

Summary
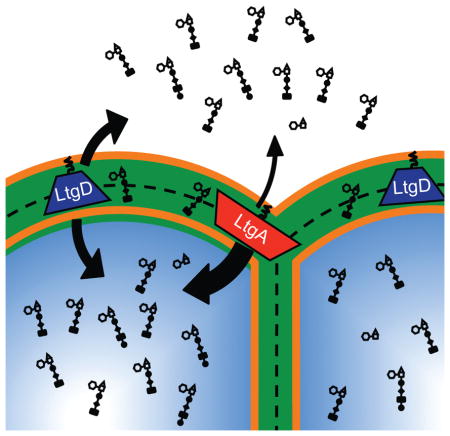
Neisseria gonorrhoeae releases peptidoglycan (PG) fragments during infection that provoke a large inflammatory response and, in pelvic inflammatory disease, this response leads to the death and sloughing of ciliated cells of the Fallopian tube. We characterized the biochemical functions and localization of two enzymes responsible for the release of proinflammatory PG fragments. The putative lytic transglycosylases LtgA and LtgD were shown to create the 1,6-anhydromuramyl moieties, and both enzymes were able to digest a small, synthetic tetrasaccharide dipeptide PG fragment into the cognate 1,6-anhydromuramyl-containing reaction products. Degradation of tetrasaccharide PG fragments by LtgA is the first demonstration of a family 1 lytic transglycosylase exhibiting this activity. Pulse-chase experiments in gonococci demonstrated that LtgA produces a larger amount of PG fragments than LtgD, and a vast majority of these fragments are recycled. In contrast, LtgD was necessary for wild-type levels of PG precursor incorporation and produced fragments predominantly released from the cell. Additionally, super-resolution microscopy established that LtgA localizes to the septum, whereas LtgD is localized around the cell. This investigation suggests a model where LtgD produces PG monomers in such a way that these fragments are released, whereas LtgA creates fragments that are mostly taken into the cytoplasm for recycling.
Graphical abstract
Neisseria gonorrhoeae releases a significant portion of cell wall-derived peptidoglycan fragments, inducing inflammation and tissue damage in the genital tract. This work describes the functions of the two lytic transglycosylases responsible for producing the peptidoglycan fragments tracheal cytotoxin and GlcNAc-anhydro-MurNAc-tripeptide, a Nod1 agonist. Although these two enzymes can perform the same biochemical reaction, each lytic transglycosylase has different substrate specificity and cellular localization that leads to differing contributions to peptidoglycan remodeling, recycling, and release.
Introduction
Neisseria gonorrhoeae is the causative agent of the sexually-transmitted infection gonorrhea, which is usually manifested as urethritis in men and cervicitis in women. The World Health Organization has estimated the rate of new gonococcal infections to be over 100 million per year (WHO, 2014). In women, between 50–80% of gonorrhea infections are asymptomatic, resulting in failure to seek treatment and increased risk of complications (Pariser, 1972). Untreated infection in women can progress to pelvic inflammatory disease (PID) and result in tubal-factor infertility, chronic pelvic pain, and ectopic pregnancies (Westrom, 1991). The process for these manifestations is complex, but the release of cell wall-derived natural products during the normal course of growth of the organism is at the root of these complications (Melly et al., 1984). As the organism grows, cell-wall homeostasis requires remodeling of the major cell-wall constituent, the peptidoglycan (PG). In the course of these events, PG fragments are released from the polymeric cell wall. Some of these fragments are internalized for recycling, yet others are released to the medium (Garcia & Dillard, 2008, Rosenthal, 1979, Johnson et al., 2013). These fragments act together with lipooligosaccharide to trigger a large inflammatory response, which kills ciliated cells of the Fallopian tube as demonstrated using an organ culture model of PID (Melly et al., 1981, Melly et al., 1984).
Gram-negative bacteria, such as N. gonorrhoeae, have a thin PG sacculus composed of glycan strands cross-linked by peptides that forms a large molecule necessary for maintaining cell shape and providing structural integrity against the internal osmotic pressure of the cell. About half of the PG of the cell wall is turned over during each round of cell division. The majority of degraded PG fragments are transported to the cytoplasm by the permease AmpG for recycling (Goodell, 1985, Jacobs et al., 1994). The PG fragments not taken up for recycling are released from the bacterium by an unknown mechanism. N. gonorrhoeae releases more PG fragments than most other Gram-negative bacteria, even more than other Neisseria species (Woodhams et al., 2013). The most abundant PG fragments released by N. gonorrhoeae are comprised of the N-acetylglucosaminyl-β-1,4-anhydro-N-acetylmuramyl (GlcNAc-anhMurNAc) species, with the muramyl moiety covalently linked to a tripeptide or tetrapeptide, collectively referred to as “PG monomers.” When applied to human Fallopian tube tissue organ culture, this mixture of disaccharide-tripeptide and disaccharide-tetrapeptide PG monomers from N. gonorrhoeae was shown to recapitulate the sloughing of ciliated cells caused by gonococcal infections (Melly et al., 1984). Death and sloughing of ciliated epithelial cells is also observed during Bordetella pertussis infections, and this tissue damage is attributed to the anhydrodisaccharide tetrapeptide, also known as tracheal cytotoxin or TCT (Goldman et al., 1982, Rosenthal et al., 1987).
The class of enzymes known as lytic transglycosylases (LTs) is necessary to create PG monomers from the polymeric cell wall. These lyases cleave the β-1,4-glycosidic bond between N-acetylmuramic acid and the N-acetylglucosamine residues and create a 1,6-anhydromuramyl bond. The LTs have been classified into four established protein families based on sequence similarities and consensus motifs, with family 1 being further classified into five subfamilies (Blackburn & Clarke, 2001). Structural studies demonstrated that LTs from families 1, 3, and 4 have a catalytic domain with a goose-type lysozyme fold (Scheurwater et al., 2008). E. coli has 8 known LTs consisting of the soluble Slt70 and the membrane-bound MltA-G (Vollmer et al., 2008, Yunck et al., 2016). E. coli also encodes putative LT RlpA, although LT activity for this protein has not yet been demonstrated in vitro (Jorgenson et al., 2014).
Previous studies described and characterized 5 putative LTs in all N. gonorrhoeae strains and an additional 1 or 2 LTs encoded in the Gonococcal Genetic Island (Kohler et al., 2007, Cloud-Hansen et al., 2008, Chan et al., 2012). Newly described LTs RlpA and MltG are also encoded in the gonococcal chromosome [RefSeq: NC_022240.1] (Jorgenson et al., 2014, Yunck et al., 2016). Of these LTs, only LtgA and LtgD are necessary for release of PG monomers, since a deletion of both ltgA and ltgD results in the elimination of released PG monomers (Cloud & Dillard, 2002, Cloud-Hansen et al., 2008). Both LtgA and LtgD are predicted to be exolytic LTs that process the PG sugar backbone by removing two sugars at a time while creating 1,6-anhydro-MurNAc-containing monomers consisting of a disaccharide with MurNAc-linked peptides. Modification of PG by O-acetylation of MurNAc blocks the activities of both lysozyme and LTs, but does not change the abundance of monomers released from growing cells (Dillard & Hackett, 2005, Scheurwater et al., 2008).
This work investigates the biochemical activities, localization, and contributions to PG turnover of LtgA and LtgD in order to determine characteristics of these enzymes responsible for the increased release of inflammatory PG monomers. Biochemical characterization of LtgA and LtgD demonstrated that these enzymes possess LT activity in vitro and this activity was abolished by introducing mutations affecting the predicted active-site residues. Both enzymes were able to degrade purified whole PG and were able to digest a synthetic tetrasaccharide dipeptide into two monomeric units. The ability of LtgA to digest this synthetic tetrasaccharide dipeptide was unexpected since homologs are known to lack this activity (Suvorov et al., 2008). This additional activity may be one of the causes of high levels of PG monomers being released from gonococci. The predicted subcellular localization of LtgA and LtgD are different than the localization of many homologs, which is commonly as soluble proteins in the periplasm. Sequence characteristics suggested that these enzymes might be lipidated, outer-membrane proteins, and localization of both LTs was confirmed by subcellular fractionation. Mutation inhibiting the lipidation of LtgD caused a reduction in the amount of released PG monomers. PG fragment release from ampG deletion strains revealed significantly less PG monomer release from ΔltgA ΔampG mutants than from either ΔampG or ΔltgD ΔampG strains, indicating LtgA produces a majority of the PG fragments that are recycled by the gonococcal cell. Cellular localization studies by super-resolution microscopy demonstrated that LtgA localizes to the septal region of diplococci, whereas LtgD was found localized to points around the cell. The different localization of LtgA and LtgD further suggest that they have different cellular functions. LtgA and LtgD were also found to have different substrate specificities within the gonococcal sacculus and to produce different products, as determined by analysis of PG fragments from in vitro digests.
Results
LtgA and LtgD have Lytic Transglycosylase Activity
LtgA and LtgD have been thought to be LTs due to activities of characterized homologs, in addition to ltgA and ltgD being necessary for the release of anhydrodisaccharide PG monomers by gonococci (Cloud-Hansen et al., 2008). However, direct evidence of their activities in vitro had not been characterized. Recombinant N-terminal hexahistidine-tagged LtgA and LtgD were produced in E. coli, and the purified proteins were tested for the ability to digest purified PG. Gonococcal sacculi, from both a wild-type strain and from a strain that does not acetylate PG were used to examine LT activity. Both LtgA and LtgD digested gonococcal sacculi into soluble PG fragments. Reversed-phase HPLC analysis of the digested products identified the presence of a variety of GlcNAc-anhMurNAc peptide monomers, and GlcNAc-anhMurNAc-containing peptide crosslinked muropeptides. The active site residues of these enzymes were identified by comparing catalytic sites of E. coli Slt70 and MltB (van Asselt et al., 1999b, van Asselt et al., 1999a). Site-directed mutagenesis of the putative catalytic glutamic acid residues was followed by purification of the recombinant proteins LtgA(E481A) and LtgD(E158A).
The major soluble products of both LtgA and LtgD digests were anhydrodisaccharide tripeptide and -tetrapeptide monomers. LtgA was able to liberate more of the soluble PG products under the conditions tested, creating significantly more PG monomers and larger products. For both LTs, non-O-acetylated sacculi yielded significantly more soluble products than were liberated from wild-type sacculi (Fig. 1). The modification of the peptidoglycan by O-acetylation at the C-6 hydroxyl on approximately half of the muramic acid residues is a regulatory mechanism for the activities of LTs in N. gonorrhoeae (Swim et al., 1983). O-Acetylation at C-6 blocks the C-6 hydroxyl such that the formation of the 1,6-anhydromuramyl moiety by LTs is prevented (Dillard & Hackett, 2005, Scheurwater et al., 2008). These results demonstrate that LtgA and LtgD both create 1,6-anhydromuramyl bonds, whose reactions are abrogated by the presence of PG O-acetylation at the C-6 hydroxyl. Both putative active-site mutants were unable to produce any detectable PG products from gonococcal sacculi.
