

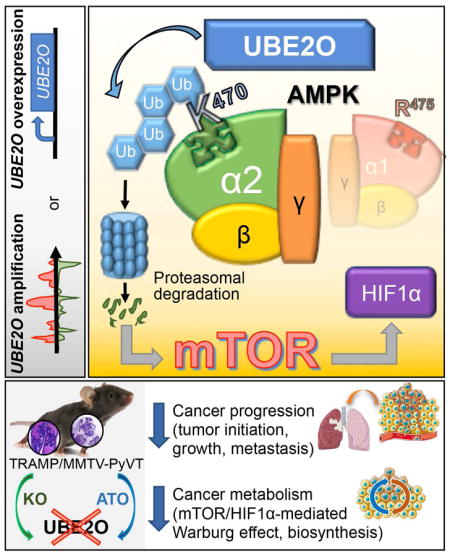
SUMMARY
UBE2O is localized in the 17q25 locus, which is known to be amplified in human cancers, but its role in tumorigenesis remains undefined. Here we show that Ube2o deletion in MMTV-PyVT or TRAMP mice profoundly impairs tumor initiation, growth and metastasis, while switching off the metabolic reprogramming of tumor cells. Mechanistically, UBE2O specifically targets AMPKα2 for ubiquitination and degradation, and thereby promotes activation of the mTOR-HIF1α pathway. Notably, inactivation of AMPKα2, but not AMPKα1, abrogates the tumor attenuation caused by UBE2O-loss, while treatment with rapamycin or inhibition of HIF1α ablates UBE2O-dependent tumor biology. Finally, pharmacological blockade of UBE2O inhibits tumorigenesis through the restoration of AMPKα2, suggesting the UBE2O-AMPKα2 axis as a potential cancer therapeutic target.
Keywords: UBE2O, AMPK, AMPKα2, mTOR, HIF1α, Breast cancer, Prostate cancer, Cancer metabolism, Ubiquitination, Arsenite
Graphical abstract
Vila et al. show that UBE2O, which is overexpressed in many human cancers, targets AMPKα2 for ubiquitination and degradation thereby promotes activation of the mTOR-HIF1α pathway. Genetic deletion or pharmacological blockade of UBE2O inhibits tumorigenesis through the restoration of AMPKα2.
INTRODUCTION
Ubiquitin-conjugating enzyme E2O (UBE2O) is a relatively large E2 ubiquitin-conjugation enzyme in comparison with other E2s. Previous studies have suggested that UBE2O could function as an E2/E3 hybrid ubiquitin-protein ligase that displays both E2 and E3 ligase activities (Berleth and Pickart, 1996; Klemperer et al., 1989). Recent biochemical and cell-based studies have showed that UBE2O ubiquitinates SMAD6 during bone morphogenetic protein signaling (Zhang et al., 2013a), induces cytoplasmic sequestration of nuclear BAP1 (Mashtalir et al., 2014), and coordinates endosomal protein trafficking (Hao et al., 2013). However, in vivo evaluation of UBE2O has been limited by a lack of appropriate animal models. Intriguingly, UBE2O is localized in the 17q25 chromosome region, which is amplified in a subset of human cancers (Briffa et al., 2015; Lin et al., 2006; Rice et al., 2011; Toffoli et al., 2014; Wang et al., 2015), but its role in tumorigenesis has yet to be fully described.
AMP-activated protein kinase (AMPK) is a critical sensor of cellular energy and nutrient levels. Loss of AMPK or deregulation of its activity has been linked to cancer. Reduced AMPK activity has been detected in human breast and kidney cancers (Cancer Genome Atlas Research Network, 2013; Hadad et al., 2009), and reduced expression of AMPKα2 has been associated with human breast, kidney, ovarian and gastric cancers (Hallstrom et al., 2008; Kim et al., 2012; Tong et al., 2011). The tumor suppressive functions of AMPK that have been described so far include: (1) inhibition of the synthesis of most cellular macromolecules by inactivating the mTOR signaling pathway (Gwinn et al., 2008); (2) downregulation of the glycolytic pathway to exert an anti-Warburg effect (Faubert et al., 2013; Faubert et al., 2014); (3) arrest of the cell cycle in concert with the stabilization of p53 and p27Kip1 (Hardie and Alessi, 2013); and (4) opposition of the epithelial-mesenchymal transition (EMT) associated with tumor invasion and metastasis (Qu et al., 2014). However, engaging AMPK signaling has also been reported to aid tumor cell survival and to provide an advantage to tumor cells by promoting mitochondrial pathways that mitigate metabolic stress and apoptosis (Jeon et al., 2012; Kishton et al., 2016; Saito et al., 2015). Thus AMPK may exert either a positive or negative effect on cancer cell survival, depending on the context of cellular stress.
AMPK is a heterotrimeric serine/threonine kinase composed of catalytic α and regulatory β and γ subunits. Although the regulation of its enzymatic activity by adenine nucleotides and upstream kinases (such as LKB1/STK11 and CaMKKβ) is already an active area of research (Hardie et al., 2012), there is compelling evidence suggesting additional modes of AMPK regulation (Lee et al., 2013; Pineda and Potts, 2015; Qi et al., 2008; Wang et al., 2012). However, the molecular mechanisms underlying selective modulation of AMPK subunit isoforms during tumorigenesis are unclear.
RESULTS
Generation of Ube2o knockout mice
To examine the in vivo biological functions of UBE2O, we generated Ube2o knockout mice by injecting Ube2o knockout (Ube2otm1(KOMP)Mpb) mouse embryonic stem cells (Figure S1A), generated by the Knockout Mouse Project (KOMP) consortium, into blastocysts derived from C57BL/6 mice to produce chimeras. Subsequent breeding of these chimeras resulted in germline transmission of the targeted allele (Figure S1B).
To examine potential UBE2O-mediated proliferation changes, we first attempted to generate Ube2o−/− mouse embryonic fibroblasts (MEFs). Significantly, primary Ube2o−/− MEFs proliferated slower than wild-type cells (Figures S1C and S1D). Classical focus-formation assays with adenovirus E1A and H-RasV12 revealed a decrease in the number of foci of morphologically transformed cells among Ube2o−/− cells (Figure S1E), indicating that loss of Ube2o confers resistance to cellular oncogenic transformation. Ube2o deficiency also significantly reduced the migration rates of cells, which was unlikely to be associated with differences in their proliferation rates (Figure S1F). Conversely, the transformation efficiency was greatly increased in cells overexpressing UBE2O (Figure S1G). In vivo mouse allograft generation with these cells confirmed the pro-growth activity of UBE2O (Figure S1H).
Ablation of Ube2o impairs mammary tumor progression and lung metastasis in a mouse model of breast cancer
To explore the role of UBE2O in tumorigenesis in vivo, we introduced the Ube2o deletion into a series of transgenic mouse models of spontaneous cancer. As MMTV-PyVT transgenic mice are known to develop mammary tumors that metastasize to the lung (Guy et al., 1992), we crossed these mice with our Ube2o−/− mice and analyzed the resultant compound mutants. Ablation of Ube2o provided a significant survival benefit for MMTV-PyVT mice (Figure 1A). Furthermore, Ube2o knockout profoundly delayed onset of PyVT-driven mammary tumors (Figure 1B). It should be noted that PyVT;Ube2o+/− mice also exhibited improved survival and delayed disease onset, indicating that UBE2O is haplo-insufficient with respect to mammary tumor development in MMTV-PyVT mice.
Figure 1. Ube2o ablation impairs mammary tumor growth and metastasis in PyVT mice.

(A) Overall survival (OS) analysis of the PyVT;Ube2o+/+ (n = 6, median OS 135 days), PyVT;Ube2o+/− (n = 16, median OS 152 days) or PyVT;Ube2o−/− (n = 14, median OS 174 days) mice.
(B) Tumor-free survival (TFS) analysis of the PyVT;Ube2o+/+ (n = 43, median TFS 73 days), PyVT;Ube2o+/− (n = 49, median TFS 86 days) or PyVT;Ube2o−/− (n = 17, median TFS 111 days) mice.
(C) Mammary tumor weight isolated from PyVT;Ube2o+/+ (n = 120), PyVT;Ube2o+/− (n = 70) and PyVT;Ube2o−/− (n = 30) mice were quantified (left). Representative images of mammary tumors isolated from mice are shown in right.
(D) H&E and anti-Ki-67 stained sections of mammary tumors isolated from PyVT;Ube2o+/+, PyVT;Ube2o+/− and PyVT;Ube2o−/− mice. Scale bars for H&E, 300 μm; scale bars for anti-Ki-67, 75 μm.
(E) Sections of mammary tumors isolated from 105 days old PyVT;Ube2o+/+ and PyVT;Ube2o−/− mice stained with anti-CD31 (left) and quantification of intra-tumoral vessel numbers (right). n = 3. Scale bars, 75 μm.
(F) The expression indicated neovascularization genes in mammary tumors of 105 days old PyVT;Ube2o+/+ and PyVT;Ube2o−/− mice measured by RT-qPCR. n = 3.
