Google Scholar]
- Yang JC, Sequist LV, Geater SL, Tsai CM, Mok TS, Schuler M, Yamamoto N, Yu CJ, Ou SH, Zhou C, et al. Clinical activity of afatinib in patients with advanced non-small-cell lung cancer harbouring uncommon EGFR mutations: a combined post-hoc analysis of LUX-Lung 2, LUX-Lung 3, and LUX-Lung 6. Lancet Oncol. 2015;16:830–838. doi: 10.1016/S1470-2045(15)00026-1. [DOI] [PubMed] [Google Scholar]
- Yao Z, Torres NM, Tao A, Gao Y, Luo L, Li Q, de Stanchina E, Abdel-Wahab O, Solit DB, Poulikakos PI, Rosen N. BRAF mutants evade ERK-dependent feedback bydifferent mechanisms that determine their sensitivity to pharmacologic inhibition. Cancer Cell. 2015;28:370–383. doi: 10.1016/j.ccell.2015.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu M, Bardia A, Wittner BS, Stott SL, Smas ME, Ting DT, Isakoff SJ, Ciciliano JC, Wells MN, Shah AM, et al. Circulating breast tumor cells exhibit dynamic changes in epithelial and mesenchymal composition. Science. 2013;339:580–584. doi: 10.1126/science.1228522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaretsky JM, Garcia-Diaz A, Shin DS, Escuin-Ordinas H, Hugo W, Hu-Lieskovan S, Torrejon DY, Abril-Rodriguez G, Sandoval S, Barthly L, et al. Mutations associated with acquired resistanceto PD-1 blockade in melanoma. N Engl J Med. 2016;375:819–829. doi: 10.1056/NEJMoa1604958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Walsh MF, Wu G, Edmonson MN, Gruber TA, Easton J, Hedges D, Ma X, Zhou X, Yergeau DA, et al. Germline mutations in predisposition genes in pediatric cancer. N Engl J Med. 2015;373:2336–2346. doi: 10.1056/NEJMoa1508054. [DOI] [PMC free article] [PubMed] [Google Scholar]