Figure 1.
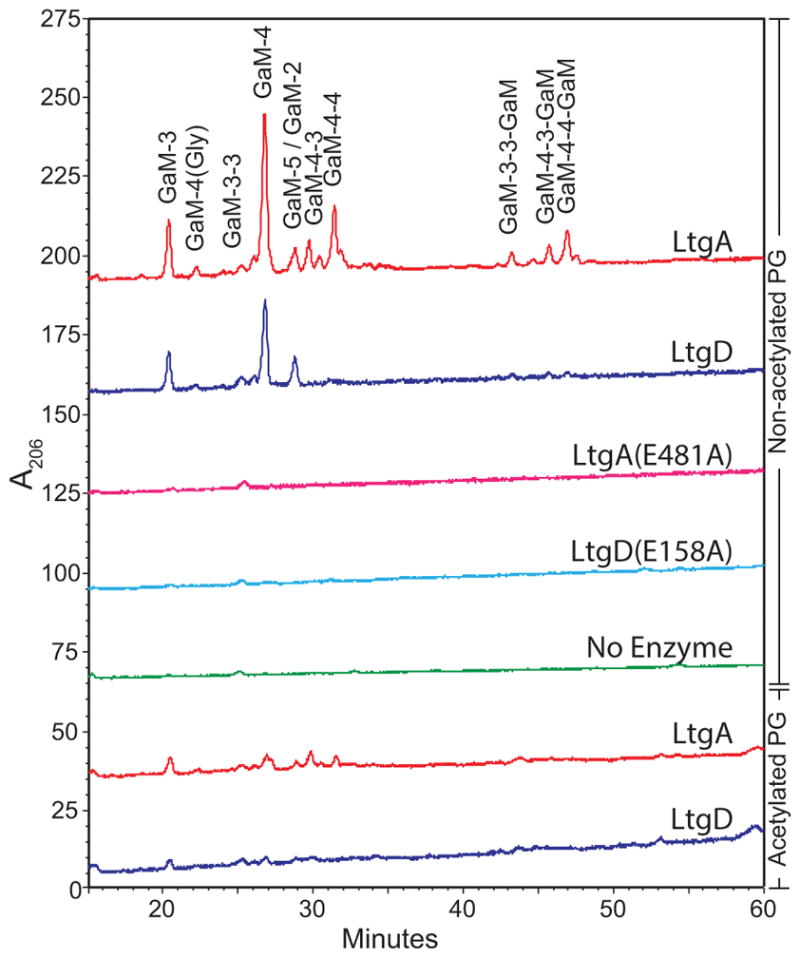
Soluble cell wall components liberated by LtgA and LtgD separated by HPLC. Reactions were performed by incubating 500 μg of purified gonococcal sacculi with purified LtgA, LtgD, or corresponding active site mutants. Soluble products were separated by reversed-phase HPLC using an ACN gradient. Sacculi from strains lacking O-acetylation and wild-type strains were digested. Peaks were identified by LC-MS of similarly digested sacculi. G, N-acetylglucosamine; aM, 1,6-anhydro-N-acetylmuramic acid; numbers represent the number of peptide stem amino acids (L-Ala, D-Glu, meso-DAP, D-Ala, D-Ala).
LtgA and LtgD have different PG substrates within the cell wall
LtgA was able to liberate both more total PG fragments and more unique species of PG fragments from sacculi than LtgD (Fig. 1). Soluble PG fragments were identified by LC-MS of products from LtgA or LtgD digests (Table 1). All muramic acids detected had 1,6-anhydromuramyl moieties, as would be expected for the reaction products of LTs. PG fragments released from LtgA-digested sacculi included all fragments detected from LtgD-digested sacculi, in addition to fragments with cross-linked peptide stems. This result indicates that LtgA is better able to act on PG with cross-linked peptides than LtgD.
Table 1.
LC-MS of soluble fragments released by digests of sacculi with LtgA or LtgD. Parentheses indicate detector counts were less than 0.5% of the total run. G, N-acetylglucosamine; aM, 1,6-anhydro-N-acetlymuramic acid; numbers represent the number of peptide stem amino acids. GaM-4(Gly) denotes a glycine in the fourth position.
| LtgA | LtgD | ||||||
|---|---|---|---|---|---|---|---|
|
| |||||||
| PG Fragment | Calculated m/z | Calculated Exact Mass | Ion | Observed m/z | Observed Mass | Observed m/z | Observed Mass |
| GaM | 479.18 | 478.18 | [M+H] | 479.19 | 478.18 | (479.19) | (478.18) |
| GaM-3 | 851.34 | 850.34 | [M+H] | 851.35 | 850.34 | 851.35 | 850.34 |
| GaM-3-1 | 512.21 | 1022.43 | [M+2H]2+ | (512.22) | (1022.43) | (512.23) | (1022.44) |
| GaM-4(Gly) | 908.37 | 907.37 | [M+H] | 908.37 | 907.37 | (454.69) | (907.37) |
| GaM-3-3 | 612.25 | 1222.51 | [M+2H]2+ | 612.26 | 1222.51 | (612.27) | (1222.52) |
| GaM-4 | 922.38 | 921.38 | [M+H] | 922.39 | 921.39 | 922.39 | 921.38 |
| GaM-4-3 | 647.77 | 1293.55 | [M+2H]2+ | 647.78 | 1293.55 | (647.78) | (1293.55) |
| GaM-5 | 993.42 | 992.42 | [M+H] | 993.43 | 992.42 | 993.43 | 992.42 |
| GaM-2 | 679.26 | 678.26 | [M+H] | 679.27 | 678.26 | 679.27 | 678.26 |
| GaM-4-2 | 583.25 | 1164.50 | [M+2H]2+ | (583.26) | (1164.51) | (583.26) | (1164.51) |
| GaM-4-4 | 683.29 | 1364.58 | [M+2H]2+ | 683.30 | 1364.59 | (683.30) | (1364.59) |
| GaM-4-4-4 | 603.60 | 1807.79 | [M+3H]3+ | (603.60) | (1807.79) | - | - |
| GaM-3-3-GaM | 842.34 | 1682.68 | [M+2H]2+ | (842.35) | (1682.68) | (842.35) | (1682.68) |
| GaM-4-3-3-GaM | 1064.44 | 2125.88 | [M+3H]2+ | (1064.45) | (2125.88) | - | - |
| GaM-4-4-3-GaM | 733.64 | 2196.92 | [M+4H]3+ | (733.65) | (2196.92) | - | - |
| GaM-4-4-4-GaM | 757.32 | 2267.95 | [M+4H]3+ | (757.33) | (2267.96) | - | - |
| GaM-4-3-GaM | 877.86 | 1753.72 | [M+2H]2+ | 877.87 | 1753.72 | (877.87) | (1753.72) |
| GaM-4-4-GaM | 913.38 | 1824.75 | [M+2H]2+ | 913.38 | 1824.75 | (913.39) | (1824.76) |
LtgA or LtgD digestion of gonococcal sacculi produced both anhydrodisaccharide tripeptide and tetrapeptide PG monomers. Similar proportions of tripeptide to tetrapeptide monomers were produced by both enzymes. To determine if LtgA and LtgD use the same macromolecular substrate to create PG monomers, purified sacculi were digested sequentially with equal molar quantities of LtgA and LtgD. PG was incubated with one enzyme, and the reaction was then heat-killed before a second enzyme incubation. Each reaction was incubated for 18 hours with the intention of digesting all available substrate. Sequential incubations with the same enzyme were done to account for differences in enzyme stability. Soluble PG fragments released from each reaction were separated by HPLC and quantified using the area under the peak (AUP) of corresponding monomers peaks from three independent experiments.
Significantly more monomers were released from digests with both LtgA and LtgD compared to a single digest with either enzyme (Fig. 2). Both combinations of LtgA and LtgD digests released significantly more PG monomer than digesting twice with the same enzyme. The order of enzyme addition affected the amount of released PG fragments. Digesting first with LtgA and then LtgD released more monomer than when the order was reversed. This result is likely due to the removal of peptide-linked substrates liberated by LtgA allowing more available LtgD substrates. Intriguingly, incubating sacculi with LtgD and then LtgA released significantly more monomer than digesting twice with LtgA, suggesting that LtgD is able to act on PG substrates in the sacculi not accessible to LtgA, even though no novel fragments are detectable by HPLC. The differences in PG products liberated from LtgA- and LtgD-digested sacculi and the increase in soluble products seen in digests with both enzymes support the idea that LtgA and LtgD have different substrates within the cell wall.
Figure 2.
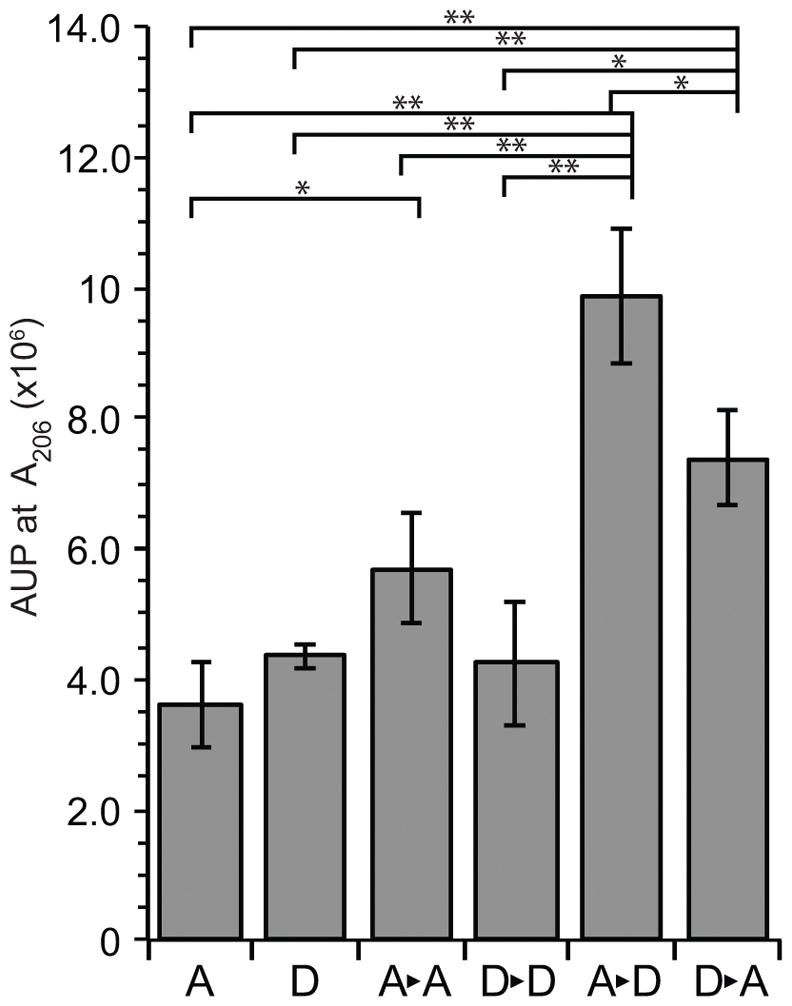
Liberated PG monomers from sequential digestion of sacculi with LtgA and LtgD. Soluble PG fragments from whole sacculi digests with LtgA or LtgD followed by boiling and a subsequent digest with LtgA or LtgD were separated by HPLC. The abundance of PG monomers (tripeptide (GaM-3) and tetrapeptide (GaM-4)) was determined by calculating the peak area (AUP) from three independent experiments. Horizontal bars indicate significance of P < 0.05, and (*) indicates P < 0.01 by Student’s t-test. Error bars represent standard deviation.