(G) H&E and anti-PyVT Ag stained sections of lungs isolated from 105 days old PyVT;Ube2o+/+, PyVT;Ube2o+/− and PyVT;Ube2o−/− mice. Arrowheads indicate clusters of metastatic cells in the lung. Scale bars, 75 μm.
(H) The number of metastatic sites in lungs of 105 days old PyVT;Ube2o+/+ (n = 11), PyVT;Ube2o+/− (n = 7) and PyVT;Ube2o−/− (n = 7) mice.
Error bars represent +/− SEM. p value was determined by Log-rank (Mantel-Cox) test (A and B) or Student’s t test (*p<0.05; **p<0.01; ***p<0.001). See also Figure S1.
At 105 day of age, PyVT;Ube2o+/+ mice had medium- to large–sized mammary tumors (median weight, 360 mg) reflecting multifocal tumor growth in response to expression of the PyVT oncogene, whereas tumor volume and weight were greatly reduced in PyVT;Ube2o+/− mice and PyVT;Ube2o−/− mice (178 mg and 28 mg, respectively) (Figure 1C). Notably, 7/13 (54%) PyVT;Ube2o−/− mice had no tumor burdens. Histological examination by H&E staining revealed severe malignant progression in PyVT;Ube2o+/+ mice, as indicated by the formation of poorly differentiated and aggressive adenocarcinoma (Figure 1D). In contrast, the mammary glands of age-matched PyVT;Ube2o−/− mice were preserved and the tissue showed a hyperplastic-like, non-malignant phenotype (Figure 1D). Moreover, Ube2o ablation markedly attenuated intra-tumoral vascularization in PyVT mammary tumors (Figure 1E). Likewise expression levels of genes involved in neovascularization such as Angpt1, Angpt2 and Kdr (Flk1/Vegfr2) and its reflecting endothelial cell number, such as Pecam1 (Cd31) and Tie1, were greatly reduced in PyVT;Ube2o−/− relative to PyVT;Ube2o+/+ mammary tumors (Figure 1F). We also noted that expression of pro-inflammatory genes such as Tnf, Nos2, Il6 and Ccl2 was significantly inhibited by Ube2o ablation in PyVT mammary tumors (Figure S1I). Furthermore, multiple tumor nodules that had metastasized to the lung surface were observed in PyVT;Ube2o+/+ mice but none was seen in PyVT;Ube2o−/− mice (Figures 1G and 1H). Taken together, these results indicate that UBE2O regulates breast cancer initiation, progression and lung metastasis.
Attenuated formation of invasive prostate carcinoma and metastasis in Ube2o-deficient TRAMP mice
We next asked what impact Ube2o deletion might have on the development of another cancer type, such as prostate cancer. We therefore crossed our Ube2o−/− mice with TRAMP (transgenic adenocarcinoma mouse prostate) mice, which develop prostate cancer due to prostate-specific expression of SV40 T antigen (Greenberg et al., 1995). To study the early effects of Ube2o ablation in the prostate, TRAMP mice of differing Ube2o backgrounds were sacrificed at 12 weeks of age, and histopathological analysis was performed. TRAMP;Ube2o−/− mice displayed normal prostate histology, whereas TRAMP;Ube2o+/+ mice consistently exhibited significantly enlarged prostate lobes and high-grade prostatic intraepithelial neoplasia (HG-PIN) (~50% of prostate glands affected) (Figures 2A–2C). We also observed that haplo-deficiency of Ube2o suppressed the development of HG-PIN, demonstrating that UBE2O is haplo-insufficient with respect to prostate cancer initiation.
Figure 2. Ube2o knockout attenuates tumor initiation, growth and metastasis in TRAMP;Ube2o mice.

(A) H&E stained sections of anterior prostate lobes isolated from 12 weeks old TRAMP;Ube2o+/+ (n = 7), TRAMP;Ube2o+/− (n = 9) and TRAMP;Ube2o−/− mice (n = 5). Scale bars, 75 μm.
(B) Quantification of prostatic intraepithelial neoplasia (PIN) in (A).
(C) Prostate tissue weight relative to body weight (BW) of 12 weeks old TRAMP;Ube2o+/+ (n = 7), TRAMP;Ube2o+/− (n = 9) and TRAMP;Ube2o−/− mice (n = 5).
(D, E) MRI analysis of 25 weeks old TRAMP;Ube2o+/+ (n = 10), TRAMP;Ube2o+/− (n = 8) and TRAMP;Ube2o−/− mice (n = 5) (D) and quantification of prostate tumor volume using an Oxirix Imaging software (E). The dorsolateral prostate is outlined.
(F) Prostate tissue weight of 30 weeks old TRAMP;Ube2o+/+ (n = 10), TRAMP;Ube2o+/− (n = 11) and TRAMP;Ube2o−/− mice (n = 4).
(G) Sections of prostate tissues isolated from 30 weeks old TRAMP;Ube2o+/+ and TRAMP;Ube2o−/− mice stained with anti-SMA. Asterisks indicate sites of tumor invasion. Scale bars, 75 μm.
(H) Sections of livers and lymph nodes isolated from 30 weeks old TRAMP;Ube2o+/+ and TRAMP;Ube2o−/− mice stained with H&E or anti-SV40 T Ag. Scale bars, 75 μm.
Error bars represent +/− SEM. p value was determined by Student’s t test (*p<0.05; **p<0.01).
To further analyze the effects of Ube2o ablation on prostate carcinoma in TRAMP mice, we followed cohorts of TRAMP mice with differing Ube2o backgrounds by magnetic resonance imaging (MRI) analysis. MRI analysis revealed the presence of large tumor masses in the prostates of 25 weeks old TRAMP;Ube2o+/+ mice (Figures 2D and 2E). However, these tumors were markedly attenuated in the age-matched TRAMP;Ube2o+/− or TRAMP;Ube2o−/− cohorts on the basis of both tumor volume (Figures 2D and 2E) and weight (Figure 2F). Notably, smooth muscle actin (SMA) staining showed a highly penetrant invasive prostatic adenocarcinoma in TRAMP;Ube2o+/+ mice at 30 weeks of age as compared to age-matched TRAMP;Ube2o−/− mice (Figure 2G). Furthermore, multiple tumor nodules that had metastasized to the liver and lymph nodes were observed in TRAMP;Ube2o+/+ mice (~25% incidence), but not in any TRAMP;Ube2o−/− mice (Figure 2H). These results highlight UBE2O as a critical factor in prostate cancer initiation, progression, invasion and metastasis.
UBE2O targets AMPKα2 for ubiquitination and degradation
In order to identify signaling pathways regulated by UBE2O, we used anti-Flag affinity purification-mass spectrometry (MS) to identify potential UBE2O-interacting proteins in Ube2o−/− MEFs expressing Flag-tagged UBE2O (Figure 3A). Interestingly, AMPKα2 (encoded by Prkaa2) was identified as a UBE2O-associated protein (Figure S2A). Reciprocal purification of Myc-tagged AMPKα2 complexes from Prkaa1−/−Prkaa2−/− MEFs corroborated this data (Figure S2B). We confirmed the interactions between UBE2O and AMPKα2 in vivo and in vitro (Figure 3B and Figure S2C). Moreover, UBE2O, as an E2/E3 hybrid ubiquitin-protein ligase, directly ubiquitinates AMPKα2 in the presence of only the E1 enzyme whereas the catalytically inactive UBE2O C1040S (CS) mutant, which can still bind to AMPKα2, had dramatically diminished ability to ubiquitinate AMPKα2 (Figures 3C and Figures S2D and S2E). Targeted disruption of Ube2o likewise decreased the ubiquitination of the endogenous AMPKα2 (Figure S2F), suggesting that UBE2O may be the major physiological ubiquitin ligase for AMPKα2. Interestingly, previous studies had predicted that three lysine (K) residues (K364, K379 and K470) in AMPKα2 could be ubiquitinated (Chen et al., 2014; Radivojac et al., 2010; Wagner et al., 2012). When wild-type or lysine mutant AMPKα2 was expressed in Prkaa1−/−Prkaa2−/− MEFs, unlike wild-type or other mutants, AMPKα2 K470R failed to be ubiquitinated (Figure 3D), indicating that K470 could be a major site for AMPKα2 ubiquitination by UBE2O. It should be noted that the K470 residue is conserved in AMPKα2 among different vertebrate species, but is replaced by an arginine (R475) in AMPKα1. RNA interference (RNAi) targeting UBE2O decreased the ubiquitination levels of AMPKα2, but not AMPKα1 (Figure 3E), further confirming the AMPKα2-selective ubiquitination by UBE2O.
Figure 3. UBE2O specifically targets AMPKα2 for ubiquitination and degradation.

(A) Silver stained gel of proteins recovered after immunoprecipitation of lysates prepared from Ube2o−/− MEFs expressing Flag-tagged UBE2O. Proteins captured with anti-Flag were eluted with Flag peptide. Shown are proteins present in the Flag eluate. Asterisk indicates the heavy chain of IgG.
(B) Lysates from HeLa cells were immunoprecipitated (IP) with IgG and anti-UBE2O (top) or anti-AMPKα2 (bottom) then immunoblotted as indicated.
