

SUMMARY
Cystic fibrosis (CF) is an autosomal recessive disease caused by mutations in the cystic fibrosis transmembrane regulator (CFTR) gene. About 2000 mutations have been described so far. We setup an ex vivo model of human nasal epithelial cells (HNECs) to study CF patients testing the effect of novel mutations and molecular therapies. We performed sampling (by brushing), followed by culture and analysis of HNECs using a series of molecular techniques. We performed 50 brushings from CF patients and controls. Using cultured cells, we: i) demonstrated the widely heterogeneous CFTR expression in patients and in controls; ii) defined the splicing effect of a CFTR mutation; iii) assessed the CFTR gating activity in patients bearing different mutations; iv) demonstrated that butyrate significantly enhances CFTR expression. Based on our data, we can conclude: 1) HNEC brushing is performed without anaesthesia and is well tolerated in all CF patients (children and adults); 2) HNECs can be preserved for up to 48 hours before culture allowings multicentre studies; 3) HNECs culture can be considered a suitable model to study the molecular effects of new CFTR gene mutations and/or uncertain meaning specific mutations of carriers; 4) an ex vivo model of HNECs may be used to evaluate, before human use, the effect of new drugs on patients’ cells bearing specific CFTR mutations; 5) the methodology is adequate for a quantitative measurement, by fluorescence, of the CFTR gating activity of the HNECs from patients with different genotypes identifying: a) CF patients bearing two severe mutations with an activity < 10% (compared to controls – 100%); b) CF patients bearing at least a mild mutation with an activity of 10-20%; c) CF carriers (heterozygous subjects) with an activity between 40-70%.
KEY WORDS: CFTR, Nasal brushing, Cystic Fibrosis, CF, Mutations
RIASSUNTO
La fibrosi cistica (FC) è una malattia autosomica recessiva causata da mutazioni nel gene CFTR (Cystic Fibrosis Transmembrane conductance Regulator). Finora sono state descritte circa 2000 mutazioni, ma per la maggior parte di esse è difficile definirne l'effetto senza complesse procedure in vitro. Abbiamo effettuato il campionamento (mediante brushing), la cultura e l'analisi di cellule epiteliali nasali umane (HNEC) utilizzando una serie di tecniche che possono aiutare a testare l'effetto delle mutazioni CFTR. Abbiamo eseguito 50 brushing da pazienti FC e controlli, e in 45 casi si è ottenuta una coltura positiva. Utilizzando cellule in coltura: i) abbiamo dimostrato l'espressione ampiamente eterogenea del CFTR nei pazienti e nei controlli; ii) abbiamo definito l'effetto di splicing di una mutazione sul gene CFTR; iii) abbiamo valutato l'attività di gating di CFTR in pazienti portatori di differenti mutazioni; iv) abbiamo dimostrato che il butirrato migliora in modo significativo l'espressione di CFTR. I dati provenienti dal nostro studio sperimentale dimostrano che l'uso del modello ex-vivo di cellule epiteliali nasali è un importante e valido strumento di ricerca e di diagnosi nella studio della FC e può anche essere mirato alla sperimentazione ed alla verifica di nuovi farmaci. In definitiva, in base ai nostri dati è possibile esprimere le seguenti conclusioni: 1) il prelievo delle cellule epiteliali nasali mediante brushing è applicabile senza alcuna anestesia ed è ben tollerato da tutti i pazienti affetti da FC (bambini e adulti), è scarsamente invasivo e facilmente ripetibile, è anche in grado di ottenere una sufficiente quantità di HNECs rappresentative, ben conservate, idonee allo studio della funzionalità di CFTR; 2) la conservazione delle cellule prelevate è possibile fino a 48 ore prima che si provveda all'allestimento della coltura e ciò permette di avviare studi multicentrici con prelievi in ogni sede e quindi di ottenere una ampia numerosità campionaria; 3) la coltura di cellule epiteliali nasali può essere considerata un modello adatto a studiare l'effetto molecolare di nuove mutazioni del gene CFTR e/o mutazioni specifiche di pazienti "carriers" dal significato incerto; 4) il modello ex-vivo delle HNECs consente inoltre di valutare, prima dell'impiego nell'uomo, l'effetto di farmaci (potenziatori e/o correttori) sulle cellule di pazienti portatori di mutazioni specifiche di CFTR; tali farmaci possono modulare l'espressione genica del canale CFTR aprendo così nuove frontiere terapeutiche e migliori prospettive di vita per pazienti affetti da una patologia cronica come la Fibrosi Cistica; 5) la metodologia da noi istituita risulta essere idonea alla misura quantitativa, mediante fluorescenza, dell'attività di gating del canale CFTR presente nelle membrane delle cellule epiteliali nasali prelevate da pazienti portatori di differenti genotipi; in tal modo è possibile individuare: a) pazienti FC portatori di 2 mutazioni gravi con un'attività < 10% (in rapporto ai controlli -100%), b) soggetti FC portatori contemporaneamente di una mutazione grave e di una lieve con un'attività tra 10-30%, c) i cosiddetti portatori "carriers"- eterozigoti - con un'attività tra 40-70%.