LtgA and LtgD are able to digest a short glycan strand PG into monomers
Compared to wild-type, ltgA and ltgD mutants release more tetrasaccharide dipeptide PG fragments, and less PG monomers from the cell, with an ltgA ltgD double mutant releasing more tetrasaccharide dipeptide than single mutants and releasing no PG monomers (Cloud & Dillard, 2002, Cloud-Hansen et al., 2008). These results suggest that both LtgA and LtgD are able to cleave tetrasaccharide dipeptide into monomers. However, to date, only homologs of E. coli MltB/Slt35 family 3 LT, such as LtgD, have been shown to digest a synthetic tetrasaccharide dipeptide (Suvorov et al., 2008). The ability of LtgA and LtgD to digest a tetrasaccharide dipeptide was tested and the resulting products were analyzed by reversed-phase HPLC with the identities of the products confirmed by mass spectrometry.
Both LtgA and LtgD were able to digest the synthetic tetrasaccharide dipeptide (compound iii in Fig. 3), and the masses of the products were consistent with LT activity, with one of the products having a 1,6-anhydroMurNAc moiety (Fig. 3). By contrast, an N. gonorrhoeae LT not involved in the release of monomers, LtgC, was not able to digest compound iii. LtgC is a family 2 exolytic LT and homolog of E. coli MltA. The activity of LtgA is the first known instance of a family 1 LT being able to cleave the tetrasaccharide dipeptide and may be responsible for N. gonorrhoeae releasing appreciably more PG monomers than other bacterial species.
Figure 3.

Digestion of a synthetic tetrasaccharide dipeptide. (A) A tetrasaccharide dipeptide was incubated with the indicated LT, and the resulting products were separated by reversed-phase HPLC using an ACN gradient. Incubation with LtgA and LtgD yielded two peaks corresponding to (i) reducing pentapeptide monomer and (ii) anhydro pentapeptide monomer. Digestion with LtgC and the no enzyme control yielded a single peak (iii) corresponding to undigested tetrasaccharide dipeptide. (B) Chemical structure of synthetic compound iii and observed digestion products with exact masses confirmed by mass spectrometry. The arrow indicates the site of LtgA and LtgD activity that results in the formation of a 1,6-anhydro bond indicative of LT activity.
LtgA and LtgD are lipoproteins that localize to the outer membrane
The primary sequence of LtgA and LtgD predicts a lipobox motif (LXXC) in the signal sequence that mediates lipidation of the cysteine and anchoring of lipidated proteins to cell membranes (Cloud & Dillard, 2002, Cloud-Hansen et al., 2008, Kovacs-Simon et al., 2011, Okuda & Tokuda, 2011). LtgA homologs, except in Neisseriales and Burkholderiales species, do not contain a lipobox motif and are predicted to be soluble periplasmic proteins. Many species have LtgD homologs that are predicted to lack a lipobox motif, including members of the genera, Burkholderia, Acinetobacter, Vibrio, Brucella, and Legionella. The E. coli homolog, MltB/Slt35, is reported to have two forms, both a 37 kDa membrane-bound form and a 35 kDa cleaved form lacking its membrane attachment site (Ehlert et al., 1995). Since LtgA and LtgD are predicted to localize differently from many homologs in other bacteria, we investigated the possibility that the lipidation and localization of these two LTs may be responsible for the increased release of PG monomers.
LtgA and LtgD are both predicted to be localized to the outer membrane due to their lipid-attachment sequence and the absence of an amino-acid residue following the lipidated cysteine that would cause retention in the inner membrane (Yamaguchi et al., 1988, Tokuda & Matsuyama, 2004). To establish the localization of LtgA and LtgD in N. gonorrhoeae, subcellular fractionation was done to separate the outer membrane (OM), total membrane (TM), and soluble fractions (SOL) using ultracentrifugation methods. Subcellular fractionation of bacteria producing C-terminal 3XFLAG- tagged LtgA or LtgD expressed from the native loci revealed that LtgA and LtgD both localize to the outer membrane and total membrane fractions and were not present in soluble fractions containing both cytoplasmic and periplasmic content (Fig. 4A). C-terminally tagged LtgA and LtgD were both shown to be functional as assessed by the release of soluble PG fragments (Fig. S1). LtgA and LtgD occurred in the same fractions as the outer-membrane control PilQ. Antiserum against the inner-membrane protein SecY was used to show that inner-membrane proteins did not contaminate outer-membrane fractions. Chloramphenicol acetyltransferase (CAT) was produced from a construct inserted between lctP and aspC on the chromosome allowing detection of CAT to identify fractions containing soluble proteins. The presence of LtgA and LtgD in the outer membrane and total membrane fractions indicates that both LTs localize to the outer membrane, as predicted by their signal sequences.
Figure 4.

Localization and lipidation of LtgA and LtgD. (A) Strains containing C-terminal 3XFLAG at the native site of ltgA or ltgD were fractionated into outer membrane (OM), total membrane (TM), and soluble (SOL) fractions using ultracentrifugation. Anti-FLAG antibodies were used to determine the subcellular localization of LtgA and LtgD, which localized to the same fractions as the OM control, PilQ. The strains used had a chromosomal cat gene to identify the cytoplasmic fraction and the presence of SecY was used as an inner membrane control. (B) The incorporation of the lipid [3H]-palmitate into recombinant LTs producing wild-type (WT), cysteine to alanine lipidation mutations (LM), or proteins lacking signal sequences (SM). The (*) indicates an active site mutant was used.
The signal sequence and lipobox motif of LtgA or LtgD were mutated to investigate the importance of protein localization to the release of PG monomers in N. gonorrhoeae. A mutation was made to substitute the critical cysteine residue of the lipobox motif with an alanine. The substitution of the lipobox cysteine blocks glycerylcysteine formation and subsequent fatty acid acetylation required for membrane localization but still allows for translocation to the periplasm (Kovacs-Simon et al., 2011). Mutation of lipobox cysteine codon (TGT) to alanine (GCA) was done to create the non-lipidated proteins variants LtgA(C21A) and LtgD(C19A). Mutations were also made to remove the entire signal peptide including the cysteine to create cytoplasmic versions of these proteins.
To confirm that wild-type LtgA and LtgD are lipidated, the ability of these LTs to incorporate radiolabeled fatty acids was tested, as was the lipid incorporation of lipidation and signal-sequence mutants. LtgA and LtgD were produced in E. coli strains in the presence of [3H]-palmitate. Total protein was extracted, and fluorography was used to visualize lipidated proteins. Radiolabeled bands in full-length LtgA and LtgD lanes indicate lipidated proteins. The observed bands are consistent with the predicted masses of mature proteins at 65.5 kDa and 37.9 kDa for LtgA and LtgD, respectively (Fig. 4B). Strains expressing constructs with mutated lipidation sites or lacking the entire signal sequence had no detectable [3H]-palmitate-labeled LtgA or LtgD. These data demonstrate that both LtgA and LtgD are lipidated at canonical lipobox motifs, and the lipidation of these proteins can be disrupted by mutation of the lipobox cysteine.
Outer-membrane localization of LtgD is necessary for abundant release of PG monomers
To determine the relative abundance of PG-derived fragments released by various ltgA or ltgD mutants, growing gonococcal cultures were pulse-labeled with [3H]-glucosamine to metabolically label PG. Cultures were normalized to the total CPM in bacteria in each culture, and released PG fragments were collected during a chase period. Collected PG fragments were separated by size-exclusion chromatography, and fractions were collected and quantified by scintillation counting to identify and compare fractions containing released radiolabeled PG fragments between strains.
PG fragment release profiles of LtgA or LtgD active-site mutants (Fig. 5A) were nearly identical to the previously reported profiles of ltgA (Cloud & Dillard, 2002) and ltgD (Cloud-Hansen et al., 2008) deletion mutants. The signal sequences of both LtgA and LtgD are also necessary for their ability to function in the release of PG fragments, since mutants that lack LtgA or LtgD signal sequences released PG fragment analogous to corresponding active-site mutants (Fig. S2). The importance of signal sequences for these LTs is consistent with their function as periplasmic sacculi-degrading enzymes.
Figure 5.

PG fragments released from growing ltgA and ltgD mutants. Cultures of gonococci were pulsed with [3H]-glucosamine to label PG, and released fragments were collected during a chase period. Radiolabeled fragments were separated by size-exclusion chromatography and measured by liquid scintillation counting. (A) Active site point mutants release PG fragments nearly identical to gene deletions with increased (1) tetrasaccharide dipeptide and (2) tetrasaccharide peptide, and decreased (3) anhydrodisaccharide peptide monomers and (4) free anhydrodisaccharide. (B) Disrupting the lipidation of LtgA had no effect on the amount of peptidoglycan fragments released, but disrupting lipidation of LtgD decreased the amount of released PG monomers. (C) Chemical structure of detected PG fragments released by N. gonorrhoeae. 1, tetrasaccharide dipeptide; 2, tetrasaccharide peptide; 3, anhydrodisaccharide peptide monomer; 4, free anhydrodisaccharide
Disrupting lipidation of LtgA had no detectable effect on the release of PG fragments (Fig. 5B). However, inhibiting lipidation of LtgD did affect PG fragment release and resulted in 39% less PG monomer released than wild-type, and it also caused a slight reduction in released tetrasaccharide dipeptide, tetrasaccharide peptide, and free anhydrodisaccharide PG fragments. The change in the abundance of PG fragments released from the LtgD lipidation mutant is predominantly limited to the abundance of PG monomers. The modest reduction in released PG tetrasaccharide dipeptide and tetrasaccharide peptide is in contrast to the significant increase of released fragments observed from an ltgD active site mutant.
