

Abstract
Stress during adolescence is a risk factor for schizophrenia, and medial prefrontal cortex (mPFC) dysfunction is proposed to interfere with stress control, increasing the susceptibility to stress. We evaluated the impact of different stressful events during adolescence (restraint stress [RS], footshock [FS], or the combination of FS and RS) on behaviors correlated with schizophrenia in rats as adults. At adulthood, animals were tested for anxiety responses (elevated plus-maze), cognitive function (novel-object recognition test) and dopamine (DA) system responsivity (locomotor response to amphetamine and DA neuron activity in the ventral tegmental area (VTA) using in vivo electrophysiology). All adolescent stressors impaired weight gain and induced anxiety-like responses in adults. FS and FS + RS also disrupted cognitive function. Interestingly, only the combination of FS and RS induced a DA hyper-responsivity as indicated by augmented locomotor response to amphetamine and increased number of spontaneously active DA neurons which was confined to the lateral VTA. Additionally, prelimbic (pl) mPFC lesions triggered a DA hyper-responsivity in animals exposed to FS alone during adolescence. Our results are consistent with previous studies showing long-lasting changes induced by stressful events during adolescence. The impact on DA system activity, however, seems to depend on intense multiple stressors. Our data also suggest that plPFC dysfunction increases the vulnerability to stress in terms of changes in the DA system. Hence, stress during adolescence could be a precipitating factor for the transition to schizophrenia, and stress control at this vulnerable period may circumvent these changes to prevent the emergence of psychosis.
Keywords: stress, adolescence, psychosis, dopamine, prefrontal cortex
Introduction
Adolescence is a period that involves a large number of age-related dynamic alterations in physiological processes and social environment.1,2 When combined with genetically influenced developmental changes, these processes shape the neurobiological features that underlie maturation of the adolescent brain. Importantly, these dynamics of brain maturation also make the developing brain highly vulnerable to environmental factors that can lead to the emergence of psychiatric disorders, including schizophrenia.3,4
Schizophrenia is believed to arise from a combination of genetic predisposition and environmental insult.4,5 Stress can play a major role in susceptibility to schizophrenia. Indeed, exposure to childhood trauma has been consistently identified as a predictor of psychosis onset, which typically manifests during late adolescence and early adulthood.6 Furthermore, adolescents that are at high risk for schizophrenia experience abnormally high reactivity to stress and are more likely to develop psychosis if they have decreased tolerance to stress. Elevated basal cortisol secretion has been correlated with this impaired stress sensitivity7,8 and to be predictive of psychosis onset in the North American Prodrome Longitudinal Study consortium.9 However, the neurobiological substrate for this susceptibility remains unclear.
The medial prefrontal cortex (mPFC), hippocampus, and amygdala are the primary integrators of the stress response.10 Additionally, stressful events affect the activity of dopamine (DA) neurons in the ventral tegmental area (VTA), an effect mediated by stress-induced primary changes in the activity of mPFC, ventral hippocampus, and basolateral amygdala.11 However, little is known about the effects of adolescent stress exposure on VTA DA neuron activity in animals as adults.
Repeated stress is known to damage the hippocampus,10,12 a region commonly reported to be altered in schizophrenia patients and which is proposed to underlie the DA system overdrive in schizophrenia patients and in animal models.13–16 Moreover, the prelimbic (pl) subdivision of the mPFC is involved in attenuating stress responses, in part via inhibition of stress-evoked responses in the amygdala and hippocampus.17–20 Therefore, a failure of the plPFC to regulate stress can cause the individual to be more susceptible to the deleterious effects of stress and, consequently, contribute to the emergence of psychiatric disorders, including schizophrenia.
Based on these evidences, we evaluated the long-lasting changes induced by repeated exposure to different stressors during adolescence on behavioral responses and DA system activity in the VTA of rats as adults that are consistent with schizophrenia. Given that a plPFC dysfunction is proposed to lead an individual to be more vulnerable to stress consequences, we tested whether the disruption of the plPFC during adolescence affects stress-induced pathology that emerges in adulthood.
Methods
Animals
Sixteen timed pregnant Sprague–Dawley rats (Harlan Research Models, Frederick, MD) were obtained at gestational day 15 and housed individually in plastic breeding tubs. On postnatal day (PD) 24, litters were weaned and housed 2 or 3 per cage. Only male offspring (a total of 101 rats) were used in this study. All experimental protocols were conducted according to National Institutes of Health guidelines and were approved by the Institutional Animal Care and Use Committee of the University of Pittsburgh.