In conclusione la possibilità di misurare l'attività del canale CFTR in HNECs fornisce un importante contributo alla diagnosi di FC, mediante individuazione di un "cut-off diagnostico", ed anche alla previsione della gravità fenotipica della malattia; quindi quanto rilevabile dalla misura del suddetto canale permette di prospettare per il futuro la possibilità di valutare meglio i pazienti per i quali il test del sudore ha dato risultati ambigui (borderline o negativi).
La metodica da noi sperimentata consente anche di monitorare i pazienti durante il trattamento farmacologico, valutando in tal modo i reali effetti delle nuove terapie.
Introduction
Cystic fibrosis (CF) is an autosomal recessive disease caused by mutations in the cystic fibrosis transmembrane regulator (CFTR) gene that encodes the CFTR membrane cAMP-activated chloride (Cl-) channel. To date, about 2000 mutations have been reported in the disease gene in patients with classic CF and in those with milder, atypical CF that are sometimes difficult to diagnose, particularly in adults 1. However, only for a few CFTR mutations has a molecular effect been defined. Molecular analysis contributes to confirm diagnosis, identify asymptomatic carriers and perform prenatal diagnosis in high risk couples 2. It is based on the analysis of a panel of the most frequent mutations that identifies about 80% of CF alleles using commercial kits 3. The subsequent sequencing of the entire coding regions of the CFTR gene 4 that includes the study of large rearrangements 5 is available. However, these procedures frequently identify novel mutations for which it is difficult to define the effect and pathogenicity without complex in vitro procedures 6.
Furthermore, only symptomatic therapies are available for CF patients, even if, recently, novel drugs that may potentiate the CFTR protein activity or may correct mislocalisation of the mutated protein have become available 7. Such therapies have an effect only in patients bearing mutations with specific effects, so it is difficult to select patients that may benefit from novel molecular drugs.
We established the sampling, culture and analysis of human nasal epithelial cells (HNEC) using a series of techniques that may help to test the effect of CFTR mutations. This ex vivo model may contribute either to study the effect of novel mutations and to assess the effect of novel molecular therapies on cells from patients bearing specific mutations.
Materials and methods
Nasal brushing
Informed consent was obtained from patients (legal guardians for minors) before sampling, after a complete description of the aims of the study. All subjects underwent complete ear-nose-throat evaluation. Freshly-isolated HNECs were collected by nasal brushing. After nasal washings with physiological saline to remove mucus (two washings per day in the week before and one washing immediately before sampling), nasal brushing was performed by a soft sterile interdental brush with 2.5 to 3 mm bristles (Paro-Isola, Switzerland) by scraping (Fig. 1a) along the middle portion of the inferior turbinate using gentle backward–forward and rotatory movements (circular movement) in each nostril, under direct visualisation, using a headlamp without decongestant or local anaesthesia (Fig. 1b). Patients were carefully monitored for vital and minor signs, comfort and pain. They were discharged on the same day.
Fig. 1.

A) an example of scraping using a soft sterile interdental brush with 2.5 to 3 mm bristles (Paro-Isola, Switzerland); B) ENT specialist during execution of nasal brushing in a patient.
Culture of nasal cells
The sample obtained from each nostril was immediately conserved in a 15 mL tube containing 2.5 mL of RPMI 1640 medium, with 3% antibiotics. Cells were placed on an Eppendorf Thermomixer and agitated at 700 rpm for one hour to remove all cells from the bristles. Once the brush was removed, cells were centrifuged at 2000 rpm for 20 minutes, the supernatant were discarded and cells were resuspended in serum-free bronchial epithelial cell growth medium BEGM (Clonetics, MD). Next, cells were placed in CELL T 25 flasks (Sarstedt Ltd, UK). At confluence of 60%, cells were passed in new flasks after counting using Invitrogen (Italy) Cell Countess. A trypan blue exclusion test was used to establish the total number of viable cells and percentage of viability. Nasal cells can be stored at 4°C up to 48 h before culturing (using the RPMI 1640 medium), and this permits collecting samples from patients followed in other centres. At a confluence of > 80%, cells were treated with 5 mM sodium butyrate for 24 hours.
Nasal cytology
Epithelial cells were classified into three major categories (ciliated, non-ciliated, striated and basal) on the basis of described criteria 8. Ciliated cells (target of our study) represent more than 80% of cells obtained from nasal brushing; they have tall columnar shapes with distinct cilia; no ciliated cells, including secretory goblet cells, have similar shape but no cilia; striated and basal cells are smaller with dense, round nuclei, strongly stained cytoplasms, and a high nuclear-to-cytoplasmic ratio. In addition, leukocytes or inflammatory cells may be found in the brushing sample if the patient had an inflammatory condition.