Mutation of ltgD results in the accumulation of PG precursors
The decrease observed for released PG fragments in the LtgD lipidation mutant could be caused by changes in protein localization that lead to either an overall decrease in the amount of PG fragments being produced, export of PG fragments, or a disruption of protein interactions necessary for proper enzymatic activity. To determine if the decreased release of PG monomers in the LtgD lipidation mutant caused changes in recycled PG fragments, the cellular, soluble PG fragments were measured by hot water extract. Cells were labeled with [3H]-glucosamine, and after a chase period, the total cytoplasmic and periplasmic PG was extracted by boiling and separated from the insoluble PG sacculus by centrifugation. These fragments were analyzed by size-exclusion chromatography, as was done for released PG fragments. To differentiate between recycling and release of PG fragments, mutants were made carrying an ampG deletion to disrupt uptake of PG fragments into the cytoplasm. AmpG is an integral inner-membrane permease that imports GlcNAc-anhMurNAc-containing PG fragments from the periplasm to the cytoplasm (Cheng & Park, 2002). These imported PG fragments can then be recycled back into the cell wall or used for other metabolic outcomes.
Two PG precursors were detectable in wild-type N. gonorrhoeae, UDP-MurNAc-pentapeptide and UDP-MurNAc-tripeptide (Fig. 6A), and the identity of these PG precursors was confirmed by comparing the HPLC retention times of radiolabeled precursors with synthesized precursors [BaCWAN] (Fig. S3). UDP-MurNAc-pentapeptide and UDP-MurNAc-tripeptide are the final two cytoplasmic precursors before the formation of lipid intermediates and subsequent translocation to the periplasm. Deletion of ltgD increased the cellular levels of UDP-MurNAc-pentapeptide two-fold, while having no effect on UDP-MurNAc-tripeptide accumulation (Fig. 6A). A strain containing only the LtgD lipidation mutation had no differences in PG precursor accumulation (Fig. 6B and 6D).
Figure 6.

Cellular PG fragments extracted from LT and ampG mutants. Hot water extracts from cultures labeled with [3H]-glucosamine were separated by size-exclusion chromatography to determine the abundance of PG precursors and fragments in the cell. (A) Mutation of ltgD resulted in increased cellular UDP-MurNAc-pentapeptide compared to wild-type. (B) Mutation of ampG decreased the amount of detectable PG precursors, but an ltgD ampG mutant increased UDP-MurNAc-pentapeptide and UDP-MurNAc-tripeptide compared to wild-type. (C) A double ltgA ampG mutant resulted in a decrease in the amount of radiolabeled PG fragments in the cell. (D) The ltgD(C19A) lipidation mutant contained wild-type levels of PG precursors, but the ltgD(C19A) ampG mutant contained high levels of UDP-MurNAc-tripeptide and -pentapeptide like the ltgD ampG mutant. UM5, UDP-MurNAc-pentapeptide; UM3, UDP-MurNAc-tripeptide.
The abundance of labeled, soluble intracellular PG products was greatly reduced in an ampG mutant (Fig. 6B and 6C) as has been reported previously (Garcia & Dillard, 2008). Contrarily, deletion of both ltgD and ampG caused a large accumulation of radiolabeled UDP-MurNAc-pentapeptide to the same levels as in the ltgD mutant, as well as accumulation of UDP-MurNAc-tripeptide to equally high levels (Fig. 6B). In contrast to the pool of precursors present in ltgD deletion strains, no PG precursors were detectable in a double ltgA ampG deletion (Fig. 6C). Although an ltgD lipidation mutant strain did not affect cellular levels of PG precursors in conjunction with mutation of ampG, both UDP-MurNAc-pentapeptide and UDP-MurNAc-tripeptide accumulated to the high levels in the double ltgD ampG mutant (Fig. 6D). These results suggest that LtgD and its lipidation are important for the incorporation of new PG in N. gonorrhoeae.
Release of PG monomers by LT mutants in the absence of PG recycling
In the absence of AmpG, PG fragments generated during growth are unable to be recycled into the cytoplasm, causing a release of PG fragments from the cell. Compared to PG fragments released by wild-type gonococci, mutation of ampG resulted in an 8- to 11-fold increase in released PG monomers and an increase in the amount of free anhydrodisaccharide (Fig. 7A and (Garcia & Dillard, 2008)). A double ltgD ampG deletion mutant also released more free anhydrodisaccharide than wild-type, but also releases more tetrasaccharide dipeptide and more tetrasaccharide-peptide, with release levels equivalent to that of ltgD mutants (Fig. 7A and 7B). There was also a 9-fold increase in the amount of PG monomers in the ltgD ampG mutant, which was slightly less than what is released when LtgD is present in an ampG mutant. This result suggests that LtgD produces only a small amount of the PG fragments that are normally recycled by the gonococcal cell.
Figure 7.

PG fragments released from strains deficient in AmpG-mediated PG recycling. Strains were labeled with [3H]-glucosamine. (A) Deletion of ampG causes the release of significantly more PG monomers and free anhydrodisaccharide compared to wild-type. (B) The ltgA ampG and ltgD ampG mutants released 84.3% and 36.2% less PG monomer, respectively, compared to an ΔampG mutant. (C) Mutations affecting the lipidation of LtgA or LtgD had no significant effect on the amount of PG monomers released in an ampG deletion background. 1, tetrasaccharide dipeptides; 2, tetrasaccharide peptides; 3, anhydrodisaccharide peptide monomers; 4, free anhydrodisaccharide.
An ltgA ampG double mutant similarly released tetrasaccharide dipeptide and tetrasaccharide peptide at levels equivalent to an ltgA mutant, as well as releasing more free anhydrodisaccharide. However, the amount of released PG monomers from the ltgA ampG mutant was comparable to levels released by wild-type strains and greatly reduced compared to either ampG or ltgD ampG mutants (Fig. 7B). The lipidation of either LtgA or LtgD did not affect the amount of released fragments in ampG mutants. Together, these results suggest that PG fragments produced by LtgA and LtgD are not equivalent in propensity to be released or recycled. LtgA is producing more of the PG monomers that are destined for recycling in wild-type cells.
Strains carrying epitope-tagged LTs were used to verify that the differences in released PG fragments of mutants were due to differences in the activities of LtgA and LtgD and not due to differences caused by changes in protein abundance (Fig. S4). Quantitative western blot analysis confirmed that cellular levels of LtgA were not changed by the absence of LtgD, nor were levels of LtgD changed by the absence of LtgA. Neither levels of LtgA or LtgD were changed due to deletion of ampG.
Cellular Localization of LtgA and LtgD
Both LtgA and LtgD are PG monomer-producing exolytic LTs, but analysis of released PG fragments and cytoplasmic PG precursors reveals that these LTs have different capacities within the cell. Stochastic optical reconstruction microscopy (STORM) was used to reveal if LtgA and LtgD function in different regions of the cell to further demonstrate potential differences between these proteins. N. gonorrhoeae, often characterized as a diplococcus, is present as about 38% diplococci and 52% monococci during exponential growth in liquid culture (Tobiason & Seifert, 2006). The presence of N. gonorrhoeae as a diplococcus reveals cellular polarity and allows for the relative site of septation to be determined. Strains producing inducible C-terminal 3XFLAG-tagged versions of LtgA or LtgD were used to determine cellular localization of each of these LTs by STORM. Cells were counterstained with DAPI to determine relative boundaries of the cell and the orientation of diplococci.
LtgA was predominantly localized at the septum of diplococci and tetracocci (Fig. 8A). The monococci lacked regions of localized signal. LtgA-dependent signal appears to form a plane perpendicular to that of the diplococci, which is consistent with the site of ongoing cell division. The signal from LtgD was localized around the cell with some diplococci having multiple clusters of aggregated signal (Fig. 8B). A regular pattern could not be recognized for the clusters of signal, but LtgD did not consistently localize to the septum. The differences in cellular localization of these two LTs could explain how two enzymes with very similar enzymatic activity and ability to produce PG monomers from tetrasaccharide dipeptides contribute differently to the ultimate destination of PG fragments.
Figure 8.

Cellular localization of LtgA and LtgD. LT deletion strains were complemented with chromosome insertions of inducible LtgA3XFLAG or LtgD3XFLAG. Localization of LTs was determined using an anti-FLAG antibody, and cells were counterstained with DAPI. (A) LtgA localized to the cell septum perpendicular to the plane of the diplococci. Monococci lacked localized signal. (B) LtgD is expressed at discrete focal points around the diplococci and is not predominantly localized at the cell septum. Scale bar = 2μm.
Discussion
Although LtgA and LtgD both contribute to the release of PG monomers and are able to digest tetrasaccharide dipeptide, the cellular localization and contributions to PG incorporation and recycling of these two enzymes reveal overall different cellular functions. We demonstrated that LtgA and LtgD have the predicted LT activity. Purified recombinant proteins were able to digest PG sacculi and release soluble PG fragments containing 1,6-anhydro-N-acetylmuramyl moieties. Mutations of predicted active-site residues were effective at eliminating LT activity on whole sacculi. Although the active-site residues of LtgA may be similar to those of E. coli Slt70, LtgA was able to digest a tetrasaccharide dipeptide, unlike Slt70 or other known homologs (Suvorov et al., 2008, Lee et al., 2013). The PG monomer-producing ability of LtgA by digestion of tetrasaccharide dipeptides is likely an important difference that allows N. gonorrhoeae to release high levels of PG monomers.
PG fragments liberated from digestion of sacculi from a WT strain and a non-acetylating mutant demonstrated the LT-inhibiting effects of PG acetylation on LtgA and LtgD. Both LtgA and LtgD released the same relative abundance of PG species from sacculi with or without O-acetylation, and they were able to degrade more PG from sacculi lacking PG acetylation, as would be expected. Digestion of sacculi lacking acetylation yielded over 3-fold more PG monomer than partially acetylated wild-type sacculi when digested with either LtgA or LtgD. Digestion of non-acetylated PG by LtgA released significantly more of the larger PG fragments. These data indicate that O-acetylation blocks the function of LtgA and LtgD and suggests the esterase ApeI, known to remove O-acetylation in N. gonorrhoeae, likely acts on PG prior to LT activity (Moynihan & Clarke, 2014).
Lipidation and resulting localization of LtgA and LtgD to the outer membrane was confirmed, as predicted by their primary sequences. No soluble forms of either of these enzymes were detected, as has been noted for some LtgD homologs (Ehlert et al., 1995). Lipidation of LtgA was anticipated to contribute to increased release of PG monomers due to most homologs being soluble, but the lipobox mutation inhibiting lipidation of LtgD, and not of LtgA, caused a reduction of released PG monomers. The decrease in released fragments suggests the lack of lipidation does not simply inhibit normal LtgD activity. Unlipidated LtgD resulted in a reduction in release of tetrasaccharide dipeptide and tetrasaccharide peptide and not the considerable increase of these two products seen in ltgD deletion or active-site mutation strains (Fig. 5A and S1). This result could mean that unlipidated LtgD, existing as a soluble protein in the periplasm, is able to degrade tetrasaccharide dipeptides and other multimeric PG released by other enzymes but is unable to allow for the insertion of new PG strands. Overall, this result indicates that the localization of LtgD at the outer membrane is necessary for its function in PG monomer release.