Experimental Design
Adolescent male rats were submitted to different stressful events (restraint stress [RS]; 1-hour session at PD 31, PD32, and PD40); footshock [FS; daily through PD31–40]; or a combination of FS and RS). Naïve animals were left undisturbed in their home cages. At adulthood, animals were tested for anxiety responses (elevated plus-maze, EPM; PD65), cognitive function (novel-object recognition test, NOR; PD66-67) and DA system responsivity (locomotor response to amphetamine [PD68-69] and recordings of VTA DA neurons [PD77-102]) (figure 1A). We also evaluated the impact of the combination of FS exposure (from PD65-74) and RS (PD65, PD66, and PD74) administered in adulthood.
Fig. 1.

Long-lasting changes induced by adolescent stress exposure in adult rats. (A) Adolescent male rats (n = 10–12/group) were submitted to restraint stress (RS; at postnatal day [PD]31, PD32, and PD40), footshock (FS; daily through PD31–40); or a combination of FS + RS. At adulthood, animals were tested in the elevated plus-maze (EPM) (PD65), novel-object recognition (NOR) test (PD66–67), and locomotor response to amphetamine (PD68–69). Extracellular recordings of ventral tegmental area (VTA) dopamine (DA) neurons started 1 week after the behavioral experiments (PD77–102). (B) All stressors induced impairment in body weight gain and (C) anxiety-like responses in the EPM. (D) FS and FS + RS also disrupted cognitive function in the NOR test as indicated by a decrease in the discrimination index. Only the combination of FS + RS induced a DA hyper-responsivity as indicated by (E) an augmented locomotor response to amphetamine (0.5 mg/kg; injection is indicated by the dashed line) and (F) an increased number of spontaneously active DA cells which was (G) confined to the lateral VTA. In the VTA recordings, data from 5 animals (3 naïve animals, 1 exposed to RS, and 1 exposed to FS) were excluded due to electrode misplacement. Data are presented as mean ± SEM. *P < .05 vs naive rats.
In another set of experiments, we examined whether a lesion of the plPFC would increase the vulnerability to FS exposure during adolescence in rats as adults. The plPFC lesion was induced by infusing ibotenic acid bilaterally into the plPFC in rats at PD25. Six days after surgery, adolescent rats were submitted to FS (daily through PD31–40). At adulthood, they were tested in the EPM (PD65), NOR test (PD66–67), locomotor response to amphetamine (PD68), and activity patterns of VTA DA neurons (PD77–94) (figure 2A).
Fig. 2.

plPFC disruption increases vulnerability to adolescent stress in adult rats. (A) plPFC lesion was induced in rats at PD25. Six days after surgery, adolescent rats (n = 6–9/group) were submitted to footshock (FS). At adulthood, they were tested in the elevated plus-maze (EPM) (PD65), novel-object recognition (NOR) test (PD66–67), locomotor response to amphetamine (PD68) and for ventral tegmental area (VTA) dopamine (DA) cells activity (PD77–94). (B) Photomicrographs show the adolescent plPFC lesion in an adult animal. The same region is also shown in sham animals; cg, cingulate PFC; pl, plPFC; il, infralimbic PFC. (C) FS exposure induced impairment in body weight gain. (D) FS by itself also induced anxiety-like responses in the EPM and (E) a cognitive impairment in the NOR test. Similar changes were observed in animals with plPFC lesion and exposed to FS. The lesion by itself induced an anxiety-like response but did not induce any change in the NOR test. (F) plPFC lesioned animals exposed to FS also showed an enhanced locomotor response to amphetamine (0.5 mg/kg; injection is indicated by the dashed line) and (G) an increased VTA DA population activity. (H) This increased DA population activity was observed throughout the VTA with significant differences in both medial and lateral VTA. In the VTA recordings, data from 1 plPFC lesion + naïve animal were excluded due to electrode misplacement. Data are presented as mean ± SEM. *P < .05 vs naive rats.
Stress Procedures
Footshock (FS).
Similar to our previous study,21 adolescent rats were exposed to one session of FS daily (through PD31–40). In each session, 25 scrambled FS (1.0 mA, 2 s) were delivered every 60 ± 20 seconds.
Restraint Stress (RS).
Animals were exposed to 3 RS sessions. Animals were submitted to restraint by placing each rat in a Plexiglas cylindrical size-adjusted restraint tube. Cylinders measured 14 cm × 3.9 cm (length × diameter) for rats at PD31–32, 20.3 cm × 5.1 cm for rats at PD40, and 23 cm × 6.3 cm for adult rats. Each RS session lasted 1 hour. For the combination of stressors, RS will be done immediately after the FS session.