We used May-Grunwald-Giemsa staining of freshly obtained nasal cells to verify the presence of an adequate amount of ciliated cells, and the possible presence of inflammatory cells. The freshly isolated human cells recovered from nasal brushings and spread on silane glass slides were stained with May-Grunwald-Giemsa. After 5 minutes of fixation in methanol, slides were immersed for 5 min in May-Grunwald's standard stain (Fluka Chemie, Switzerland), freshly diluted with an equal volume of phosphate buffer pH 6.8, and then, without washing, immersed for 10 to 15 min in Giemsa stain (Merck, Germany) diluted with 9 volumes of phosphate buffer pH 6.8. After 3-4 rapid washes in phosphate buffer pH 6.8 and 2 to 5 min in water, slides were mounted with Entellan (Merck), covered with glass coverslips and dried for at least 1 hour before analysis. Samples on slides were evaluated for cell differential count and morphology on a conventional light microscope (Zeiss, Germany).
Furthermore, the culture of nasal epithelial cells helps to selectively expand epithelial ciliate cells. To verify that such cells maintain their phenotype after prolonged culture (> 20 days) we used cytokeratin staining. Anti- KRT18/ cytokeratin-18 (CK- 18; Abcam, Italy, ab52948) antibody was used to confirm epithelial cell purity, and that with anti-CD3 +, (Abcam, ab5690), CD4 + (Abcam, ab51312), or CD19 + (Abcam, ab25232) 1:500 antibodies was used to exclude the presence of lymphocytes or inflammatory cells. Moreover, cells were treated with anti-Pan-cytokeratin (C5992, Sigma Aldrich, Italy) 1:500 and MUC5AC (Abcam, ab3649) or MUC3B (Abcam, ab85006) 1:200 antibodies to exclude mucipar differentiation.
Real-time PCR for quantitative analysis of CFTR mRNA
Total RNA was isolated from nasal epithelial cells using TRIzol reagent (Invitrogen, Italy) as previously described 9. RNA concentration and purity was determined with a Nano- Drop ND-1000 spectrophotometer; reverse transcription was carried out on 1 μg of total RNA resuspended in DEPC-treated nano-pure water using a QuantiTect Rev Transcription Kit (Qiagen, CA) using the protocol supplied by the manufacturer. To evaluate levels of CFTR transcript in nasal epithelial cells, relative quantification by real-time PCR was performed in duplicates using LightCycler 480 Probes Master containing CFTR primers (Roche, Italy) and a TaqMan CFTR probe (ID. Assay 102716). Amplification was carried out with a LightCycler 480 System for real-time PCR (Roche) with a two-step PCR protocol (preincubation for 10 min at + 95° C followed by 45 cycles of amplification: 95°C for 10 sec, 60°C for 25 sec, 72°C for 1 sec). mRNA quantification results were normalised using glyceraldehyde 3-phosphate dehydrogenase (GAPDH) gene (Roche, ID. Assay 101128) as an endogenous control.
RT-PCR analysis to assess the effect of splicing mutations
All mutations that are located in the exon-intron boundary were, first of all, analysed by prediction software such as Alamut or NetGene2. Next, if in silico analysis predicted an alteration of the splicing pattern, we performed an electrophoretic analysis on cDNA obtained by RT-PCR from CFTR mRNA extracted from cultured nasal cells. We used different pairs of primers complementary to two (or more) subsequent exonic sequences. Using these primers, intronic DNA sequences retained in the mRNA due to the altered splicing (if present) were amplified, giving rise to one or more bands of greater size compared to wild type.
Quantitative analysis of CFTR channel activity
To test the functionality of the CFTR protein, we used the halide-sensitive fluorescent system. The iodide-sensitive fluorescent indicator, SPQ (Molecular Probes, Invitrogen, M440), was introduced into cells in a hypotonic solution of iodide buffer (130 mM NaI, 4 mM KNO3, 1 mM Ca(NO3)2, 1 mM Mg(NO3)2, 10 mM glucose and 20 mM HEPES, pH 7.4) diluted 1:1 with water and containing a final concentration of 10 μm SPQ. Nasal cells were loaded for 20 min at 37°C in a humidified chamber with 5% CO2. SPQ-loaded cells were then mounted on a LSM510 meta-confocal microscope with a 37°C heated stage and perfused with iodide buffer. Changes in CFTR-mediated SPQ fluorescence were monitored at 445 nm in response to excitation at 340 nm. Fluorescent was constantly measured by the passage between different solutions containing halide anions. Cells were initially perfused with iodide buffer followed by perfusion with nitrate buffer (NaI replaced with 130 mM NaNO3) with the addition of specific activators of CFTR channel as forskolin (20 μM) (Sigma Aldrich) and genistein (50 μM) (Sigma Aldrich). The peak iodide efflux rate was calculated in accordance with the Stern-Volmer relationship as follows:
where F is the observed fluorescence, Fo is the fluorescence in the absence of a quenching anion, CQ is the concentration of the quenching anion, and K is the Stern-Volmer quench constant. The rates were calculated using SigmaPlot Version 7.1 for each mean fluorescence trace generated from the 50 cells examined per population per coverslip.