(C) Recombinant AMPKα2 proteins were subjected to in vitro ubiquitination assay in the presence of in vitro-translated wild-type (WT) or C1040S (CS) mutant UBE2O.
(D) Lysates from Prkaa1−/−Prkaa2−/− MEFs expressing WT or various indicated lysine-to-arginine (KR) mutants of AMPKα2 treated with MG132 (10 μM) for 4 hr were subjected to metal-affinity purification for His-tagged ubiquitin then immunoblotting for ubiquitinated AMPKα2. Ni-NTA, Ni2+-nitrilotriacetic acid.
(E) Lysates from HCT116 cells expressing UBE2O shRNA together with Myc-tagged ubiquitin treated with MG132 (10 μM) for 4 hr were immunoprecipitated (IP) with IgG or anti-Myc and analyzed for ubiquitination with anti-AMPKα2 or anti-AMPKα1.
(F) Lysates from HCT116 cells expressing UBE2O shRNA treated with cycloheximide (CHX) for the indicated times were subjected to immunoblotting (top). AMPKα2 or AMPKα1 protein levels were quantified by normalizing to the intensity of the Actin band (bottom).
(G) Lysates from Prkaa1−/−Prkaa2−/− MEFs expressing WT or K470R mutant AMPKα2 treated with CHX for the indicated times were subjected to immunoblotting (top). AMPKα2 protein levels were quantified by normalizing to the intensity of the Actin band (bottom).
(H) Lysates from mammary tissues of 13 weeks old Ube2o+/+, Ube2o+/− and Ube2o−/− mice were subjected to immunoblotting.
(I) Lysates from primary MEFs expressing UBE2O shRNA together with WT or CS mutant UBE2O were subjected to immunoblotting.
See also Figures S2 and S3.
UBE2O promoted K48-linked, rather than K63-linked, ubiquitination of AMPKα2 (Figure S2G). Ubiquitination through K48 of the ubiquitin chain generally targets proteins for degradation. Indeed, proteasomal inhibition (MG132, 6 hr) increased AMPKα2 protein levels (Figure S3A), and cycloheximide treatment revealed that UBE2O-mediated AMPKα2 ubiquitination greatly alters its protein turnover rate (Figures 3F and 3G). In Ube2o−/− mouse tissues, the levels of endogenous AMPKα2 (but not AMPKα1) protein rose strikingly, with no effect on mRNA levels or cellular compartmentalization (Figure 3H and Figures S3B and S3C). Likewise, downregulation of UBE2O by RNAi in AMPKα2-positive HCT116 human colon carcinoma cells resulted in increased AMPKα2 (but not AMPKα1) protein levels, accompanied by elevated expression of additional subunits of the AMPK holoenzyme (Figure S3D). These effects were likely dependent on the expression of the AMPKα2, because UBE2O-loss did not significantly alter the AMPK holoenzyme in AMPKα2-deficient DLD-1 human colon carcinoma cells (Figure S3D).
Knockdown of UBE2O also increased phosphorylation of AMPK substrates, such as acetyl CoA carboxylase 1 (ACC1) and Raptor, the regulatory-associated protein of mTOR, along with suppression of mTORC1 signaling as determined by phosphorylation of S6 (Figure 3I). This effect was reversed, however, by reconstitution with exogenous wild-type (but not CS mutant) UBE2O from a Ube2o ORF transcript lacking the 3′-UTR sequence targeted by shRNA (Figure 3I). Together these data indicate that UBE2O downregulates AMPKα2 through ubiquitination.
UBE2O-mediated tumorigenesis is AMPKα2 dependent
Next, we examined whether the mechanism for UBE2O dependent tumorigenesis operates specifically through the AMPKα2 axis. While most cell types express both AMPKα1 and AMPKα2, B and T lymphocytes express only AMPKα1 (Stapleton et al., 1996; Tamas et al., 2006) (Figure S4A). We therefore generated two mouse models of lymphoma, in which AMPKα2 is undetectable, that harbored a mutation in Ube2o (Figure S4B): An Eμ-Myc mouse model of B cell lymphoma (BCL) (Adams et al., 1985) (Eμ-Myc;Ube2o−/−) and a Pten-deficient mouse developing T cell lymphoma (Di Cristofano et al., 1999; Podsypanina et al., 1999) and germinal center subtype BCL (Pfeifer et al., 2013) (Pten+/−;Ube2o−/−). To our surprise, after a maximum of ~400 days, we found no significant difference in disease onset and mortality rates between Eμ-Myc control mice and Eμ-Myc animals lacking Ube2o (Figures 4A–4C). Similarly, we found that ablation of Ube2o does not impair the progression of lymphoma in cohorts of Pten+/− mice (Figures S4C and S4D). It is also noteworthy that loss of Ube2o did not lead to alterations in levels of AMPKα1 in lymph node tumors isolated from mice (Figure 4D and Figure S4E). These data suggest that Ube2o ablation does not provide a significant anti-tumor effect or survival advantage in mouse models of lymphoma in which AMPKα2 is undetectable, and the AMPKα1 may not play a role in UBE2O-dependent tumorigenesis.
Figure 4. UBE2O-mediated tumorigenesis is AMPKα2 dependent.

(A, B) Overall (A) or tumor-free (B) survival analysis of Eμ-Myc;Ube2o+/+ (n = 15), Eμ-Myc;Ube2o+/− (n = 21) or Eμ-Myc;Ube2o−/− (n = 4) mice. p value was determined by Log-rank (Mantel-Cox) test (n.s., non-significant).
(C) Sections of lymph nodes isolated from 16 weeks old Eμ-Myc;Ube2o+/+, Eμ-Myc;Ube2o+/− and Eμ-Myc;Ube2o−/− mice stained for H&E or Ki-67. Scale bars, 75 μm.
(D) Lysates from lymphomas of 16 weeks old Eμ-Myc;Ube2o+/+, Eμ-Myc;Ube2o+/− and Eμ-Myc;Ube2o−/− mice were subjected to immunoblotting.
(E) Growth curves of HAP1 control (left) or HAP1 cells knocked out for PRKAA1 (middle) or PRKAA2 (right) using CRISPR/Cas9 technology expressing UBE2O shRNA. n = 3, p value was determined by ANOVA (n.s., non-significant; ***p<0.001).
(F) Lysates from (E) were subjected to immunoblotting.
(G) Tumor volumes of mouse xenograft implanted with HCT116 cells expressing shRNAs designed against the indicated genes were measured at different day. n = 7, p value was determined by Student’s t test (***p<0.001, shUBE2O;shAMPKα2 vs. shUBE2O;shCont.; $$p<0.01, $$$p<0.001, shUBE2O;shAMPKα2 vs. shUBE2O;shAMPKα1).
(H) Representative images, immunohistochemical analysis of Ki-67, and quantification of Ki-67 positive cells of mouse xenograft tumors from (G). Scale bars, 75 μm. n = 7, p value was determined by Student’s t test (n.s., non-significant; *p<0.05).
(I) Lysates from (G) were subjected to immunoblotting.
(J) Tumor volumes of mouse allograft implanted with E1A+H-RasV12 MEFs overexpressing UBE2O together with WT or K470R mutant AMPKα2 were measured at different day. n = 6, p value was determined by Student’s t test (***p<0.001, UBE2O;Cont. vs. Cont.;Cont.; $$p<0.01, $$$p<0.001, UBE2O;Cont. vs. UBE2O;α2K470R).
(K) Representative images, immunohistochemical analysis of Ki-67, and quantification of Ki-67 positive cells of mouse allograft tumors from (J). Scale bars, 75 μm. n = 6, p value was determined by Student’s t test (n.s., non-significant; **p<0.01).
(L) Lysates from (J) were subjected to immunoblotting. Error bars represent +/− SEM. See also Figure S4.
To further study the contribution of AMPKα1 and AMPKα2 to UBE2O dependent tumorigenesis, we used a human haploid cancer cell line (HAP1) in which the single allele of PRKAA1 or PRKAA2 had been knocked out using CRISPR/Cas9 technology. Critically, even in HAP1 cells knocked out for PRKAA1, knockdown of UBE2O resulted in decreased cell growth and increased phosphorylation of AMPK substrates such as ACC1 and Raptor (Figures 4E and 4F). In contrast, UBE2O depletion did not affect the rates of cellular proliferation and AMPK signaling in PRKAA2 knockout cells (Figures 4E and 4F). Likewise UBE2O silencing in AMPKα2-negative DLD-1 cells did not significantly alter cell growth rates (Figure S4F).
We also attempted to distinguish between AMPKα1 and AMPKα2 using isoform-specific knockdowns. Notably, HCT116 xenograft demonstrated that knockdown of AMPKα2, but not AMPKα1, markedly reverses the phenotype of UBE2O loss-induced tumor attenuation (Figures 4G–4I), underscoring that AMPKα2 (but not AMPKα1) is very likely to be a major factor in reliable UBE2O function in tumorigenesis.
Furthermore, either expression of ubiquitination-defective K470R AMPKα2 mutant or treatment with the allosteric AMPK activator A769662 reversed tumor growth in mice bearing E1A and H-RasV12 transformed MEFs overexpressing UBE2O (Figures 4J–4L and Figures S4G–S4I), suggesting that AMPKα2 ubiquitination by UBE2O promotes tumor growth, and pharmacological activation of AMPK could show promise as a treatment for UBE2O-positive tumors.