Elevated Plus-Maze
The EPM consisted of 2 opposite open arms (50 cm × 10 cm), crossed at right angle by 2 arms enclosed by 40 cm high opaque walls. The maze was positioned 50 cm above the floor. During the test, each rat was placed in the center of the EPM facing an enclosed arm, and its movement was recorded for 5 minutes. The percentage of time spent in the open arms was calculated as index of anxiety.
Novel-Object Recognition (NOR) Test
The NOR test was conducted in a rectangular test box (L70 cm × W40 cm × H30 cm). One day before the test session, animals were habituated to the arena for 10 minutes. On the test day, animals were submitted to 2 trials separated by 1 hour. During the first trial (acquisition trial, T1), rats were placed in the arena containing 2 identical objects for 5 minutes. For the second trial (retention trial, T2), one of the objects presented in T1 was replaced by an unknown (novel) object. Animals were then placed back in the arena for 5 minutes. Object exploration was defined as situations where the animal is directing its face to the object in a distance of approximately 2 cm while watching, licking, sniffing, or touching it with the forepaws while sniffing. Recognition memory was assessed using the discrimination index (discrimination index = [novel – familiar / novel + familiar]), corresponding to the difference between the time exploring the novel and the familiar object, corrected for total time exploring both objects.
Locomotor Response to Amphetamine
Rats were tested in an open-field chamber in which locomotor activity was determined by beam breaks and recorded with TruScan software (Coulbourn Instruments). Spontaneous activity was recorded for 30 minutes. After that, rats were injected with D-amphetamine sulfate (0.5 mg/kg, i.p.; Sigma) and their locomotor activity was recorded for another 60 minutes. Total distance traveled was computed for each 5 minutes.
VTA Extracellular Recordings
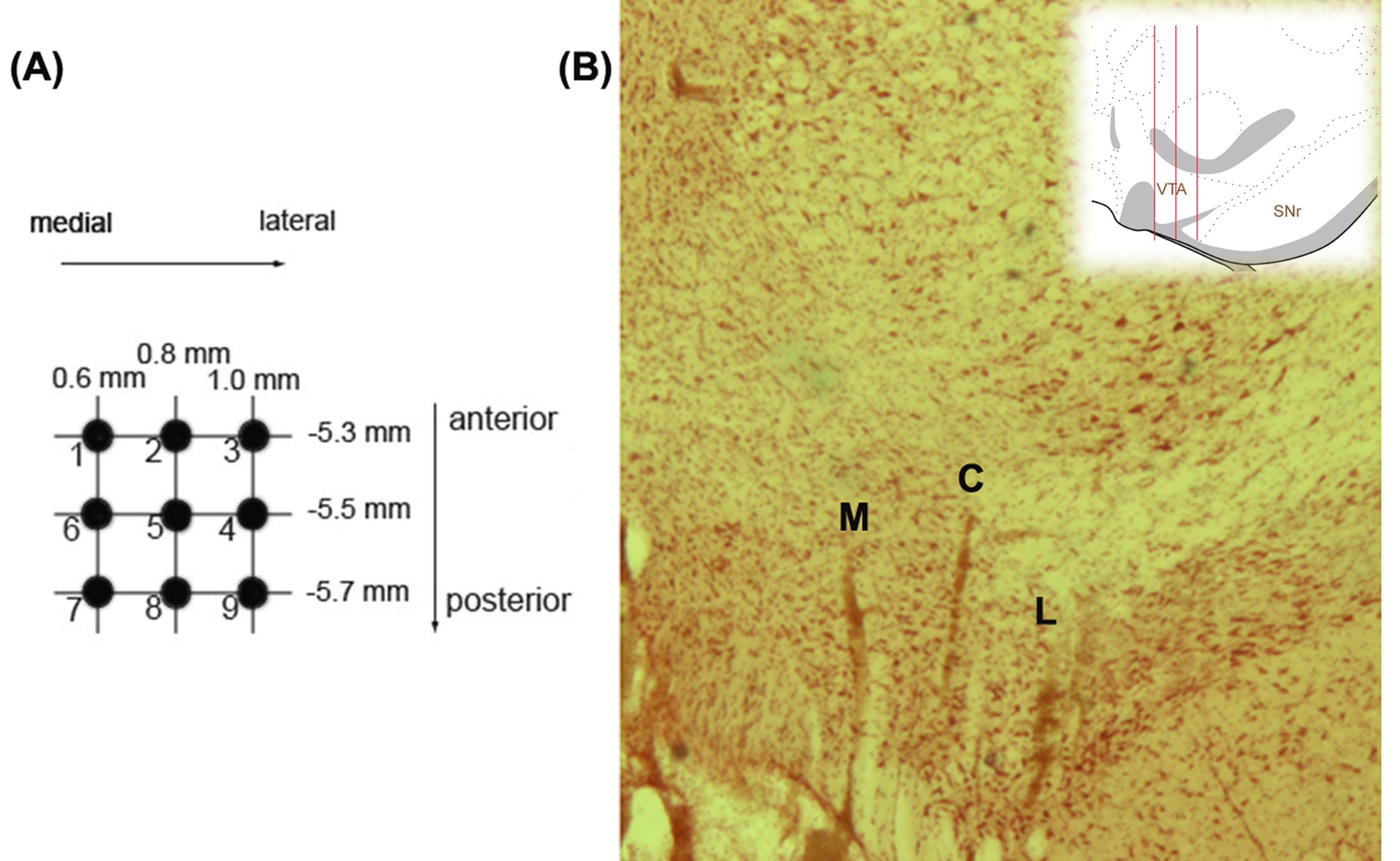
Rats were anesthetized with chloral hydrate (400 mg/kg, i.p.; Sigma) and mounted on a stereotaxic frame. The coordinates for the VTA were 5.3 mm posterior from bregma, 0.6 mm lateral to the midline, and 6.5–9.0 mm ventral from the brain surface. Electrodes were lowered through 6–9 vertical tracks in a predetermined pattern within the VTA of each rat to enable the comparison between DA neurons located throughout the medial, central, and lateral areas of the VTA. DA neurons were identified according to well-established electrophysiological features.22–24 Three parameters were measured: population activity, ie, the number of spontaneously active DA neurons per electrode track; average firing rate; and the percentage of action potentials occurring in bursts. At the end of recordings, the recording sites were marked via iontophoretic ejection of Chicago Sky Blue dye from the tip of the electrode (20 μA constant negative current, 20 min) for posterior histological confirmation of the electrode sites.
plPFC Lesion
Rats at PD25 were anesthetized with isoflurane and placed in a stereotaxic frame. Ibotenic acid (5 µg/0.5 µl delivered over 2.5 min; Sigma) or an equal volume of saline was stereotaxically administered bilaterally using a 10 μl Hamilton syringe at coordinates: +2.9 mm anteroposterior, ±0.6 mm mediolateral from the bregma and −3.0 mm ventral from the skull surface targeting the plPFC. At adulthood, immediately after the VTA recordings, the brains were removed and sectioned for histological evaluation of the lesion extension.
Statistical Analysis
All data were represented as mean ± SEM. Appropriated ANOVA (1, 2, or 3-way ANOVA and repeated measures 2- or 3-way ANOVA) was employed based on the factors (ie., stress, plPFC lesion, VTA subregions, time). Post hoc analysis was performed using the Tukey’s test. Results of statistical tests with P < .05 were considered significant. For details, see supplementary material.
Results
Impact of Stress Exposure During Adolescence on DA System Activity at Adulthood
All stressors induced impairment in weight gain which persisted until adulthood (F3,42 = 8.95, P = .0001, repeated measures 2-way ANOVA; P < .05 vs naïve from PD41–76, Tukey’s test; figure 1B). They also induced anxiety-like responses indicated by a decrease in the exploration of the open arms of the EPM (F3,42 = 5.34, P = .004, 1-way ANOVA; P < .05 vs naïve, Tukey’s test; figure 1C). In the NOR test, a disrupted cognitive function was observed in animals exposed to FS or the combination of FS + RS. No significant difference in time spent exploring the 2 identical objects was observed among groups in the acquisition trial (supplementary figure S1A). In contrast, in the retention trial, all groups explored the novel object significantly longer than the familiar object (P < .05, Tukey’s test). However, the difference in time spent exploring novel vs. familiar object was markedly decreased in animals exposed to FS or to the combination of FS + RS (supplementary figure S1B), reflecting in a decreased discrimination index when compared to naïve animals (F3,42 = 7.10, P = .0006, 1-way ANOVA; P < .05 vs naïve, Tukey’s test; figure 1D).