Statistical analysis
For real-time PCR, the values of CFTR mRNA are reported as means +/- SD ratio to GAPDH housekeeping mRNA. Rate of chloride efflux was measured in at least 50 cells for experiment. Mean ± SD are those of three experiments. Statistical significance was defined as a p value of < 0.05 vs control subjects.
Results
We collected samples of nasal epithelial cells from 20 healthy volunteers and from 30 CF patients (or carriers) with different CFTR mutations. In all 50 cases, the sampling was obtained successfully, with any complication or discomfort for subjects; May-Grunwald-Giemsa staining (performed in 20 cases) confirmed the presence of adequate amounts of ciliated epithelial cells. In all cases we cultured cells and in 45/50 (90.0%) cases we obtained a positive culture. Figure 2 shows an example of the culture of nasal epithelial cells at different days. In 5/50 cases the cells did not expand due to the low number of cells obtained by sampling because of the strong contamination of cells with mucus or with a high number of keratinocytes. In fact, we modified our original protocol, and before sampling we now: i) carefully verify the absence of any clinical condition potentially associated with high mucus production; ii) perform washings with physiological solutions (see Materials and methods).
Fig. 2.
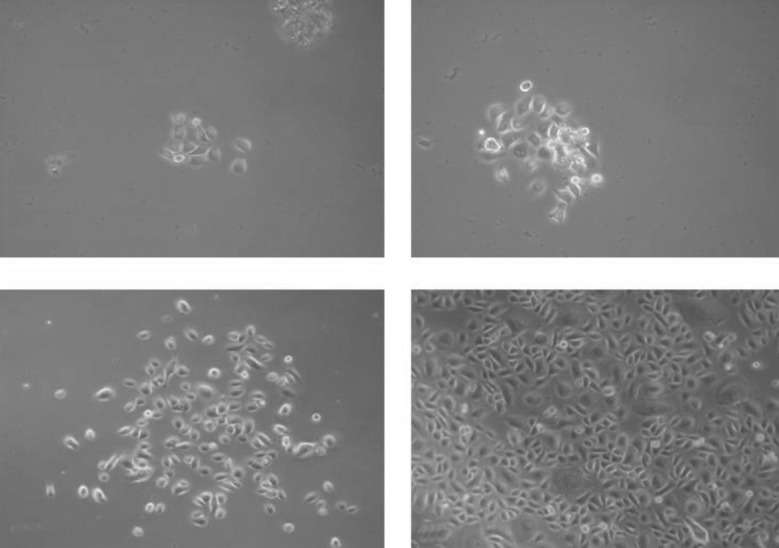
An example of human nasal epithelial cell expansion at different days of culture. A: 3 days; B: 4 days; C: 7 days; D: 10 days.
In order to verify that the culture did not modify the phenotype of cultured cells, we used a panel of anti-cytokeratin antibodies, specific for epithelial cells; furthermore, we assessed, by quantitative RT-PCR, the levels of CFTR transcript in cells before culture and at different days of culture until day 20 (in 10 different experiments), and no significant changes were observed (data not shown). Finally, we assessed the effect of storage of cells in transport medium before culture, and in 10 different experiments we demonstrated that cells can be stored at least 48 hr at 4°C before culture.
Next we analysed, by quantitative RT-PCR, the levels of CFTR mRNA as a ratio with the GAPDH gene transcript in a control sample from a healthy subject and in CF patients with different mutations (Fig. 3); this analysis can be performed either on RNA from cultured cells or on RNA extracted from fresh sampled nasal cells entrapped in the brush, avoiding culture. The analysis showed a very heterogeneous basal expression of CFTR mRNA. We then treated cultured nasal epithelial cells from 5 controls and 20 patients with sodium butyrate. The treatment caused the enhancement of CFTR mRNA in all cases (see an example in Fig. 4).
Fig. 3.