Investigations of intracellular PG fragments and precursors in the presence and absence of AmpG-mediated PG recycling suggest an unexpected role for LtgD. Deletion of ltgD caused an increase in the accumulation of UDP-MurNAc-pentapeptide, the final cytoplasmic PG precursor (Fig. 6A). A double ampG ltgD deletion mutant accumulated high levels of UDP-MurNAc-pentapeptide in addition to high levels of UDP-MurNAc-tripeptide, the penultimate cytoplasmic PG precursor (Fig. 6). The reason for the exacerbation of PG precursor build-up by the ampG mutation in the ltgD background is unknown, but this result might indicate that PG precursor synthesis is increased when PG recycling is diminished. Overall, these results suggest that LtgD is at least in part necessary for normal levels of PG incorporation into the sacculus. During growth, UDP-MurNAc-tripeptide is converted to UDP-MurNAc-pentapeptide by MurF. UDP-MurNAc-pentapeptide is then transferred to an undecaprenyl phosphate in the inner membrane by MraY to form Lipid I. N-acetylglucosamine is added to the N-acetylmuramic acid residue by MurG to form Lipid II, which is then flipped across the inner membrane by MurJ or FtsW (Mohammadi et al., 2014, Sham et al., 2014). Lipid II polymerizes to a linear chain of alternating MurNAc-GlcNAc by the action of synthetic transglycosylase PBP1. This linear strand is cross-linked to the growing sacculus by transpeptidases, either PBP1 or PBP2. The accumulation of UDP-MurNAc-pentapeptide can also be caused by certain antibiotics, such as moenomycin and vancomycin, which block polymerization of Lipid II (Lara et al., 2005, de Kruijff et al., 2008). Since deletion of ltgD also causes an accumulation of cytoplasmic PG precursors, LtgD is seemingly influencing the polymerization of Lipid II, although the mechanism remains unclear.
Mutation of ampG, or ampG and ltgA, did not lead to accumulation of appreciable levels of radiolabeled cytoplasmic PG precursors. In the absence of AmpG-mediated PG recycling, the only [3H]-glucosamine that enters the PG synthesis pathway is that used in the pulse-labeling and it is incorporated into the cell wall. The radiolabeled PG incorporated during the pulse period is then unable to be recycled during the chase and subsequent analysis of cytoplasmic PG fragments. LtgA must not be required for the incorporation of new PG since no accumulation of precursors is readily detectable in an ampG ltgA mutant. Consequently, the PG precursors in ampG ltgD mutants must be assumed to have accumulated during the pulse period and remained unincorporated during a chase period. The necessity of LtgD to allow efficient incorporation of PG precursors into the cell wall suggests that LtgD is involved in nascent PG incorporation as well as PG monomer release. Although it is possible that LtgD interacts directly with PBP1 and affects its activity, it seems more likely that LtgD activity is simply necessary to open a space for new PG incorporation in the non-septal area of the coccus. Gonococci cannot be said to have an elongosome complex of PG breakdown and synthesis proteins since they are round in shape, but they may have an analogous set of proteins for expanding the coccus into a bigger ball. LtgD may make the spaces in the cell wall necessary for the biosynthetic machinery to implement this cell wall enlargement.
Both LtgA and LtgD contribute to monomer release in wild-type cells, since single mutation of ltgA or ltgD result in a 40% and 60% reduction of released PG monomers, respectively (Cloud-Hansen et al., 2008). The release of PG fragments in the absence of AmpG-mediated PG recycling indicates the contribution of LtgA and LtgD to monomer release. PG monomer release in ltgA ampG mutants is only 16% the amount released in ampG mutants. In contrast, ltgD ampG mutants release four-fold more PG monomers than ltgA ampG mutants. This result demonstrates that LtgA is responsible for liberating the majority of PG monomers from the sacculus. Since there was no change in LtgA or LtgD levels in reciprocal deletion or in ΔampG strains, the contribution to PG monomer release of each LT can reasonably be attributed to their activity (Fig. S4). The ΔltgA ΔampG strains releases considerably less monomer than the ΔampG strain. PG monomers that would have otherwise been taken up by AmpG and recycled are instead released from the cell. These data show LtgA is the LT responsible for creating a vast majority of the PG monomers recycled by AmpG, while LtgD is responsible for producing the majority of released PG fragments (Cloud-Hansen et al., 2008).
The cellular localization of LtgA and LtgD by immunofluorescence microscopy shows overall different localization for each of these LTs (Fig. 8). LtgA is localized at the cell septum between the diplococcal cells. Localization of LtgA to the septum implies a role in PG degradation and cell-wall expansion at the septum, which is supported by notably less LtgA detected around monococci. The association of LtgA at the septum is consistent with most LtgA-produced PG monomers being recycled, since in cocci most cell growth is thought to occur at the septum (Margolin, 2009). LtgD is found at discrete points around cells. The localization of LtgD in addition to data showing the need for LtgD for efficient incorporation of UDP-MurNAc-pentapeptide suggests that a certain portion of nascent PG incorporation occurs at regions of LtgD localization around the cell. Localization of both LtgA and LtgD in association with their cellular functions implies that each of these LTs has different cellular functions and both may associate with different PG-associated complexes.
Experimental Procedures
Bacteria and growth conditions
E. coli cultures were grown at 37°C with aeration in Luria-Bertani broth (LB) and supplemented with appropriate antibiotics (500 μg mL−1 erythromycin, 40 μg mL−1 kanamycin, or 25 μg mL−1 chloramphenicol) to maintain plasmids. TAM1 competent cells (Active Motif, Carlsbad, CA, USA) were used for molecular cloning and propagation of plasmid DNA for gonococcal transformations. BL21(DE3) (Invitrogen, Carlsbad, CA, USA) was used for expression in pET based vectors.
N. gonorrhoeae stains were derived from strain MS11 and grown on GCB agar (Difco) plates with Kellogg’s supplements in 5.0% CO2, or in gonococcal base liquid (GCBL) with Kellogg’s supplements, 0.042% sodium bicarbonate, and aeration. Transformation of naturally competent N. gonorrhoeae was performed by spotting 5–20 μg of linearized plasmid DNA on GCB agar plates and growing piliated N. gonorrhoeae on the spots (Dillard, 2006). Potential transformants containing unmarked mutations were screened by isolating single colonies and identifying transformants by PCR and restriction enzyme digestion, followed by sequencing.
Plasmid construction
The plasmids used in this study are listed in Table 2. N. gonorrhoeae strain MS11 chromosomal DNA was used as a PCR template unless otherwise noted. To create plasmids for purifying proteins lacking signal sequences, ltgA was PCR amplified using ltgA/5′/NheI (5′ - ACA GCT AGC CTG CCA GCC GGC AAG ACC) and LtgA/3′/XhoI (5′ - GGC CTC GAG ACG TCA GCG TGC GGG AAC), followed by digestion with NheI and XhoI, and ligated with pET28b to create plasmid pET28b-LtgA. The coding sequence of ltgD was amplified using ltgD/5′/NdeI (5′ - CGC ATA TGG AGG CCC GCA CAC CC) and ltgD/3′/XhoI (5′ - GCC CTC GAG CGG GAA ATC CCC ATA AAA), digested with NdeI and XhoI, and ligated with pET28b to create pET28b-LtgD.
Table 2.
Strains and plasmids used in this study. ErmR, erythromycin-resistant; CmR, chloramphenicol-resistant; KanR, kanamycin-resistant.