UBE2O promotes metabolic reprogramming of cancer cells through inactivation of AMPKα2
Metabolic reprogramming toward aerobic glycolysis (or the Warburg effect) and biomass accumulation is known to accompany tumorigenesis (Vander Heiden et al., 2009). We therefore decided to explore the possible role of UBE2O in cancer cell metabolism. Notably, UBE2O-depleted HCT116 cells exhibited decreased glucose consumption and lactate production, a metabolic signature consistent with the ‘anti-Warburg effect’ (Figure S5A). Conversely, forced expression of wild-type (but not CS mutant) UBE2O in the ABC-1 NSCLC cell line with the lowest UBE2O expression [due to the lower copy number of UBE2O, according to the Cancer Cell Line Encyclopedia (CCLE) (http://www.broadinstitute.org/ccle)], led to increases in glucose consumption and lactate production (Figure S5B). Moreover, using targeted capillary electrophoresis (CE) mass spectrometry, we extracted 116 metabolites and found a significant reduction in levels of glycolytic metabolites in UBE2O-silenced cells compared to control cells (Figure 5A). We further examined the metabolic fate of 13C-labeled glucose through a quantitative isotope analysis, and found that the proportion of glycolytic metabolites containing the 13C label was markedly reduced by UBE2O loss (Figure S5C). Our metabolomic analysis also revealed that UBE2O-depleted cancer cells had a significantly reduced biosynthetic capacity of purines and amino acids, which is closely linked to tumor cell growth and size (Vander Heiden et al., 2009) (Figures S5D and S5E). Likewise UBE2O-depleted HCT116 cells displayed a 15% decrease in median cell size whereas ectopic expression of UBE2O in ABC-1 cells increased cell size (Figure 5B), implying that UBE2O plays a pivotal role in the biomass regulation of cancer cells. Because the commonly used HCT116 cell line contains a PIK3CA kinase domain mutation (PIK3CAH1047R) that could directly affect the metabolic state of the cells, we used a PIK3CA wild-type cancer cell line, MDA-MB-231, and found that knockdown of UBE2O still resulted in marked reductions in glucose consumption as well as lactate and biomass production (Figures S5F and S5G). Pre-treatment of HCT116 cells with the PI3K inhibitor BKM120 corroborated our conclusion that UBE2O-induced metabolic rewiring is unlikely to be associated with the PIK3CA status of cancer cells (Figures S5H and S5I).
Figure 5. Inhibition of AMPKα2 by UBE2O impacts metabolic reprogramming.

(A) Principal component (PC) analysis was conducted to compare the overall metabolic profiles in HCT116 cells expressing UBE2O shRNA using a HMT SampleStat software (left). Representative heat map of metabolome profiles was analyzed by hierarchical clustering analysis (middle). Heat map colors represent relative metabolite levels as indicated in the color key. Average absolute concentrations (pmol) of glycolytic metabolites, as indicated, per 106 cells were measured by CE-MS (right). G-6-P, glucose 6-phosphate; F-6-P, fructose 6-phosphate; F-1,6-P, fructose 1,6-phosphate; GAP, glyceraldehyde 3-phosphate; 3-PG, 3-phosphoglyceric acid; 2-PG, 2-phosphoglyceric acid; PEP, phosphoenolpyruvic acid. n = 3.
(B) Forward scatter (FSC) levels of HCT116 cells expressing UBE2O shRNA (top) or ABC-1 cells ectopically expressing UBE2O (bottom) were determined by flow cytometry.
(C) Glucose consumption and lactate production by HAP1 control (left) or HAP1 cells knocked out for PRKAA1 (middle) or PRKAA2 (right) using CRISPR/Cas9 technology expressing UBE2O shRNA were determined by enzymatic assay. n = 3.
(D) FSC levels of HAP1 control (top) or HAP1 cells knocked out for PRKAA1 (middle) or PRKAA2 (bottom) expressing UBE2O shRNA were determined by flow cytometry.
(E) Glucose (Glc) consumption and lactate (Lac) production by HCT116 cells expressing AMPKα1 or AMPKα2 shRNA together with UBE2O shRNA were determined by enzymatic assay. n = 3.
(F) FSC levels of HCT116 cells expressing AMPKα1 or AMPKα2 shRNA together with UBE2O shRNA were determined by flow cytometry.
(G) Glucose consumption and lactate production by E1A+H-RasV12 MEFs overexpressing UBE2O together with K470R AMPKα2 mutant were determined by enzymatic assay. n = 3.
(H) FSC levels of E1A+H-RasV12 MEFs overexpressing UBE2O together with K470R AMPKα2 mutant were determined by flow cytometry.
Error bars represent +/− SEM. p value was determined by Welch’s t test (A) or Student’s t test (n.s., non-significant; *p<0.05; **p<0.01; ***p<0.001). See also Figure S5.
AMPK is known to suppress the rewiring of metabolism toward glycolytic and biosynthetic pathways in cancerous contexts (Faubert et al., 2013; Faubert et al., 2014). We therefore asked whether the effect of UBE2O on the metabolic reprogramming of tumor cells occurs through AMPKα2. Knockdown of UBE2O markedly reduced rates of glucose consumption as well as lactate and biomass production in AMPKα2(+) HCT116, but not AMPKα2(−) DLD-1, human colon carcinoma cells (Figures S5J and S5K). Moreover, RNAi targeting of UBE2O reduced the rates of glucose consumption and the production of lactate and biomass in both HAP1 control and HAP1 cells knocked out for PRKAA1, whereas these rates were not affected in PRKAA2 KO cells (Figures 5C and 5D). Knockdown of AMPKα2, but not AMPKα1, with isoform-specific shRNAs attenuated the effects of UBE2O loss on metabolic reprogramming (Figures 5E and 5F). Critically, expression of ubiquitination-defective K470R mutant AMPKα2 reversed the glycolytic and biosynthetic phenotypes observed in UBE2O-overexpressing tumor cells (Figures 5G and 5H). Taken together, these results suggest that UBE2O-dependent enhanced metabolic flux into glycolytic and biosynthetic pathways of tumor cells is specifically due to AMPKa2 degradation.
An AMPKα2-mTOR-HIF1α axis is critical to UBE2O dependent tumor biology
Numerous studies have revealed that the mTOR pathway is one of the major growth regulatory pathways controlled by AMPK (Hardie and Alessi, 2013; Shackelford and Shaw, 2009). In alignment with these findings, we observed that UBE2O promoted heightened activation of mTORC1 signaling, marked by increased S6 phosphorylation in an AMPKα2-dependent manner (Figures 3I, 4D, 4F, 4I and 4L and Figures S4E and S6A). We also noted that downregulation of UBE2O did not significantly alter the protein levels of any other components of the mTORC1 pathway (Figure S6A), suggesting that the effects of UBE2O are largely due to AMPKα2 regulation.
To examine the contribution of mTORC1 signaling to UBE2O-induced tumor growth in vivo, we subcutaneously implanted transformed MEFs ectopically expressing UBE2O into Swiss-nu/nu mice, which were then treated with the mTORC1 inhibitor rapamycin every other day. We found that tumor growth in mice bearing cells overexpressing UBE2O was profoundly suppressed by rapamycin (Figures 6A–6C). Moreover, rapamycin treatment ablated the enhanced levels of glucose consumption, lactate production (Figures 6D and 6E) and biomass accumulation (Figure 6F) displayed by UBE2O-overexpressing cells.
Figure 6. AMPKα2/mTOR/HIF1α axis is key to UBE2O dependent tumor biology.

(A) Tumor volumes of mouse allograft implanted with E1A+H-RasV12 MEFs overexpressing UBE2O treated with rapamycin (i.p. 4 mg/kg, indicated by arrows) were measured at different day. n = 7.
(B) Representative images, immunohistochemical analysis of Ki-67, and quantification of Ki-67 positive cells of mouse allograft tumors from (A). Scale bars, 75 μm. n = 6.
(C) Lysates from (A) were subjected to immunoblotting.
(D) Lysates from transformed MEFs overexpressing UBE2O treated with rapamycin at the indicated concentrations for 24 hr were subjected to immunoblotting.
(E) Glucose consumption and lactate production by transformed MEFs overexpressing UBE2O treated with rapamycin (100 nM) for 24 hr were determined by enzymatic assay. n = 3.
(F) FSC levels of transformed MEFs overexpressing UBE2O treated with rapamycin (100 nM) for 24 hr were determined by flow cytometry.
(G) Lysates from mammary tissues of 90 days old PyVT;Ube2o+/+ and PyVT;Ube2o−/− mice (left) or anterior prostate lobes of 12 weeks old TRAMP;Ube2o+/+ and TRAMP;Ube2o−/− mice (right) were subjected to immunoblotting.
(H) The mRNA level of HIF1α target genes in mammary tumors of 105 days old PyVT;Ube2o+/+ and PyVT;Ube2o−/− mice or in prostate tumors of 30 weeks old TRAMP;Ube2o+/+ and TRAMP;Ube2o−/− mice were measured by RT-qPCR. n = 3.