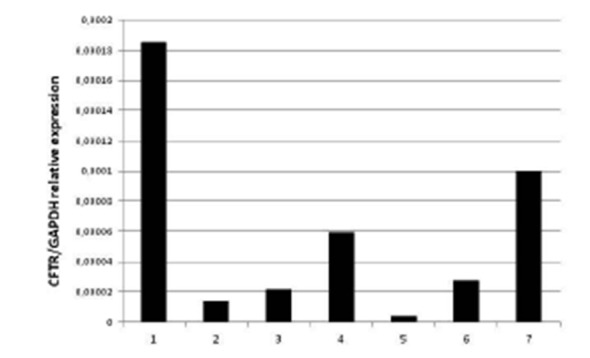
Quantitative RT-PCR analysis of CFTR mRNA levels expressed as a ratio to the housekeeping GAPDH mRNA. 1: control sample from a healthy subject; 2 to 7: samples obtained from CF patients with different CFTR genotypes.
Fig. 4.
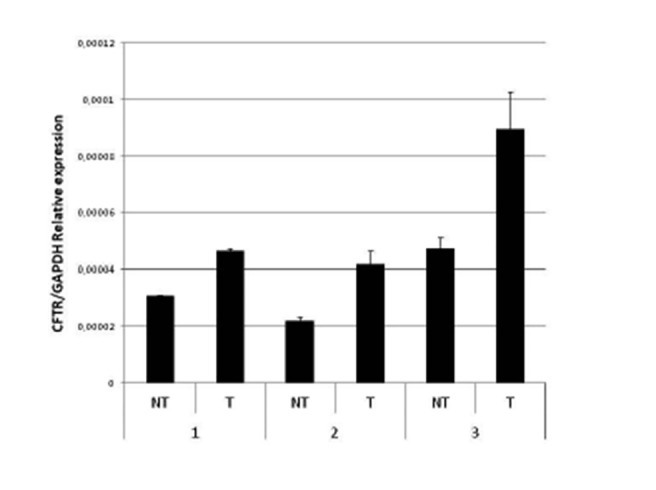
Effect of butyrate on CFTR mRNA expression. The figure shows the quantitative RT-PCR analysis of CFTR mRNA levels expressed as a ratio to the housekeeping GAPDH mRNA in three samples of nasal epithelial non-treated (NT) and butyrate- treated cells (T).
Furthermore, we studied a CF patient heterozygous for the 711 + 1G > A mutation, that was predicted to cause the altered splicing of exon 5. The analysis was performed by RT-PCR, using primers that included CFTR exons 4, 5 and 6. Electrophoretic analysis of the cDNA amplicon clearly showed that the 711 + 1G > A mutation caused the retention of intron 5 due to altered splicing (Fig. 5).
Fig. 5.

RT-PCR analysis of CFTR mRNA from a healthy control subject (1) and from a CF patient heterozygous for the 711+1G>A mutation (2). The mutation has a potential effect of altered splicing of the CFTR mRNA causing the retention of an intronic sequence that appears as an electrophoretic band with a higher molecular weight in addition to the normal band also present in the healthy subject.
Finally, we analysed the quantitative gating activity of CFTR in all 30 CF patients or carriers. In all cases, the analysis provided a clear result, and Figure 6 shows several examples: # 1 is a healthy control subject (its activity is considered 100%); # 2 and 3 are two CF patients with two severe mutations each (i.e., F508del/F508del for case # 2 and G542X/4016insT for case # 3): they show an activity of 9.9% and 10.4% compared to the control, respectively; case # 4 is a CF patient with a severe and a mild CF mutation (i.e., W1282X/D1152H), which showed an activity of about 20.3%. Finally, case # 5 is a heterozygous carrier of the severe G542X mutation, which showed an activity of 76.8%.
Fig. 6.
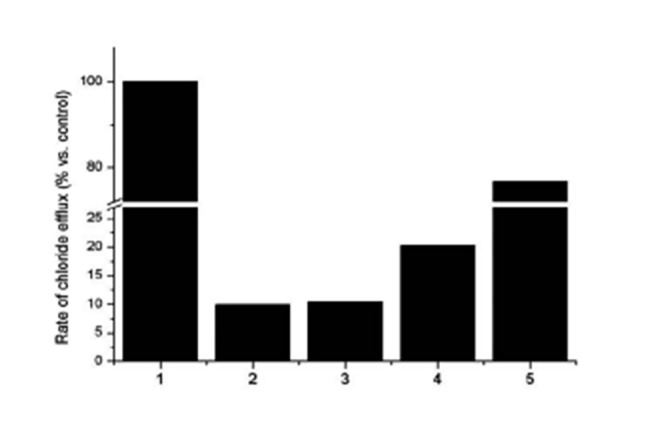
Quantitative gating activity of CFTR. #1 is a healthy control subject (its activity is considered 100%); #2 and #3 are two CF patients compound heterozygous for two severe mutations each (i.e., F508del/F508del for case #2 and G542X/4016insT for case #3): they show an activity of 9.9% and 10.4% compared to the control, respectively; case #4 is a CF patient with a severe and a mild CF mutation (i.e., W1282X/D1152H) with 20.3% of activity. Finally, case #5 is a heterozygous carrier of the severe G542X mutation with an activity of 76.8%.