| Plasmid or Strain | Description | Reference | |
|---|---|---|---|
| Plasmid | |||
|
| |||
| pMR100 | C-terminal 3XFLAG vector, ErmR | (Ramsey et al., 2014) | |
| pKH37 | Complementation vector, CmR | (Kohler et al., 2007) | |
| pIDN1 | Insertion-duplication and cloning vector, ErmR | (Hamilton et al., 2001) | |
| pDG132 | ampG deletion vector, ErmR | (Garcia & Dillard, 2008) | |
| pET28b | IPTG inducible expression vector, KanR | Novagen | |
| pKH84 | ltgA deletion vector, ErmR | (Cloud-Hansen et al., 2008) | |
| pET28b-ltgA | His6-LtgA expression vector, KanR | This study | |
| pET28b-ltgA(E481A) | His6-LtgA(E481A) expression vector, KanR | This study | |
| pET28b-ltgD | His6-LtgD expression vector, KanR | This study | |
| pET28b-ltgD(E158A) | His6-LtgD(E158A) expression vector, KanR | This study | |
| pRS44/pET28b-ltgC | His6-LtgC expression vector, KanR | This study | |
| pRS09/pIDN1-ltgA(C21A) | LtgA lipidation mutant recombination vector, ErmR | This study | |
| pRS10/pIDN1-ltgD(C19A) | LtgD lipidation mutant recombination vector, ErmR | This study | |
| pRS53/pIDN1-ltgA3XFLAG | LtgA3XFLAG construct, ErmR | This study | |
| pRS61/pIDN1-ltgA3XFLAG+DS | LtgA3XFLAG chromosomal recombination vector, ErmR | This study | |
| pRS66/pKH37-ltgA3XFLAG | IPTG inducible LtgA3XFLAG complementation vector, CmR | This study | |
| pRS65/pIDN1-ltgD3XFLAG | LtgD3XFLAG construct, ErmR | This study | |
| pRS41/pIDN1-ltgD3XFLAG+DS | LtgD3XFLAG chromosomal recombination vector, ErmR | This study | |
| pRS67/pKH37-ltgD3XFLAG | IPTG inducible LtgD3XFLAG complementation vector, CmR | This study | |
| pRS42/pIDN1-ltgA(NoSig) | Construct to remove the signal sequence of LtgA, ErmR | This study | |
| pRS43/pIDN1-ltgD(NoSig) | Construct to remove the signal sequence of LtgD, ErmR | This study | |
| pRS91/pIDN1-ltgA(E481A) | LtgA active site mutation construct, ErmR | This study | |
| pRS92/pIDN1-ltgD(E158A) | LtgD active site mutation construct, ErmR | This study | |
| pRS93/pET-ltgA(E481A)-FL | IPTG inducible full-length LtgA(E481A) overexpression construct, KanR | This study | |
| pRS94/pET-ltgA(C21A) | IPTG inducible LtgA lipidation mutant overexpression construct, KanR | This study | |
| pRS95/pET-ltgA(No Sig) | IPTG inducible signal sequence deletion LtgA overexpression construct, KanR | This study | |
| pRS96/pET-ltgD-FL | IPTG inducible full-length LtgD overexpression construct, KanR | This study | |
| pRS97/pET-ltgD(C19A) | IPTG inducible LtgD lipidation mutant overexpression construct, KanR | This study | |
| pRS98/pET-ltgD(No Sig) | IPTG inducible signal sequence deletion LtgD overexpression construct, KanR | This study | |
| Strain | |||
|
| |||
| Escherichia coli | |||
| TAM1 | mcrA Δ(mrr-hsdRMS-mcrBC) Φ80lacZΔM15 ΔlacX74 recA1 araD139 Δ(ara-leu)7697 galU galK rpsL endA1 nupG | Active Motif | |
| BL21(DE3) | F- ompT hsdSB(rB- mB-) gal dcm (DE3) | Novagen (EMD) | |
| Neisseria gonorrhoeae | |||
| MS11 | Wild-type | (Segal et al., 1985) | |
| DG132 | MS11 ΔampG | (Garcia & Dillard, 2008) | |
| KH530 | MS11 ΔpacA | (Dillard & Hackett, 2005) | |
| KC119 | MS11 ΔltgD | (Cloud-Hansen et al., 2008) | |
| KH565 | MS11 ΔltgA | MS11 x pKH84 | This study |
| RS501 | MS11 ltgA(C21A) | MS11 x pRS9 | This study |
| RS502 | MS11 ltgD(C19A) | MS11 x pRS10 | This study |
| RS503 | MS11 ltgD3XFLAG | MS11 x pRS41 | This study |
| RS504 | MS11 ltgA(NoSignal) | MS11 x pRS42 | This study |
| RS505 | MS11 ltgD(NoSignal) | MS11 x pRS43 | This study |
| RS507 | MS11 ltgA3XFLAG | MS11 x pRS61 | This study |
| RS518 | MS11 ΔltgA comp ltgA3XFLAG, CmR | KH565 x pRS66 | This study |
| RS520 | MS11 ΔltgD comp ltgD3XFLAG, CmR | pKC119 x pRS67 | This study |
| RS522 | MS11 ΔltgA ΔampG | KH565 x pDG132 | This study |
| RS523 | MS11 ΔltgD ΔampG | KC119 x pDG132 | This study |
| RS524 | MS11 ltgA(C21A) ΔampG | RS501 x pDG132 | This study |
| RS525 | MS11 ltgD(C19A) ΔampG | RS502 x pDG132 | This study |
| RS545 | MS11 ltgA3XFLAG comp, CmR | RS507 x pKH37 | This study |
| RS546 | MS11 ltgA(C21A)3XFLAG comp, CmR | RS542 x pKH37 | This study |
| RS555 | MS11 ltgA(E481A) | MS11 x pRS91 | This study |
| RS557 | MS11 ltgD(E158A) | MS11 x pRS92 | This study |
| RS563 | MS11 ΔltgD ltgA3XFLAG | KC119 x pRS61 | This study |
| RS565 | MS11 ΔltgA ltgD3XFLAG | KH565 x pRS41 | This study |
| RS567 | MS11 ΔltgD ltgA3XFLAG ΔampG | RS563 x pDG132 | This study |
| RS569 | MS11 ΔltgA ltgD3XFLAG ΔampG | RS565 x pDG132 | This study |
| RS571 | MS11 ltgA3XFLAG ΔampG | RS507 x pDG132 | This study |
| RS572 | MS11 ltgD3XFLAG ΔampG | RS503 x pDG132 | This study |
Active site mutations were made using KOD DNA Polymerase (Novagen) amplifying pET28b-LtgA with slt70ltgA/A1469C/5′ (5′ - TCA TCC GTC AGG CAA GCC GCT TCG TT) and slt70ltgA/A1469C/3′ (5′ - TAA CGA AGC GGC TTG CCT GAC GGA TGA), and pET28b-LtgD with ltgD/5′/E158A (5′ - GCG ATT ATC GGG ATT GCA ACG AAT TAC GGC AAA) and ltgD/3′/E158A (5′ - TTT GCC GTA ATT CGT TGC AAT CCC GAT AAT CGC) followed by digestion with DpnI and self-ligations to create pET28B-LtgA(E481A) and pET28b-LtgD(E158A), respectively. LtgC-NS-NdeI-F (5′ - TTC ATA TGA GCA GGA GCA TCC AAA CCT TTC CG) and LtgC-XhoI-R (5 - TTC TCG AGT CAC GGG CGG TAT TCG GGC) were used to amplify ltgC which was digested with NdeI and XhoI and ligated with pET28b to create pET28b-LtgC. Active site mutations were transferred from protein production to transformation vectors using internal restriction sites. pET28b-LtgA(E481A) was digested with NdeI and XhoI and ligated into pIDN1-LtgA3XFLAG+DS to create pIDN1-LtgA(E481A). pET28b-LtgD(E158A) was digested with AgeI and XhoI and ligated into pIDN1-LtgD3XFLAG to create pIDN1-LtgD(E158A).
Lipidation mutants were created using overlap extension PCR. The PCR product of Fslt70 (5′ - GCA TCT AGA GGG CAA CCA TTT CGG ACA A) and LtgA-C21A-R (5′ - GTG TTT GTC GAA GAT GCC GCG GCA AGC ACC) and the PCR product of LtgA-C21A-F (5′ - GGT GCT TGC CGC GGC ATC TTC GAC AAA CAC) and Rslt70 (5′ - TTA GAA TTC GCC GTC AAT GCC GTT) were used as a PCR template using Fslt70 and Rslt70. The resulting PCR product was digested with XbaI and EcoRI and ligated into pKC3 (Cloud & Dillard, 2002), and the BamHI and EcoRI digested fragment was then subcloned into pIDN1 (Hamilton et al., 2001) to create pIDN1-LtgA(C21A). To create pIDN1-LtgD(C19A) the PCR product of LtgDF1 (5′ - AAA CCC TCG CCA CGG AAT ACA CTT) and ltgD-C19A-R (5′ - CCT CCA TCG CCG TTG CGG CAG ACA AAG CC) and the product of LtgD-C19A-F (5′ - CGG CTT TGT CTG CCG CAA CGG CGA TG) and LtgD/3/dwnstrm/XhoI (5′ - AAC CTC GAG TAT GCC GCC GTC AGC) were used as a PCR template using LtgDF1 and LtgD/3/dwnstrm/XhoI. The resulting PCR product was digested with EcoRV and XhoI and ligated into pIDN1.
C-terminal 3XFLAG sequences were added to genes by amplifying with LtgA-RBS-SacI-F (5′ - TTG AGC TCG TTC AGG GTT CCG ATT CTT AAG G) and LtgA-EcoRI-R (5′ - TTG AAT TCG CGT GCG GGA ACG GTG CCC), and with LtgD-RBS-SacI-F (5′ - TTG AGC TCC CTC ACC CTT GTG CCG CTT TCC) and LtgD-EcoRI-R (5′ - GAA CCG AAT TCC AAT CCC GGG CCG CCG AGC). These products were digested with SacI and EcoRI, then ligated into pMR100 (Ramsey et al., 2014) to create pIDN1-LtgA3XFLAG and pIDN1-LtgD3XFLAG, respectively. To amplify the region 3′ of ltgA for additional homology, the PCR product of LtgA-DS-HindIII-F (5′ - TTA AGC TTG CCG ATG CCG TCT GAA ACC C) and LtgA-DS-XhoI-R (5′ - AAC TCG AGT TGC GTT TGC GTT CGG TAG TCG G) was digested with HindIII and XhoI and ligated with pIDN1-LtgA3XFLAG to create pIDN1-LtgA3XFLAG+DS. Similarly, LtgD-DS-HindIII-F (5′ - TTT AAG CTT ATG TGT TTT TTA AAA TGC TGT CTG AAC C) and LtgD/3/dwnstrm/XhoI were used to amplify the region 3′ of ltgD, digested with HindIII and XhoI, and ligated with pIDN1-LtgD3XFLAG to create pIDN1-LtgD3XFLAG+DS. To add 3XFLAG constructs to complementation plasmid pKH37 (Kohler et al., 2007), pIDN1-LtgA3XFLAG and pIDN1-LtgD3XFLAG were digested with SacI and XhoI and ligated into pKH37 to create pKH37-LtgA3XFLAG and pKH37-LtgD3XFLAG, respectively.
Protein over-production plasmids for palmitate labeling incorporation were made by inserting genes with different signal sequences into pET28b without the addition of a hexahistidine sequence. After numerous abortive attempts to clone LtgA with its native signal sequence, it was assumed to be too toxic in this vector and the active-site point mutant of LtgA was cloned. LtgA-F-NcoI (5′ - TCC ATG GAC CTA CCC TCT ATG AAG CAT TCC C) and LtgA/3′/XhoI were used to amplify wild-type chromosomal DNA and pET28b-LtgA(E481A). The wild-type product was digested with NcoI and MluI, and the pET28b-LtgA(E481A) PCR product was digested with MluI and XhoI. The NcoI/MluI of the wild-type product and the MluI/XhoI point mutant product were ligated into pET28b to create pET-LtgA(E481A)-FL. LtgA-F-NcoI and LtgA/3′/XhoI were used to amplify pIDN1-LtgA(C21A), and the product was digested with NcoI and XhoI and inserted into pET28b to create pET-LtgA(C21A). The PCR product of LtgA-NS-NcoI-F (5′ - ACC ATG GCT TCG ACA AAC ACA CTG CCA GCC) and LtgA/3′/XhoI was digested with NcoI and XhoI to create pET-LtgA-NS. To make LtgD production plasmids, the product of LtgD-F-NcoI (5′ - TCC ATG GAA AAG AGA AAA ATA CTG CCG CTG G) and LtgD/3′/XhoI and the product of LtgD-NS-NcoI-F (5′ - ACC ATG GCG GCG ATG GAG GCC CGC ACA CCC C) and LtgD/3′/XhoI were digested with NcoI and XhoI, then ligated into pET28b to make pET-LtgD-FL and pET-LtgD-NS, respectively. LtgD-F-NcoI and LtgD/3′/XhoI were used to amplify pIDNI-LtgD(C19A), and the resulting product was digested with NcoI and XhoI and ligated into pET28b to create pET-LtgD(C19A).