(I) Total RNAs from HAP1 control or HAP1 cells knocked out (KO) for PRKAA1 or PRKAA2 using CRISPR/Cas9 technology expressing UBE2O shRNA were subjected to RT-qPCR. % reduction formula is the decrease in mRNA levels of UBE2O-depleted cells over those of control cells. n = 3. n.s. (non-significant), *p<0.05, PRKAA1 KO vs. WT; $$p<0.01, $$$p<0.001, PRKAA2 KO vs. WT.
(J) Total RNAs from E1A+H-RasV12 MEFs overexpressing UBE2O together with K470R AMPKα2 mutant were subjected to RT-qPCR. n = 3. *p<0.05, **p<0.01, ***p<0.001, UBE2O vs. Cont.; $$p<0.01, $$$p<0.001, UBE2O vs. UBE2O;α2K470R.
(K) Immunoblots and growth curves of transformed MEFs overexpressing UBE2O together with HIF1α shRNA.
(L) Glucose consumption and lactate production by transformed MEFs overexpressing UBE2O together with HIF1α shRNA were determined by enzymatic assay. n = 3.
(M) FSC levels of transformed MEFs overexpressing UBE2O together with HIF1α shRNA were determined by flow cytometry.
Error bars represent +/− SEM. p value was determined by ANOVA (A) or Student’s t test (n.s., non-significant; *p<0.05; **p<0.01; ***p<0.001). See also Figures S6.
mTORC1 signaling has been linked to control of hypoxia inducible factor 1α (HIF1α) expression, which in turn promotes cell growth and biosynthesis (Laplante and Sabatini, 2012). Consistently, loss of Ube2o has led to marked reductions in HIF1α levels in cells and mice through the AMPKα2-mTOR axis (Figures 4D, 4F, 4I, 4L, 6C, 6D and 6G and Figures S4E, S6A and S6B). Our microarray analysis revealed that UBE2O-depleted cells exhibited reduced HIF1α gene enrichment signatures (Figure S6C). Indeed, expression of HIF1α target genes such as Flt1, Vegfa, Tgfb3, Ccng2, Hes6, Idha, Sox9, Pdk1, Jmjd1a and Plod2 was significantly attenuated in Ube2o-deficient MMTV-PyVT or TRAMP mice (Figure 6H). Additionally, it is likely that the regulation of HIF1α target gene expression signatures by UBE2O is AMPKα2 dependent (Figures 6I and 6J and Figure S6D). We next sought to determine the contribution of HIF1α to the pro-growth and glycolytic phenotypes observed in UBE2O-dependent tumors. Expression of HIF1α shRNA significantly decreased cellular proliferation in UBE2O-overexpressing cells (Figure 6K), and was associated with reductions in glucose consumption, lactate production (Figure 6L) and biomass accumulation (Figure 6M). Collectively, these data suggest that the UBE2O-dependent pro-growth, glycolytic and biosynthetic programs of tumor cells are supported by the AMPKα2-mTOR-HIF1α axis.
UBE2O regulation of AMPKα2/mTOR/HIF1α is relevant in patients
UBE2O is localized in the 17q25 locus, the amplification of which is recurrent in a subset of human cancers (Briffa et al., 2015; Lin et al., 2006; Rice et al., 2011; Toffoli et al., 2014; Wang et al., 2015), and we found that in TCGA datasets from cBioPortal (www.cbioportal.org) UBE2O is upregulated in ~20% of human breast, bladder, liver and lung carcinomas (Figures 7A and 7B and Figures S7A and S7B). Previously published microarray-based gene expression analyses have also indicated a high expression rate of UBE2O in several subsets of human cancer (Figure 7C and Figure S7C). This data was corroborated by our own immunohistochemical analysis, which revealed that UBE2O is indeed highly expressed in breast cancer samples (Figure 7D and Figure S7D). We further confirmed the clinical significance of UBE2O overexpression through a cancer patient survival analysis drawn from another available database (Figure 7E), suggesting that its expression may impact neoplastic malignancies and clinical outcomes.
Figure 7. UBE2O expression is increased and correlates with AMPKα2/mTOR/HIF1α in human cancers.

(A) TCGA database analysis available from cBioportal for UBE2O expression in human cancers.
(B) 17q25.1 copy number compared to gene expression for UBE2O mRNA versus copy number in human breast cancer using the cBioportal database. The line in the middle, upper and lower of the boxplot represents the mean, upper and lower quartile of the relative mRNA level of all samples, respectively. The lines above and below the box are the maximum and minimum values. Data points beyond the whiskers (>1.5 interquartile ranges) are drawn as individual dots.
(C) UBE2O expression in human cancers using previously published microarray database. Liu’s BCa, breast carcinoma, (GEO: GSE22820, n = 176); Stephenson’s CaP, prostate carcinoma, (Stephenson et al., 2005, n = 97); Taylor’s CaP (GEO: GSE21032, n = 179). The line in the middle, upper and lower of the boxplot represents the mean, upper and lower quartile of the relative mRNA level of all samples, respectively. The lines above and below the box are the maximum and minimum values. Data points beyond the whiskers (>1.5 interquartile ranges) are drawn as individual dots.
(D) Representative images (top) and quantification (bottom) of immunohistochemical analysis of human breast tissue microarrays for UBE2O protein. Scale bars, 200 μm. n = 72.
(E) Online analysis of survival in breast cancer (n = 1746), lung adenocarcinoma (n = 1926) or gastric cancer (n = 876) patients with high or low UBE2O expression. The number of surviving patients at different time points is indicated below the graphs. HR, hazard ratio.
(F) Immunohistochemical analysis of UBE2O, AMPKα2, AMPKα1, P-S6 and HIF1α proteins in human breast tumor samples (left). Scale bars, 50 μm. Correlation between the indicated protein levels was determined by the PASS Pearson Chi-Square test (right). n = 99. R, correlation coefficient.
Error bars represent +/− SEM. p value was determined by Log-rank (Mantel-Cox) test (E), Chi-square test (F) or Student’s t test (*p<0.05; **p<0.01; ***p<0.001). See also Figures S7.
We also sought to determine the clinical relevance of UBE2O regulation of the AMPKα2-mTOR-HIF1α axis. Importantly, an immunohistochemical analysis of human breast tumors demonstrated a statistically significant inverse correlation between UBE2O and AMPKα2 but not AMPKα1 protein levels (Figure 7F). In sharp contrast, we found a high degree of positive correlations between UBE2O and phospho-S6 or HIF1α protein levels (Figure 7F). Taken together, these results suggest that in a significant fraction of human cancers, UBE2O is upregulated, which may contribute to regulation of the AMPKα2-mTOR-HIF1α pathway.
Pharmacological blockade of UBE2O inhibits tumorigenesis through AMPKα2 restoration
The biological and clinical relevance of UBE2O in tumorigenesis suggests that UBE2O may be a promising target for anti-cancer therapeutics. Interestingly, UBE2O is known to be susceptible to inhibition by arsenite, which can crosslink adjacent cysteines within its catalytic domain (Berleth and Pickart, 1996; Klemperer et al., 1989) (Figure 8A). Critically, we found that in a clinically achievable concentration (0.5~1 μM), arsenic trioxide (ATO) diminished the heavy ubiquitination of AMPKα2 induced by UBE2O (Figure 8B and Figure S8A), and increased the protein levels of AMPKα2, but not AMPKα1, in a UBE2O-dependent manner (Figure 8C and Figures S8B and S8C). It was also shown many years ago that arsenite activated AMPK and were associated with large increases in the AMP:ATP ratio at concentrations far higher (500~1000 fold) (Corton et al., 1994) than those examined in our study. These suggest arsenite could upregulate AMPK through activation as well as overexpression of AMPK.
Figure 8. Blockade of UBE2O with ATO reduces tumorigenesis through AMPKα2 restoration.

(A) A diagram depicting the mechanism by which arsenite inhibits UBE2O (top). Recombinant UBE2O proteins were subjected to in vitro self-ubiquitination assay in the presence of ATO (10 μM) (bottom).
(B) Lysates from 293T cells expressing the indicated plasmids treated with ATO (1 μM) together with MG132 (10 μM) for 6 hr were subjected to metal-affinity purification for His-tagged ubiquitin then immunoblotting for ubiquitinated AMPKα2. Ni-NTA, Ni2+-nitrilotriacetic acid. # indicates nonspecific band.
(C) Lysates from HCT116 cells expressing UBE2O shRNA treated with ATO (0.5 μM) for the indicated times were subjected to immunoblotting (top). AMPKα2 or AMPKα1 protein levels were quantified by normalizing to the intensity of the Actin band (bottom).
(D) Tumor volumes of mouse allograft implanted with E1A+H-RasV12 MEFs overexpressing UBE2O treated with ATO (i.p. 2 mg/kg, daily) were measured at different day. n = 7, p value was determined by ANOVA (n.s., non-significant; ***p<0.001). Scale bars, 75 μm.