Discussion
Culture of HNECs is a suitable model to study the molecular effect of CFTR mutations and to assess the effects of novel drugs in cells from patients bearing specific CFTR mutations. We already used this model to study the effect of butyrate on the expression of the SLC26A3 gene in patients with congenital chloride diarrhoea 10. In the present study, we improved and validated the procedures for sampling and culture on a large number of cases, and established procedures for molecular analysis of the CFTR gene. The sampling was well tolerated by all 50 subjects studied. Interestingly, the use of the transport medium permits storing sampled cells before culture for up to 48 hours, allowing the analysis of sampled cells from other centres.
Staining with May-Grunwald-Giemsa and anti-cytokeratin antibodies confirmed that we effectively obtained and cultured HNECs without contamination of the culture with inflammatory cells. HNECs can be effectively cultured up to 15 days, as we also demonstrated in a previous study in which we assessed the effect of mannose binding lectin on several types of cells 11. So far, only a few studies have been performed on the use of human ex vivo models for CF. This is mainly because of the invasiveness and the risk of most techniques used to collect human cells, the small number of cells collected, and the limited number, poor quality, and non representative nature of samples resulting from surgery (such as nasal polypectomies or lung transplants). Brushing of the respiratory tract allows easy sampling of numerous, representative, well-preserved and dissociated cells from the superficial mucosa. The group by Garratt et al., recently described the bronchial brushing technique as a possible gold standard model of airway disease in CF, but such sampling requires anaesthesia of patients, and less that 50% of samples could be successfully cultured 12. Another study examined the use of porcine nasal epithelial cells in culture as a model to study the pathogenesis of sinusitis, but a such model is limited by the possibility to study transgenic pigs with only a single CFTR genotype 13.
Other studies used cultured cells from nasal polyps. Recently, for example, a very elegant study carried out proteomic analysis of nasal epithelial cells obtained from nasal polyps 14. However, the limit of such approach is that only a few percentage of CF patients undergo surgery for rhinosinusitis with nasal polyps 15 16. The availability of cells from patients bearing specific mutations permits study of the molecular effects of mutations of uncertain significance. For example, mutations within exon-intron boundaries may affect the splicing process (more than two dozen CFTR mutations described so far impair the splicing process), also in addition to silent mutations (i.e., missense mutations that do not change the amino acid) may impair the splicing 17. The study of the splicing effect of novel mutations would require a complex procedure to express the mutation in vitro, followed by the mini-gene assay 17. This is a rather complex and expensive procedure, and not available for routine use. Conversely, the availability of nasal cells directly from the patient with the mutation to be characterised permits studying the splicing effect with a simple RT-PCR reaction followed by electrophoresis. This analysis can be performed on nasal epithelial cells with no need for culture. Both our group for a mutation 1 and another for two mutations 18 demonstrated that the results obtained with this novel procedure fully match with those obtained with the classic minigene assay.
Quantitative RT-PCR analysis can be performed rapidly on cultured and directly on fresh sampled nasal cells, and can be used to assess the effect of mutations in the promoter region 19 or in other regulatory regions of the gene 20 21 revealing the mutations that may cause a reduced gene expression. Also in this case, a simple quantitative RT-PCR analysis of epithelial nasal cells would avoid the complex and expensive procedure of in vitro expression and analysis of mutations in cell lines 19. However, our study demonstrated that the levels of CFTR gene expression are highly heterogeneous in normal subjects and in CF patients, and it would be necessary to study a larger number of healthy subjects to obtain reference values. Quantitative RT-PCR analysis may be used also to assess the effect of potential drugs that may enhance or reduce gene expression, like butyrate, that seems to enhance CFTR gene expression.
Finally, we also set up a functional method for the quantitative fluorescence measurement of chloride secretion to assess the gating activity of CFTR protein. Chloride transport across epithelial cell membranes can be assessed using a fluorescent microscopic assay based on the quenching of a water-soluble fluorescent dye, SPQ, by iodide. The possibility to measure the channel activity of CFTR in cells from patients bearing different genotypes is a useful contribution to either CF diagnosis and to the prediction of the phenotypic severity.
In particular, CF patients bearing two severe mutations have an activity < 10%, as in case 2 (Fig. 6; homozygous for the F508del microdeletion) and in case 3 who is heterozygous for the G542X nonsense mutation and for the 4016insT variant, a severe mutation frequently observed in CF alleles from southern Italy 22. However, CF patients bearing a mild mutation, such as D1152H 23 present in patient 4 shown in Figure 6 shows an activity between 10% and 20%. Of course, quantitative analysis of CFTR in nasal cells can also be used to assess, in the ex vivo model from patients bearing specific mutations, the effect of potential drugs like potentiators and/or correctors 7 or molecular therapies 24 25 before their use in humans.