To make plasmids for removing the signal sequence of LtgA, LtgA-Sig-NcoI-F (5′ - TTC CAT GGA TGT CTT CGA CAA ACA CAC TGC C) and LtgA-Sig-NcoI-R (5′ - TTC CAT GGT CCT TAA GAA TCG GAA CCC TGA ACG) were used to amplify around pIDN1-LtgA(C21A). The resulting PCR product was digested with NcoI and ligated to make pIDN1-LtgA-NS. Likewise, LtgD-Sig-NcoI-F (5′ - TTC CAT GGA TGA CGG CGA TGG AGG CCC GC) and LtgD-Sig-NcoI-R (5′ - TTC CAT GGA TTC CGA ACA AAT AGG GTA AGT GGG) were used to amplify pIDN1-LtgD(C21A). The PCR product was cut with NcoI and ligated to form pIDN1-LtgD-NS.
PG Purification
Macromolecular PG was isolated from wild-type and non-O-acetylating (ΔpacA) gonococcal strains grown overnight on GCB plates. Cells were harvested in PB [25 mM sodium phosphate buffer pH=6] and washed twice with PB using centrifugation at 6,000 × g for 10 minutes at 4°C. Cells were suspended in cold PB and added dropwise into an equal volume of boiling PB with 8% sodium dodecyl sulfate (SDS) and boiled for 1 hour. Cooled insoluble material was harvested by centrifugation and washed four times with PB at 45,000 × g for 30 minutes at 15°C. Insoluble material was then suspended in 500 μL PB and treated with 2 mg mL−1 Pronase (Sigma P5147) at 37°C for 16 hours. To remove the Pronase, the sample was added to 10 mLs of boiling PB with 8% SDS, and the insoluble PG sacculi were washed five times by suspension in PB followed by centrifugation at 45,000 × g for 30 minutes at 15°C. Purified PG was suspended in PB and quantified using OPA assay (Thermo Scientific).
Protein purification
E. coli harboring overexpression plasmids were suspended in LB at an optical density at 600 nm (OD600) of 0.05 from overnight cultures. Once cultures reached OD600 ~ 0.7, protein production from pET vectors was induced with a final concentration of 1.0 mM isopropyl β-D-1-thiogalactopyranoside (IPTG). After an additional incubation of 3 hours, cells were collected by centrifugation at 7000 × g and washed with phosphate buffered saline (PBS) [137 mM NaCl, 2.7 mM KCl, 8 mM Na2HPO4, and 2 mM KH2PO4; pH=7.4]. Cells were then suspended in nickel buffer (NB) [300 mM NaCl, 20 mM Tris-HCl pH = 8] with 10 mM imidazole and lysed by two passages through a French press at 12,000 psi. Insoluble material and unlysed cells were removed by centrifugation at 18,000 × g. His6-tagged proteins were purified using Ni2+-nitrilotriacetic acid (Ni2+-NTA) affinity chromatography using NB with 100 mM imidazole to elute proteins. Proteins were dialyzed into NB with 10% (v/v) glycerol.
Digestion of PG and analysis of soluble fragments
The ability of LtgA and LtgD to digest PG in vitro was determined by incubation with purified PG sacculi. Purified PG (500 μg) was digested with 1 μM purified protein in 200 μL PB at 37°C for 18 hours with agitation. Soluble products were collected using a 10 kDa MWCO Centricon filter at 17,000 × g for 20 minutes to remove insoluble larger PG and protein. 50 μL of reaction products were separated using a Prevail (Alltech) C18 HPLC column (5 μm pore, 25 × 4.6 mm) using a 0–7.5% gradient from 0 to 10 minutes and a 7.5–25% gradient from 10 to 65 minutes of Buffer B [60% acetonitrile (ACN)] at a flow of 1 mL per minute. PG fragments were identified by injecting 10 μL of sample diluted 1:1 with 0.6% trifluoroacetic acid onto a Zorbax SB-C18 2.1 × 50 mm 1.8 um [Agilent] at a flow rate of 0.25 mL min−1. Mass was detected using a LC/MSD TOF [Agilent] in positive mode to detect a mass range of 50–3200 m/z. Products of reactions were detected by incubation of 75 μg of synthetic tetrasaccharide dipeptide with 4 μM purified protein in a 100 μL reaction with of 50 mM Tris-HCl pH=7.5 buffer for 4 hours at 37°C. Products were separated using a Prevail C18 HPLC column with a 0–40% of 25% ACN over 100 minutes. The identity of the products was confirmed by the detection of M+Na ions by MALDI-TOF.
Palmitate Labeling
E. coli BL21(DE3) strains harboring ltgA and ltgD lipidation expression constructs were grown to OD540=0.5 and 5 μCi mL−1. [3H]-palmitate was added to each culture for 90 minutes. Cultures were then induced with 1.0 mM IPTG for 30 minutes before being harvested and washed in PBS. Cells were then suspended in 20 mM Tris-HCl pH=8, 1.0 mM EDTA, and 1% (w/v) SDS and boiled for 15 minutes and centrifuged at 13,000 × g for 30 minutes. The supernatant was then precipitated in acetone at −20°C overnight, and the precipitated protein was then suspended in 1% (w/v) SDS. CPM was measured by liquid scintillation counting, and 5.0 × 105 CPM of each sample was loaded and run on a 12% acrylamide gel. The gel was then soaked in 16% (w/w) 2,5-diphenyloxazole in dimethyl sulfoxide, vacuum-dried, and exposed to film for 24 hours at −80°C.
Characterization of released PG fragments
The labeling, collection, and separation of PG fragments by size-exclusion chromatography were done essentially as described by Cloud and Dillard (Cloud & Dillard, 2002). Briefly, log-phase cultures were washed and suspended at OD540=0.2 in GCBL lacking glucose and containing pyruvate as a carbon source. Cultures were pulse-labeled with 10 μCi mL−1 [6-3H] glucosamine for 45 minutes, washed, and suspended in fresh GCBL. The total CPM per culture was normalized by removing necessary culture volume to achieve an equivalent amount of 3H label in the bacteria in each culture. Released soluble PG fragments were collected during a 2.5-hour chase period and filtered using a 0.2 μm syringe filter. PG fragments were then passed through a tandem size-exclusion chromatography column and eluted with 100 mM LiCl into 3 mL fractions. The CPM of each fraction was determined by taking 500 μL of each fraction and adding to 3 mL of scintillation cocktail, followed by counting on a liquid scintillation counter. Fractions making up peaks of interest were pooled and collected.
Preparation of hot-water extracts
Gonococcal cultures were grown into log phase, washed, and used to seed a total of 6 mLs of culture at OD540 = 0.2 in GCBL media lacking glucose but containing 0.4% pyruvate as a carbon source. [6-3H]-glucosamine (10 μC mL−1) was added to cultures and grown for 40 minutes. Cells were then collected at 2,000 × g for 5 minutes and washed with GCBL to remove unincorporated label. Cultures were then suspended in GCBL, and the total CPM per culture was then normalized by the removal of culture volume and grown for 45 minutes. After growth, the cultures were immediately chilled on ice and then collected at 2,000 × g for 5 minutes at 4°C. Cells were washed once with cold water and then suspended in 2 mLs boiling water and then boiled for an additional 5 minutes. Insoluble material was removed by centrifugation at 12,000 × g for 10 minutes at 4°C, and the supernatant was saved to be separated by size-exclusion chromatography as performed for released PG fragments. HPLC analysis was carried out using a Prevail C18 HPLC reversed-phase column in the presence of 0.05% TFA using a 4–13% ACN gradient over 30 minutes using 16% B equilibration with buffer B containing 25% ACN.
Subcellular Fractionation
Fractionation was done essentially as described by Ramsey et al., 2014 (Ramsey et al., 2014). Outer membrane was isolated by inducing blebbing using a 22-gauge syringe in lithium acetate buffer [0.2 M lithium chloride, 0.1 M lithium acetate, and 10 mM EDTA pH=6], filtering out cells using a 0.22 μm filter and harvesting blebs by ultracentrifugation at 150,000 × g for 2 hours. The total membrane fraction was separated from soluble fraction by sonication of log-phase cultures after being washed and concentrated. Unbroken cells were removed by centrifugation at 15,000 × g for 10 minutes and the supernatant was ultracentrifuged at 100,000 × g for 2 hours. Supernatant was saved as the soluble fraction, while the pellet was suspended in PBS as the total membrane fraction. Western blot analysis performed as described (Ramsey et al., 2014).
Microscopy
Gonococcal cultures were inoculated from plates grown overnight to OD540 = 0.25 and grown for 3 hours before fixing washed cells with 4% formaldehyde in PBS. Cells were washed with PBS and incubated with GTE buffer [50 mM glucose, 20 mM Tris-HCl pH=7.5, 1.0 mM EDTA]. Cells were permeabilized by washing with PBS-T [PBS with 0.2% Triton X-100] followed by incubation with methanol at −20°C. Cells were washed with PBS-T and then blocked with PBS-T with 5.0% (v/v) normal goat serum. Mouse anti-FLAG M2 antibody (Sigma-Aldrich) was used at 1:150 in PBS. Washed slides were incubated with Alexa Fluor 647 goat anti-mouse IgG secondary antibody used 1:100 in PBS 5.0% (v/v) normal goat serum with 300 nM DAPI. Vectashield was used as a mounting medium. Cells were visualized using a Nikon N-STORM Microscope using NIS-Elements Ar with N-STORM analysis software. Epifluorescence was used to visualize DAPI counterstain and direct stochastic optical reconstruction microscopy (dSTORM) was used for Alexa Fluor 647. ImageJ was used adjust levels to add a Gaussian blur for smoother rendering.
Supplementary Material
Acknowledgments
We would like to acknowledge Kathleen T. Hackett for technical support related to this work. This research was supported by National Institute of Allergy and Infectious Diseases (NIAID) grants AI097157 (J.P.D) and AI090348 (S.M). The authors declare that they have no conflict of interest with work submitted in this article.
Footnotes
Author Contributions
RES, YAC, and JPD designed and conceived this study. RES designed, performed, and analyzed all experiments. YAC performed initial experiments identifying LtgA and LtgD active sites and demonstrating lytic transglycosylase activity. ML, DH, and SM designed and synthesized synthetic tetrasaccharide dipeptide. RES wrote the manuscript with assistance and advice from JPD, SM, and ML.