(E) Representative images, immunohistochemical analysis of Ki-67, and quantification of Ki-67 positive cells of mouse xenograft tumors from (D). Scale bars, 75 μm. n = 7, p value was determined by Student’s t test (n.s., non-significant; ***p<0.001).
(F) Lysates from (D) were subjected to immunoblotting.
(G) TFS analysis of PyVT;Ube2o+/+ mice treated with vehicle (n = 15) or ATO (i.p. 2.5 mg/kg, every other day for 65 days) (n = 13). TFS curves of vehicle-treated PyVT;Ube2o+/− mice (n = 15) are also shown. p value was determined by Log-rank (Mantel-Cox) test (n.s., non-significant; **p<0.01).
(H) The weight of mammary tumors isolated from 105 days old vehicle-treated PyVT;Ube2o+/+ (n = 13), ATO-treated PyVT;Ube2o+/+ (n = 13) and vehicle-treated PyVT;Ube2o+/− (n = 7) mice were quantified. p value was determined by Student’s t test (*p<0.05; ***p<0.001).
(I) Sections of lungs isolated from 105 days old vehicle-treated PyVT;Ube2o+/+, ATO-treated PyVT;Ube2o+/+ and vehicle-treated PyVT;Ube2o+/− mice were stained for H&E or PyV T Ag. Arrowheads indicate clusters of metastatic cells in the lung. Scale bars, 75 μm. The number of metastatic sites in lungs of mice was also quantified (right). n = 7, p value was determined by Student’s t test (n.s., non-significant; *p<0.05).
(J) H&E-stained sections of anterior prostate lobes isolated from 12 weeks old TRAMP;Ube2o+/+ mice treated with ATO (i.p. 2.5 mg/kg, every other day for 4 weeks). Scale bars, 75 μm.
(K) Quantification of PIN in (J). Vehicle-treated TRAMP;Ube2o+/+ mice cohort (n = 9); ATO-treated TRAMP;Ube2o+/+ mice cohort (n = 6); vehicle-treated TRAMP;Ube2o+/− mice cohort (n = 9). p value was determined by Student’s t test (n.s., non-significant; *p<0.05; **p<0.01).
(L) Prostate tissue weight of 12 weeks old vehicle-treated TRAMP;Ube2o+/+ (n = 10), ATO-treated TRAMP;Ube2o+/+ (n = 6) and vehicle-treated TRAMP;Ube2o+/− (n = 9) mice. p value was determined by Student’s t test (n.s., non-significant; *p<0.05).
Error bars represent +/− SEM. See also Figure S8.
Both inorganic and synthesized organic arsenite are currently being tested against various forms of cancer in clinical trials (https://clinicaltrials.gov). To determine the in vivo contribution of UBE2O function to the anti-tumor ability of ATO, we subcutaneously implanted transformed cells ectopically expressing UBE2O into Swiss nu/nu mice and treated them daily with ATO. ATO treatment dramatically reduced tumor growth in mice bearing cells overexpressing UBE2O, but not in mice bearing control cells, accompanied by the restoration of AMPKα2 expression and marked reductions in mTOR activity and HIF1α levels (Figures 8D–8F). Conversely, depletion of UBE2O abrogated the sensitivity of HCT116 xenografts to ATO (Figures S8D–S8F). Notably, ATO produced an effect similar in size to that of UBE2O silencing, without any further additive effect, supporting the idea that ATO may exert selective action on UBE2O. Additionally, ATO treatment resulted in an anti-Warburg metabolic state, as well as a reduced median cell size in HCT116 control cells, but not UBE2O-silenced cells (Figure S8G and S8H), suggesting that ATO impairs the UBE2O-dependent pro-growth, glycolytic and biosynthetic programs of cancer cells. ATO is also known to induce oxidative stress through the generation of reactive oxygen species (ROS) (Dilda and Hogg, 2007). However, it should be noted that while treatment with the anti-oxidant N-acetylcysteine (NAC) attenuated ATO-induced ROS production, it did not overcome ATO-mediated reduction in the growth rate of cancer cells (Figure S8I). In contrast, AMPKα2, but not AMPKα1, arbitrated the anti-cancer effect of ATO (Figure S8J), suggesting that inhibition of the UBE2O-AMPKα2 axis may be a mechanism underlying ATO-mediated tumor suppression.
Finally, we examined whether the blockade of UBE2O with ATO in the MMTV-PyVT and TRAMP models could reduce tumor incidence and progression. Critically, the blockade of UBE2O with ATO provided a significant survival benefit for PyVT;Ube2o+/+ mice (median tumor onset 84 days vs. 72 days for vehicle-treated mice, p = 0.0024) and delayed median tumor onset to similar to that of PyVT;Ube2o+/− mice (84 days) (Figure 8G), without an adverse effect on the liver (data not shown). We also found that mammary tumor volume and weight as well as histological aggressiveness were greatly reduced by the blockade of UBE2O with ATO (Figure 8H). Moreover, in contrast to multiple tumor nodules presented on the lung surface in vehicle-treated PyVT;Ube2o+/+ mice at 105 day of age, none was seen in age-matched ATO-treated mice (Figure 8I). Likewise, ATO treatment led to reduced prostate lobe enlargement and HG-PIN development in TRAMP;Ube2o+/+ mice at 12 weeks of age (Figures 8J–8L). Taken together, these results suggest that the blockade of UBE2O, as epitomized here by treatment with ATO, reduces tumorigenesis to levels comparable to that observed in cases of UBE2O deficiency.
DISCUSSION
Our findings allow us to reach a number of relevant conclusions:
Loss of one or both alleles of Ube2o results in delayed tumor initiation and diminished tumor growth and metastasis rates in mouse models of breast and prostate cancers. We have demonstrated that UBE2O is a positive regulator of both aerobic glycolysis and cellular biosynthesis in cancer cells, and that inactivation of UBE2O is sufficient to turn off the glycolytic and biosynthetic programs of tumor cells. Thus, UBE2O may act as an oncogene that triggers cancer progression and eliminates key metabolic checkpoints that antagonize pro-growth cellular metabolism.
Our initial screen for UBE2O-interacting proteins in MEFs identified AMPKα2, but not AMPKα1, even though the latter is expressed in MEFs. Furthermore, UBE2O selectively targets the AMPKα2 for ubiquitination and degradation; the major site of this ubiquitination has been identified as K470 residue that is conserved in the AMPKα2 isoforms of a variety of vertebrate species, but notably, is replaced by an arginine (R475) in AMPKα1. Furthermore, our data clearly demonstrate that the mechanism of UBE2O action in tumor biology is specific to AMPKα2 but not to AMPKα1.
AMPKα1 and AMPKα2 demonstrate some specificity in tissue distribution, subcellular localization and substrate selection (Salt et al., 1998; Stapleton et al., 1996; Woods et al., 1996). Thus it will be interesting to determine whether these differences in the two AMPKα isoforms are reflected in differing effects of UBE2O on tumor biology. Settling this question is not only important from a physiological point of view but also may facilitate the design of drugs targeting AMPK, if the precise AMPK isoform responsible for the desirable anti-tumor effect is defined.
We have demonstrated that the presence of UBE2O in cancer results in the upregulation of the mTOR-HIF1α pathway, which is closely associated with pro-growth, glycolytic and biosynthetic programs. In addition, ablation of Ube2o in our mouse cancer models attenuated intra-tumoral vascularization and expression of neovascularization genes, including HIF1α targets, highlighting that UBE2O regulation of the mTOR-HIF1α pathway is critical for the angiogenic signaling pathway and the neovascularization required for cancer growth and metastasis. UBE2O has been also shown to regulate other targets; in the case of NF-κB, depletion of UBE2O has been shown to enhance NF-κB activation (Zhang et al., 2013b). Thus one might expect that tumors developed in animals in which Ube2o has been deleted would display more innate immune cell populations. In sharp contrast, however, expression of NF-κB–dependent pro-inflammatory genes was inhibited by Ube2o ablation in mouse cancer models.
Our findings should prove highly relevant to the development of drugs designed to regulate UBE2O activity as a mode of cancer therapy, as the blockade of UBE2O with ATO produces an effect on tumor biology of similar size to that of UBE2O deficiency. Furthermore, as loss of a single allele of Ube2o results in decreased tumorigenesis, it is expected that partial inhibition by agents (e.g., small molecule inhibitors) directed against UBE2O may exert the desired anti-cancer effect.
EXPERIMENTAL PROCEDURES
Mice
To generate Ube2o knockout mice, we injected fully verified, three different Ube2otm1(KOMP)Mpb ES clones (JM8A3.N1), which were created by the CSD consortium (CSD81565) from the trans-NIH Knock-Out Mouse Project (KOMP), into blastocysts derived from C57BL/6 mice to produce chimeras. Transmitting chimeric mice were bred from EIIa–Cre transgenic mice to generate the Ube2o− alleles. Cre+;Ube2o+/− males were backcrossed twice to C57BL/6 females and progeny of these matings that were Cre− were then backcrossed to littermates to yield the experimental cohort. The details of PCR genotyping are described in the Supplemental Experimental Procedures. All animal experiments in this study were approved by and adhered to the guidelines of the MD Anderson Cancer Center Animal Care and Use Committee.