Conclusions
The ex vivo model of cultured HNECs is a very contributory tool to study the pathogenetic mechanism of specific CF mutations directly on cells from the affected patient allowing investigation of the effect of novel mutations and assessment of the effect of novel molecular therapies. Based on our data, we conclude the following:
HNEC brushing can be performed without anaesthesia and is well tolerated in all CF patients (children and adults). It is slightly invasive, easily repeatable, and allows collection of a sufficient amount of representative, well-preserved HNECs, which are suitable for studying respiratory epithelium through a wider range of cell culture techniques;
HNEC can be preserved for up to 48 hours before culture, thus allowing multicentre studies with large samples;
HNEC culture can be considered a suitable model to study the molecular effects of new CFTR gene mutations and/or uncertain meaning specific mutations in carriers;
the ex-vivo model of HNECs may be used to evaluate, before human use, the effect of new drugs (potentiators and/or correctors) on patients' cells bearing specific CFTR mutations; these drugs can modulate CFTR gene expression opening new therapeutic frontiers and better perspectives of life for these patients and others;
-
our methodology is adequate for the quantitative measurement, by fluorescence, of the CFTR gating activity of the HNECs from patients with different genotypes identifying:
CF patients bearing two severe mutations with an activity < 10% (compared to controls – 100%);
CF patients bearing a mild mutation with an activity of 10-20%;
CF carriers (heterozygous patients) with an activity between 40-70%.
In conclusion, the possibility of measuring the activity of the CFTR channel in HNECs provides an important contribution to the diagnosis of CF, by identification of a "diagnostic cut-off", and to the prediction of phenotypic severity of disease. This quantitative channel gating activity measurement improves the ability to evaluate patients with ambiguous results (borderline or negative) at the sweat test.
Moreover, our experimental method allows monitoring patients during drug treatment, and evaluating the real effects of new molecular therapies.
Acknowledgements
Grants from Italian Cystic Fibrosis Research Foundation (Verona), project call 2013, are gratefully acknowledged.
References
- 1.Amato F, Bellia C, Cardillo G, et al. Extensive molecular analysis of patients bearing CFTR-related disorders. J Mol Diagn. 2012;14:81–89. doi: 10.1016/j.jmoldx.2011.09.001. [DOI] [PubMed] [Google Scholar]
- 2.Maruotti GM, Frisso G, Calcagno G, et al. Prenatal diagnosis of inherited diseases: the 20 years experience of an Italian Regional Reference Centre. Clin Chem Lab Med. 2013;51:2211–2217. doi: 10.1515/cclm-2013-0194. [DOI] [PubMed] [Google Scholar]
- 3.Tomaiuolo R, Spina M, Castaldo G. Molecular diagnosis of Cystic Fibrosis: comparison of four analytical procedures. Clin Chem Lab Med. 2003;41:26–32. doi: 10.1515/CCLM.2003.006. [DOI] [PubMed] [Google Scholar]
- 4.Castaldo G, Polizzi A, Tomaiuolo R, et al. Comprehensive cystic fibrosis mutation epidemiology and haplotype characterization in southern Italy population. Ann Hum Genet. 2005;69:15–24. doi: 10.1046/j.1529-8817.2004.00130.x. [DOI] [PubMed] [Google Scholar]
- 5.Tomaiuolo R, Sangiuolo F, Bombieri C, et al. Epidemiology and a novel procedure for large scale analysis of CFTR rearrangements in classic and atypical CF patients: a multicentric Italian study. J Cyst Fibrosis. 2008;7:347–351. doi: 10.1016/j.jcf.2007.12.004. [DOI] [PubMed] [Google Scholar]
- 6.Castaldo G, Lembo F, Tomaiuolo R. Molecular diagnostics: between chips and customized medicine. Clin Chem Lab Med. 2010;48:973–982. doi: 10.1515/CCLM.2010.182. [DOI] [PubMed] [Google Scholar]
- 7.Bell SC, Boeck K, Amaral MD. New pharmacological approaches for cystic fibrosis: promises, progress, pitfalls. Pharmacol Ther. 2015;145C:19–34. doi: 10.1016/j.pharmthera.2014.06.005. [DOI] [PubMed] [Google Scholar]
- 8.Ventura MT, Gelardi M, D'Amato A, et al. Clinical and cytologic characteristics of allergic rhinitis in elderly patients. Ann Allergy Asthma Immunol. 2012;108:141–144. doi: 10.1016/j.anai.2012.01.013. [DOI] [PubMed] [Google Scholar]