References
- Blackburn NT, Clarke AJ. Identification of four families of peptidoglycan lytic transglycosylases. J Mol Evol. 2001;52:78–84. doi: 10.1007/s002390010136. [DOI] [PubMed] [Google Scholar]
- Chan YA, Hackett KT, Dillard JP. The lytic transglycosylases of Neisseria gonorrhoeae. Microb Drug Resist. 2012;18:271–279. doi: 10.1089/mdr.2012.0001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng Q, Park JT. Substrate specificity of the AmpG permease required for recycling of cell wall anhydro-muropeptides. J Bacteriol. 2002;184:6434–6436. doi: 10.1128/JB.184.23.6434-6436.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cloud KA, Dillard JP. A lytic transglycosylase of Neisseria gonorrhoeae is involved in peptidoglycan-derived cytotoxin production. Infect Immun. 2002;70:2752–2757. doi: 10.1128/IAI.70.6.2752-2757.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cloud-Hansen KA, Hackett KT, Garcia DL, Dillard JP. Neisseria gonorrhoeae uses two lytic transglycosylases to produce cytotoxic peptidoglycan monomers. J Bacteriol. 2008;190:5989–5994. doi: 10.1128/JB.00506-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Kruijff B, van Dam V, Breukink E. Lipid II: a central component in bacterial cell wall synthesis and a target for antibiotics. PLEFA. 2008;79:117–121. doi: 10.1016/j.plefa.2008.09.020. [DOI] [PubMed] [Google Scholar]
- Dillard JP. Genetic manipulation of Neisseria gonorrhoeae. Current protocols in microbiology. 2006;Chapter 4(Unit 4A):2. doi: 10.1002/9780471729259.mc04a02s00. [DOI] [PubMed] [Google Scholar]
- Dillard JP, Hackett KT. Mutations affecting peptidoglycan acetylation in Neisseria gonorrhoeae and Neisseria meningitidis. Infect Immun. 2005;73:5697–5705. doi: 10.1128/IAI.73.9.5697-5705.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehlert K, Holtje JV, Templin MF. Cloning and expression of a murein hydrolase lipoprotein from Escherichia coli. Mol Microbiol. 1995;16:761–768. doi: 10.1111/j.1365-2958.1995.tb02437.x. [DOI] [PubMed] [Google Scholar]
- Garcia DL, Dillard JP. Mutations in ampG or ampD affect peptidoglycan fragment release from Neisseria gonorrhoeae. J Bacteriol. 2008;190:3799–3807. doi: 10.1128/JB.01194-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldman WE, Klapper DG, Baseman JB. Detection, isolation, and analysis of a released Bordetella pertussis product toxic to cultured tracheal cells. Infect Immun. 1982;36:782–794. doi: 10.1128/iai.36.2.782-794.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodell EW. Recycling of murein by Escherichia coli. J Bacteriol. 1985;163:305–310. doi: 10.1128/jb.163.1.305-310.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamilton HL, Schwartz KJ, Dillard JP. Insertion-duplication mutagenesis of Neisseria: use in characterization of DNA transfer genes in the gonococcal genetic island. J Bacteriol. 2001;183:4718–4726. doi: 10.1128/JB.183.16.4718-4726.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobs C, Huang LJ, Bartowsky E, Normark S, Park JT. Bacterial cell wall recycling provides cytosolic muropeptides as effectors for beta-lactamase induction. EMBO J. 1994;13:4684–4694. doi: 10.1002/j.1460-2075.1994.tb06792.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson JW, Fisher JF, Mobashery S. Bacterial cell-wall recycling. Annals of the New York Academy of Sciences. 2013;1277:54–75. doi: 10.1111/j.1749-6632.2012.06813.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jorgenson MA, Chen Y, Yahashiri A, Popham DL, Weiss DS. The bacterial septal ring protein RlpA is a lytic transglycosylase that contributes to rod shape and daughter cell separation in Pseudomonas aeruginosa. Mol Microbiol. 2014;93:113–128. doi: 10.1111/mmi.12643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohler PL, Hamilton HL, Cloud-Hansen K, Dillard JP. AtlA functions as a peptidoglycan lytic transglycosylase in the Neisseria gonorrhoeae Type IV Secretion System. J Bacteriol. 2007;189:5421–5428. doi: 10.1128/JB.00531-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovacs-Simon A, Titball RW, Michell SL. Lipoproteins of bacterial pathogens. Infect Immun. 2011;79:548–561. doi: 10.1128/IAI.00682-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lara B, Mengin-Lecreulx D, Ayala JA, van Heijenoort J. Peptidoglycan precursor pools associated with MraY and FtsW deficiencies or antibiotic treatments. FEMS microbiology letters. 2005;250:195–200. doi: 10.1016/j.femsle.2005.07.005. [DOI] [PubMed] [Google Scholar]
- Lee M, Hesek D, Llarrull LI, Lastochkin E, Pi H, Boggess B, Mobashery S. Reactions of all Escherichia coli lytic transglycosylases with bacterial cell wall. Journal of the American Chemical Society. 2013;135:3311–3314. doi: 10.1021/ja309036q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margolin W. Sculpting the bacterial cell. Curr Biol. 2009;19:R812–822. doi: 10.1016/j.cub.2009.06.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melly MA, Gregg CR, McGee ZA. Studies of toxicity of Neisseria gonorrhoeae for human fallopian tube mucosa. The Journal of infectious diseases. 1981;143:423–431. doi: 10.1093/infdis/143.3.423. [DOI] [PubMed] [Google Scholar]
- Melly MA, McGee ZA, Rosenthal RS. Ability of monomeric peptidoglycan fragments from Neisseria gonorrhoeae to damage human fallopian-tube mucosa. The Journal of infectious diseases. 1984;149:378–386. doi: 10.1093/infdis/149.3.378. [DOI] [PubMed] [Google Scholar]
- Mohammadi T, Sijbrandi R, Lutters M, Verheul J, Martin NI, den Blaauwen T, de Kruijff B, Breukink E. Specificity of the transport of lipid II by FtsW in Escherichia coli. J Biol Chem. 2014;289:14707–14718. doi: 10.1074/jbc.M114.557371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moynihan PJ, Clarke AJ. Substrate specificity and kinetic characterization of peptidoglycan O-acetyltransferase B from Neisseria gonorrhoeae. J Biol Chem. 2014;289:16748–16760. doi: 10.1074/jbc.M114.567388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okuda S, Tokuda H. Lipoprotein sorting in bacteria. Annu Rev Microbiol. 2011;65:239–259. doi: 10.1146/annurev-micro-090110-102859. [DOI] [PubMed] [Google Scholar]
- Pariser H. Asymptomatic gonorrhea. The Medical clinics of North America. 1972;56:1127–1132. doi: 10.1016/s0025-7125(16)32338-0. [DOI] [PubMed] [Google Scholar]
- Ramsey ME, Hackett KT, Bender T, Kotha C, van der Does C, Dillard JP. TraK and TraB are conserved outer membrane proteins of the Neisseria gonorrhoeae type IV secretion system and are expressed at low levels in wild-type cells. J Bacteriol. 2014;196:2954–2968. doi: 10.1128/JB.01825-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenthal RS. Release of soluble peptidoglycan from growing gonococci: hexaminidase and amidase activities. Infect Immun. 1979;24:869–878. doi: 10.1128/iai.24.3.869-878.1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenthal RS, Nogami W, Cookson BT, Goldman WE, Folkening WJ. Major fragment of soluble peptidoglycan released from growing Bordetella pertussis is tracheal cytotoxin. Infect Immun. 1987;55:2117–2120. doi: 10.1128/iai.55.9.2117-2120.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheurwater E, Reid CW, Clarke AJ. Lytic transglycosylases: bacterial space-making autolysins. The international journal of biochemistry & cell biology. 2008;40:586–591. doi: 10.1016/j.biocel.2007.03.018. [DOI] [PubMed] [Google Scholar]
- Segal E, Billyard E, So M, Storzbach S, Meyer TF. Role of chromosomal rearrangement in N. gonorrhoeae pilus phase variation. Cell. 1985;40:293–300. doi: 10.1016/0092-8674(85)90143-6. [DOI] [PubMed] [Google Scholar]
- Sham LT, Butler EK, Lebar MD, Kahne D, Bernhardt TG, Ruiz N. Bacterial cell wall. MurJ is the flippase of lipid-linked precursors for peptidoglycan biogenesis. Science. 2014;345:220–222. doi: 10.1126/science.1254522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suvorov M, Lee M, Hesek D, Boggess B, Mobashery S. Lytic transglycosylase MltB of Escherichia coli and its role in recycling of peptidoglycan strands of bacterial cell wall. Journal of the American Chemical Society. 2008;130:11878–11879. doi: 10.1021/ja805482b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swim SC, Gfell MA, Wilde CE, 3rd, Rosenthal RS. Strain distribution in extents of lysozyme resistance and O-acetylation of gonococcal peptidoglycan determined by high-performance liquid chromatography. Infect Immun. 1983;42:446–452. doi: 10.1128/iai.42.2.446-452.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tobiason DM, Seifert HS. The obligate human pathogen, Neisseria gonorrhoeae, is polyploid. PLoS biology. 2006;4:e185. doi: 10.1371/journal.pbio.0040185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tokuda H, Matsuyama S. Sorting of lipoproteins to the outer membrane in E. coli. Biochimica et biophysica acta. 2004;1693:5–13. doi: 10.1016/j.bbamcr.2004.02.005. [DOI] [PubMed] [Google Scholar]
- van Asselt EJ, Dijkstra AJ, Kalk KH, Takacs B, Keck W, Dijkstra BW. Crystal structure of Escherichia coli lytic transglycosylase Slt35 reveals a lysozyme-like catalytic domain with an EF-hand. Structure (London, England: 1993) 1999a;7:1167–1180. doi: 10.1016/s0969-2126(00)80051-9. [DOI] [PubMed] [Google Scholar]
- van Asselt EJ, Thunnissen AM, Dijkstra BW. High resolution crystal structures of the Escherichia coli lytic transglycosylase Slt70 and its complex with a peptidoglycan fragment. J Mol Biol. 1999b;291:877–898. doi: 10.1006/jmbi.1999.3013. [DOI] [PubMed] [Google Scholar]
- Vollmer W, Joris B, Charlier P, Foster S. Bacterial peptidoglycan (murein) hydrolases. FEMS Microbiol Rev. 2008;32:259–286. doi: 10.1111/j.1574-6976.2007.00099.x. [DOI] [PubMed] [Google Scholar]
- Westrom L. Pelvic inflammatory disease. JAMA. 1991;266:2612. doi: 10.1001/jama.1991.03470180112046. [DOI] [PubMed] [Google Scholar]
- WHO. Global action plan to control the spread and impact of antimicrobial resistance in Neisseria gonorrhoeae. World Health Organization; Geneva, Switzerland: 2014. p. 32. [Google Scholar]
- Woodhams KL, Chan JM, Lenz JD, Hackett KT, Dillard JP. Peptidoglycan fragment release from Neisseria meningitidis. Infect Immun. 2013;81:3490–3498. doi: 10.1128/IAI.00279-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamaguchi K, Yu F, Inouye M. A single amino acid determinant of the membrane localization of lipoproteins in E. coli. Cell. 1988;53:423–432. doi: 10.1016/0092-8674(88)90162-6. [DOI] [PubMed] [Google Scholar]
- Yunck R, Cho H, Bernhardt TG. Identification of MltG as a potential terminase for peptidoglycan polymerization in bacteria. Mol Microbiol. 2016;99:700–718. doi: 10.1111/mmi.13258. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.