Human tumor TMAs analysis
Human tumor tissue microarrays (TMAs) were obtained from US Biomax and Pantomics, and work was performed in accordance with the Institutional Review Board (IRB) approval at MD Anderson Cancer Center. TMA slides were incubated with antibodies against UBE2O, AMPKα1, AMPKα2, P-S6 or HIF1α and a biotin-conjugated secondary antibody and then incubated with an avidin-biotin-peroxidase complex. Visualization was performed using 3-amino-9-ethylcarbazole (AEC) chromogen. The TMA cores were scored by the pathologist (W.X.) blind to cancer outcomes. According to histologic scoring, the intensity of staining was ranked into one of four groups: high (+++), medium (++), low (+), and negative (−).
Statistical analysis
Statistical analysis was performed with SPSS V.20.0 and GraphPad Prism 6. Two-tailed Student’s t tests were used for single comparison, and analysis of variance (ANOVA) with Bonferroni post-hoc tests was used for multiple comparisons unless otherwise specified. The correlation coefficients were calculated by the PASS Pearson Chi-Square test. p values below 0.05 were considered statistically significant.
Supplementary Material
SIGNIFICANCE.
The regulation of the enzymatic activity of AMPK by adenine nucleotides and upstream kinases is already an active area of research, but there is compelling evidence suggesting additional modes of AMPK regulation. Here we show that UBE2O and AMPKα2, but not AMPKα1, form a functional axis that elicits pro-growth, glycolytic and biosynthetic cancer programs mediated by mTOR-HIF1α. While ablation of Ube2o provides a significant anti-tumor effect and survival advantage in mouse models of breast and prostate cancers, inactivation of AMPKα2, but not AMPKα1, reverses the attenuation of tumorigenesis caused by UBE2O loss. These findings suggest that AMPKα2-selective modulation by UBE2O is a critical determinant of tumorigenesis, and a potential target of therapeutic strategies.
Highlights.
Genetic ablation of Ube2o impairs progression of breast and prostate cancers in mice.
UBE2O specifically targets AMPKα2 for ubiquitination and degradation.
UBE2O-dependent tumor biology is mediated by mTOR and HIF1α.
UBE2O blockade inhibits tumorigenesis through AMPKα2 restoration.
Acknowledgments
We thank M. G. Lee, D. Sarbassov, H. K. Lin, A. Sahin, C. Logothetis and P. P. Pandolfi for sharing reagents and critical discussions. We are grateful to T. Garvey for critical editing of the manuscript. We are thankful to J. S. Lee for technical support and advice for microarray. This work was supported by a grant of Basic Science Research Program through NRF of Korea funded by the Ministry of Science, ICT & Future Planning (2014R1A1A3051340) and a grant of the Korea Health Technology R&D Project through KHIDI funded by the Ministry of Health & Welfare (HI15C2679) to S.J.S., and a Cancer Prevention Research Institute of Texas grant (RP150084), a Department of Defense grant (W81XWH-15-1-0662) and a National Institutes of Health grant (CA196740) to M.S.S.
Footnotes
Accession number
Microarray data are deposited in the NCBI with accession number GEO: GSE92919.
AUTHOR CONTRIBUTIONS
The research were conceived and designed by I.K.V., Y.Y., S.J.S. and M.S.S. Most experiments were performed by I.K.V. and Y.Y. G.K. performed mass spectrometry analysis and cancer biostatistical analysis. W.X. and M.-C.H. performed pathological analysis. E.Z.B. performed MRI analysis. H.K, S.-J.K., M.K.P. and J.P.H. assisted in experiments. Data were analyzed by I.K.V., Y.Y., S.J.S. and M.S.S. The paper was written by I.K.V., Y.Y., S.J.S. and M.S.S.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Adams JM, Harris AW, Pinkert CA, Corcoran LM, Alexander WS, Cory S, Palmiter RD, Brinster RL. The c-myc oncogene driven by immunoglobulin enhancers induces lymphoid malignancy in transgenic mice. Nature. 1985;318:533–538. doi: 10.1038/318533a0. [DOI] [PubMed] [Google Scholar]
- Berleth ES, Pickart CM. Mechanism of ubiquitin conjugating enzyme E2-230K: catalysis involving a thiol relay? Biochemistry. 1996;35:1664–1671. doi: 10.1021/bi952105y. [DOI] [PubMed] [Google Scholar]
- Briffa R, Um I, Faratian D, Zhou Y, Turnbull AK, Langdon SP, Harrison DJ. Multi-Scale Genomic, Transcriptomic and Proteomic Analysis of Colorectal Cancer Cell Lines to Identify Novel Biomarkers. PLoS One. 2015;10:e0144708. doi: 10.1371/journal.pone.0144708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cancer Genome Atlas Research Network. Comprehensive molecular characterization of clear cell renal cell carcinoma. Nature. 2013;499:43–49. doi: 10.1038/nature12222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen T, Zhou T, He B, Yu H, Guo X, Song X, Sha J. mUbiSiDa: a comprehensive database for protein ubiquitination sites in mammals. PLoS One. 2014;9:e85744. doi: 10.1371/journal.pone.0085744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corton JM, Gillespie JG, Hardie DG. Role of the AMP-activated protein kinase in the cellular stress response. Curr Biol. 1994;4:315–324. doi: 10.1016/s0960-9822(00)00070-1. [DOI] [PubMed] [Google Scholar]
- Di Cristofano A, Kotsi P, Peng YF, Cordon-Cardo C, Elkon KB, Pandolfi PP. Impaired Fas response and autoimmunity in Pten+/− mice. Science. 1999;285:2122–2125. doi: 10.1126/science.285.5436.2122. [DOI] [PubMed] [Google Scholar]
- Dilda PJ, Hogg PJ. Arsenical-based cancer drugs. Cancer Treat Rev. 2007;33:542–564. doi: 10.1016/j.ctrv.2007.05.001. [DOI] [PubMed] [Google Scholar]
- Faubert B, Boily G, Izreig S, Griss T, Samborska B, Dong Z, Dupuy F, Chambers C, Fuerth BJ, Viollet B, et al. AMPK is a negative regulator of the Warburg effect and suppresses tumor growth in vivo. Cell Metab. 2013;17:113–124. doi: 10.1016/j.cmet.2012.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faubert B, Vincent EE, Griss T, Samborska B, Izreig S, Svensson RU, Mamer OA, Avizonis D, Shackelford DB, Shaw RJ, et al. Loss of the tumor suppressor LKB1 promotes metabolic reprogramming of cancer cells via HIF-1alpha. Proc Natl Acad Sci USA. 2014;111:2554–2559. doi: 10.1073/pnas.1312570111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenberg NM, DeMayo F, Finegold MJ, Medina D, Tilley WD, Aspinall JO, Cunha GR, Donjacour AA, Matusik RJ, Rosen JM. Prostate cancer in a transgenic mouse. Proc Natl Acad Sci USA. 1995;92:3439–3443. doi: 10.1073/pnas.92.8.3439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guy CT, Cardiff RD, Muller WJ. Induction of mammary tumors by expression of polyomavirus middle T oncogene: a transgenic mouse model for metastatic disease. Mol Cell Biol. 1992;12:954–961. doi: 10.1128/mcb.12.3.954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gwinn DM, Shackelford DB, Egan DF, Mihaylova MM, Mery A, Vasquez DS, Turk BE, Shaw RJ. AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol Cell. 2008;30:214–226. doi: 10.1016/j.molcel.2008.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hadad SM, Baker L, Quinlan PR, Robertson KE, Bray SE, Thomson G, Kellock D, Jordan LB, Purdie CA, Hardie DG, et al. Histological evaluation of AMPK signalling in primary breast cancer. BMC Cancer. 2009;9:307. doi: 10.1186/1471-2407-9-307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hallstrom TC, Mori S, Nevins JR. An E2F1-dependent gene expression program that determines the balance between proliferation and cell death. Cancer Cell. 2008;13:11–22. doi: 10.1016/j.ccr.2007.11.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hao YH, Doyle JM, Ramanathan S, Gomez TS, Jia D, Xu M, Chen ZJ, Billadeau DD, Rosen MK, Potts PR. Regulation of WASH-dependent actin polymerization and protein trafficking by ubiquitination. Cell. 2013;152:1051–1064. doi: 10.1016/j.cell.2013.01.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardie DG, Alessi DR. LKB1 and AMPK and the cancer-metabolism link - ten years after. BMC Biol. 2013;11:36. doi: 10.1186/1741-7007-11-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardie DG, Ross FA, Hawley SA. AMPK: a nutrient and energy sensor that maintains energy homeostasis. Nat Rev Mol Cell Biol. 2012;13:251–262. doi: 10.1038/nrm3311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeon SM, Chandel NS, Hay N. AMPK regulates NADPH homeostasis to promote tumour cell survival during energy stress. Nature. 2012;485:661–665. doi: 10.1038/nature11066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim YH, Liang H, Liu X, Lee JS, Cho JY, Cheong JH, Kim H, Li M, Downey TJ, Dyer MD, et al. AMPKalpha modulation in cancer progression: multilayer integrative analysis of the whole transcriptome in Asian gastric cancer. Cancer Res. 2012;72:2512–2521. doi: 10.1158/0008-5472.CAN-11-3870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kishton RJ, Barnes CE, Nichols AG, Cohen S, Gerriets VA, Siska PJ, Macintyre AN, Goraksha-Hicks P, de Cubas AA, Liu T, et al. AMPK Is Essential to Balance Glycolysis and Mitochondrial Metabolism to Control T-ALL Cell Stress and Survival. Cell Metab. 2016;23:649–662. doi: 10.1016/j.cmet.2016.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klemperer NS, Berleth ES, Pickart CM. A novel, arsenite-sensitive E2 of the ubiquitin pathway: purification and properties. Biochemistry. 1989;28:6035–6041. doi: 10.1021/bi00440a047. [DOI] [PubMed] [Google Scholar]