- 9.Castaldo G, Calcagno G, Sibillo R, et al. Quantitative analysis of aldolase A mRNA in liver discriminates between hepatocellular carcinoma and cirrhosis. Clin Chem. 2000;46:901–906. [PubMed] [Google Scholar]
- 10.Canani RB, Terrin G, Elce A, et al. Genotype-dependency of butyrate efficacy in children with congenital chloride diarrhea. Orphanet J Rare Dis. 2013;8:194–194. doi: 10.1186/1750-1172-8-194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tomaiuolo R, Ruocco A, Salapete C, et al. Activity of mannose- binding lectin (MBL) in centenarians. Aging Cell. 2012;3:394–400. doi: 10.1111/j.1474-9726.2012.00793.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Garratt LW, Sutanto EN, Foo CJ, et al. Determinants of culture success in an airway epithelium sampling program of young children with cystic fibrosis. Exp Lung Res. 2014;40:447–459. doi: 10.3109/01902148.2014.946631. [DOI] [PubMed] [Google Scholar]
- 13.Dean N, Ranganath NK, Jones B, et al. Porcine nasal epithelial cultures for studies of cystic fibrosis synusitis. Int Forum Allergy Rhinol. 2014;4:565–570. doi: 10.1002/alr.21335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jeanson L, Guerrera IC, Papon JF, et al. Proteomic analysis of nasal epithelial cells from cystic fibrosis patients. PLoS One. 2014;9:e108671–e108671. doi: 10.1371/journal.pone.0108671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Achar P, Duvvi S, Kumar BN. Endoscopic dilatation sinus surgery (FEDS) versus functional endoscopic sinus surgery (FESS) for treatment of chronic rhinosinusitis: a pilot study. Acta Otorhinolaryngol Ital. 2012;32:314–319. [PMC free article] [PubMed] [Google Scholar]
- 16.Cantone E, Castagna G, Sicignano S, et al. Impact of intranasal sodium hyaluronate on the short-term quality of life of patients undergoing functional endoscopic sinus surgery for chronic rhinosinusitis. Int Forum Allergy Rhinol. 2014;4:484–487. doi: 10.1002/alr.21310. [DOI] [PubMed] [Google Scholar]
- 17.Pagani F, Buratti E, Stuani C, et al. Splicing factors induce cystic fibrosis regulator exon 9 skipping through a nonevolutionary conserved intronic element. J Biol Chem. 2000;275:21041–21047. doi: 10.1074/jbc.M910165199. [DOI] [PubMed] [Google Scholar]
- 18.Masvidal L, Igreja S, Ramos MD, et al. Assessing the residual CFTR gene expression in human nasal epithelium cells bearing CFTR splicing mutations causing cystic fibrosis. Eur J Hum Genet. 2014;22:784–791. doi: 10.1038/ejhg.2013.238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Giordano S, Amato F, Elce A, et al. Molecular and functional analysis of the large 5' promoter region of CFTR gene revealed pathogenic mutations in CF and CFTR-related disorders. J Mol Diagn. 2013;15:331–340. doi: 10.1016/j.jmoldx.2013.01.001. [DOI] [PubMed] [Google Scholar]
- 20.Elce A, Boccia A, Cardillo G, et al. Three novel CFTR polymorphic repeats improve segregation analysis for cystic fibrosis. Clin Chem. 2009;55:1372–1379. doi: 10.1373/clinchem.2008.119545. [DOI] [PubMed] [Google Scholar]
- 21.Amato F, Seia M, Giordano S, et al. Gene mutation in Micro- RNA target sites of CFTR gene: a novel pathogenetic mechanism in cystic fibrosis? Plos One. 2013;8:e60448–e60448. doi: 10.1371/journal.pone.0060448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Castaldo G, Fuccio A, Cazeneuve C, et al. Detection of five rare cystic fibrosis mutations peculiar to Southern Italy: implications in screening for the disease and phenotype characterization for patients with homozygote mutations. Clin Chem. 1999;45:957–962. [PubMed] [Google Scholar]
- 23.Terlizzi V, Carnovale V, Castaldo G, et al. Clinical expression of patients with the D1152H CFTR mutation. J Cyst Fibros. 2015;14:447–452. doi: 10.1016/j.jcf.2014.12.012. [DOI] [PubMed] [Google Scholar]
- 24.Amato F, Tomaiuolo R, Borbone N, et al. Design, synthesis and biochemical investigation, by in vitro luciferase report system, of peptide nucleic acids as a new inhibitors of mirR- 509-3p involved in the regulation of cystic fibrosis diseasegene expression. Med Chem Comm. 2014;5:68–71. [Google Scholar]
- 25.Amato F, Tomaiuolo R, Nici F, et al. Exploitation of a very small peptide nucleic acid as a new inhibitor of miR-509-3p involved in the regulation of cystic fibrosis disease-gene expression. Biomed Res Int. 2014;2014:610718–610718. doi: 10.1155/2014/610718. [DOI] [PMC free article] [PubMed] [Google Scholar]