- Laplante M, Sabatini DM. mTOR signaling in growth control and disease. Cell. 2012;149:274–293. doi: 10.1016/j.cell.2012.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JO, Lee SK, Kim N, Kim JH, You GY, Moon JW, Jie S, Kim SJ, Lee YW, Kang HJ, et al. E3 ubiquitin ligase, WWP1, interacts with AMPKalpha2 and down-regulates its expression in skeletal muscle C2C12 cells. J Biol Chem. 2013;288:4673–4680. doi: 10.1074/jbc.M112.406009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin M, Smith LT, Smiraglia DJ, Kazhiyur-Mannar R, Lang JC, Schuller DE, Kornacker K, Wenger R, Plass C. DNA copy number gains in head and neck squamous cell carcinoma. Oncogene. 2006;25:1424–1433. doi: 10.1038/sj.onc.1209166. [DOI] [PubMed] [Google Scholar]
- Mashtalir N, Daou S, Barbour H, Sen NN, Gagnon J, Hammond-Martel I, Dar HH, Therrien M, Affar el B. Autodeubiquitination protects the tumor suppressor BAP1 from cytoplasmic sequestration mediated by the atypical ubiquitin ligase UBE2O. Mol Cell. 2014;54:392–406. doi: 10.1016/j.molcel.2014.03.002. [DOI] [PubMed] [Google Scholar]
- Pfeifer M, Grau M, Lenze D, Wenzel SS, Wolf A, Wollert-Wulf B, Dietze K, Nogai H, Storek B, Madle H, et al. PTEN loss defines a PI3K/AKT pathway-dependent germinal center subtype of diffuse large B-cell lymphoma. Proc Natl Acad Sci USA. 2013;110:12420–12425. doi: 10.1073/pnas.1305656110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pineda CT, Ramanathan S, Fon Tacer K, Weon JL, Potts MB, Ou YH, White MA, Potts PR. Degradation of AMPK by a cancer-specific ubiquitin ligase. Cell. 2015;160:715–728. doi: 10.1016/j.cell.2015.01.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Podsypanina K, Ellenson LH, Nemes A, Gu J, Tamura M, Yamada KM, Cordon-Cardo C, Catoretti G, Fisher PE, Parsons R. Mutation of Pten/Mmac1 in mice causes neoplasia in multiple organ systems. Proc Natl Acad Sci USA. 1999;96:1563–1568. doi: 10.1073/pnas.96.4.1563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi J, Gong J, Zhao T, Zhao J, Lam P, Ye J, Li JZ, Wu J, Zhou HM, Li P. Downregulation of AMP-activated protein kinase by Cidea-mediated ubiquitination and degradation in brown adipose tissue. Embo J. 2008;27:1537–1548. doi: 10.1038/emboj.2008.92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qu C, Zhang W, Zheng G, Zhang Z, Yin J, He Z. Metformin reverses multidrug resistance and epithelial-mesenchymal transition (EMT) via activating AMP-activated protein kinase (AMPK) in human breast cancer cells. Mol Cell Biochem. 2014;386:63–71. doi: 10.1007/s11010-013-1845-x. [DOI] [PubMed] [Google Scholar]
- Radivojac P, Vacic V, Haynes C, Cocklin RR, Mohan A, Heyen JW, Goebl MG, Iakoucheva LM. Identification, analysis, and prediction of protein ubiquitination sites. Proteins. 2010;78:365–380. doi: 10.1002/prot.22555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice KL, Lin X, Wolniak K, Ebert BL, Berkofsky-Fessler W, Buzzai M, Sun Y, Xi C, Elkin P, Levine R, et al. Analysis of genomic aberrations and gene expression profiling identifies novel lesions and pathways in myeloproliferative neoplasms. Blood Cancer J. 2011;1:e40. doi: 10.1038/bcj.2011.39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito Y, Chapple RH, Lin A, Kitano A, Nakada D. AMPK Protects Leukemia-Initiating Cells in Myeloid Leukemias from Metabolic Stress in the Bone Marrow. Cell Stem Cell. 2015;17:585–596. doi: 10.1016/j.stem.2015.08.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salt I, Celler JW, Hawley SA, Prescott A, Woods A, Carling D, Hardie DG. AMP-activated protein kinase: greater AMP dependence, and preferential nuclear localization, of complexes containing the alpha2 isoform. Biochem J. 1998;334(Pt 1):177–187. doi: 10.1042/bj3340177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shackelford DB, Shaw RJ. The LKB1-AMPK pathway: metabolism and growth control in tumour suppression. Nat Rev Cancer. 2009;9:563–575. doi: 10.1038/nrc2676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stapleton D, Mitchelhill KI, Gao G, Widmer J, Michell BJ, Teh T, House CM, Fernandez CS, Cox T, Witters LA, et al. Mammalian AMP-activated protein kinase subfamily. J Biol Chem. 1996;271:611–614. doi: 10.1074/jbc.271.2.611. [DOI] [PubMed] [Google Scholar]
- Stephenson AJ, Smith A, Kattan MW, Satagopan J, Reuter VE, Scardino PT, Gerald WL. Integration of gene expression profiling and clinical variables to predict prostate carcinoma recurrence after radical prostatectomy. Cancer. 2005;104:290–298. doi: 10.1002/cncr.21157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamas P, Hawley SA, Clarke RG, Mustard KJ, Green K, Hardie DG, Cantrell DA. Regulation of the energy sensor AMP-activated protein kinase by antigen receptor and Ca2+ in T lymphocytes. J Exp Med. 2006;203:1665–1670. doi: 10.1084/jem.20052469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toffoli S, Bar I, Abdel-Sater F, Delree P, Hilbert P, Cavallin F, Moreau F, Van Criekinge W, Lacroix-Triki M, Campone M, et al. Identification by array comparative genomic hybridization of a new amplicon on chromosome 17q highly recurrent in BRCA1 mutated triple negative breast cancer. Breast Cancer Res. 2014;16:466. doi: 10.1186/s13058-014-0466-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong WH, Sourbier C, Kovtunovych G, Jeong SY, Vira M, Ghosh M, Romero VV, Sougrat R, Vaulont S, Viollet B, et al. The glycolytic shift in fumarate-hydratase-deficient kidney cancer lowers AMPK levels, increases anabolic propensities and lowers cellular iron levels. Cancer Cell. 2011;20:315–327. doi: 10.1016/j.ccr.2011.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science. 2009;324:1029–1033. doi: 10.1126/science.1160809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner SA, Beli P, Weinert BT, Scholz C, Kelstrup CD, Young C, Nielsen ML, Olsen JV, Brakebusch C, Choudhary C. Proteomic analyses reveal divergent ubiquitylation site patterns in murine tissues. Mol Cell Proteomics. 2012;11:1578–1585. doi: 10.1074/mcp.M112.017905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang P, Zhang RY, Song J, Guan YF, Xu TY, Du H, Viollet B, Miao CY. Loss of AMP-activated protein kinase-alpha2 impairs the insulin-sensitizing effect of calorie restriction in skeletal muscle. Diabetes. 2012;61:1051–1061. doi: 10.2337/db11-1180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Li X, Cheng Y, Sun X, Self S, Kooperberg C, Dai JY. Copy number alterations detected by whole-exome and whole-genome sequencing of esophageal adenocarcinoma. Hum Genomics. 2015;9:22. doi: 10.1186/s40246-015-0044-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woods A, Salt I, Scott J, Hardie DG, Carling D. The alpha1 and alpha2 isoforms of the AMP-activated protein kinase have similar activities in rat liver but exhibit differences in substrate specificity in vitro. FEBS Lett. 1996;397:347–351. doi: 10.1016/s0014-5793(96)01209-4. [DOI] [PubMed] [Google Scholar]
- Zhang X, Zhang J, Bauer A, Zhang L, Selinger DW, Lu CX, Ten Dijke P. Fine-tuning BMP7 signalling in adipogenesis by UBE2O/E2-230K-mediated monoubiquitination of SMAD6. Embo J. 2013a;32:996–1007. doi: 10.1038/emboj.2013.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Zhang J, Zhang L, van Dam H, ten Dijke P. UBE2O negatively regulates TRAF6-mediated NF-kappaB activation by inhibiting TRAF6 polyubiquitination. Cell Res. 2013b;23:366–377. doi: 10.1038/cr.2013.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.