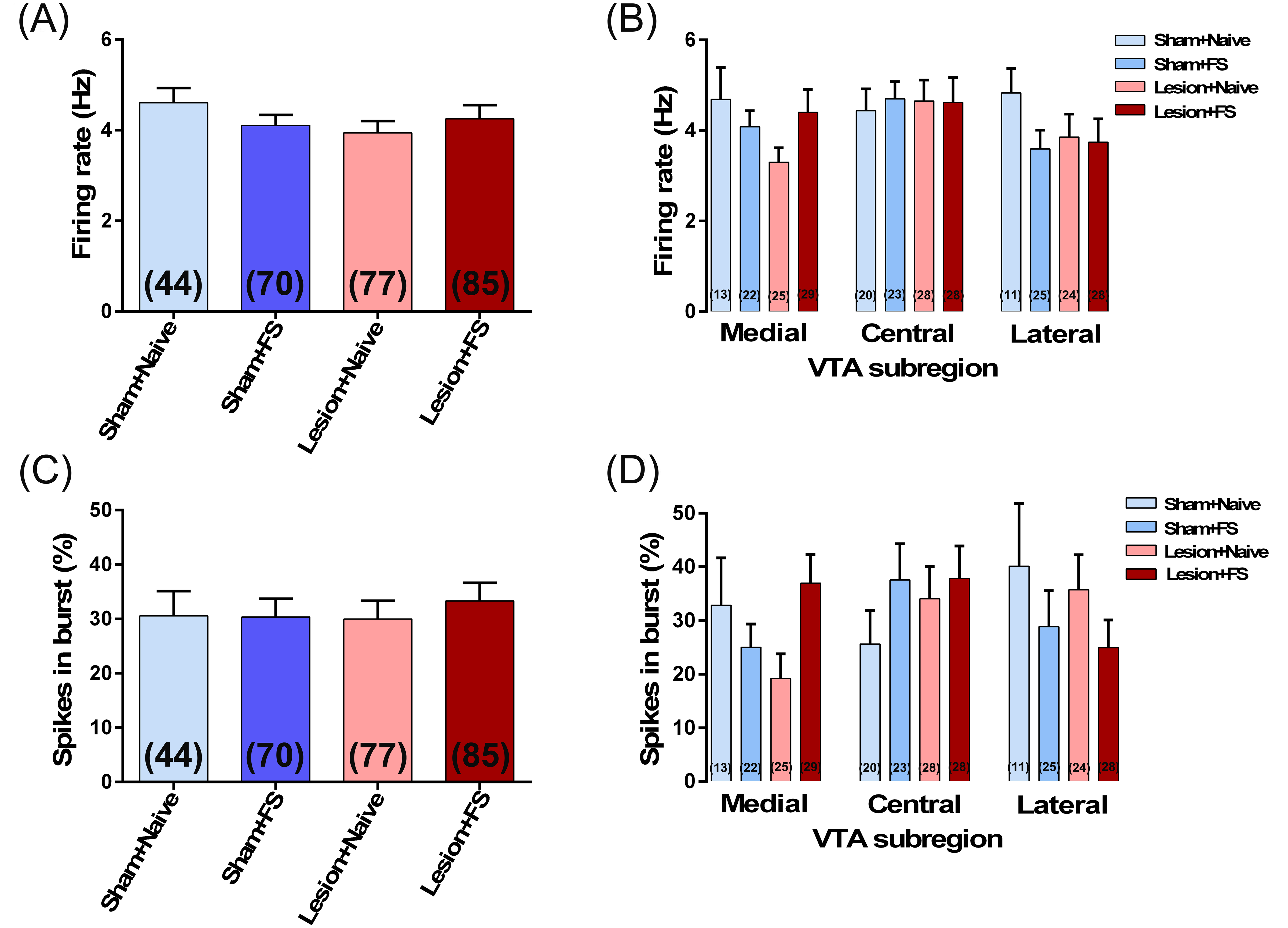
Interestingly, only animals exposed to the combination of FS + RS showed a DA hyper-responsivity as indicated by an augmented locomotor response to amphetamine and an increased number of spontaneously active DA neurons. Animals exposed to FS + RS showed significantly higher levels of locomotor activity in response to amphetamine administration (P < .05 vs naïve at 5, 10, and 35 min after amphetamine, Tukey’s test; figure 1E). One week after the behavioral experiments, we evaluated the activity of VTA DA neurons. Animals exposed to the combination of FS + RS (n = 12 rats; 107 cells; 1.45 ± 0.09 cells/track) demonstrated significant increases in the number of DA neurons firing spontaneously compared to naïve rats (n = 9 rats; 58 cells; 1.04 ± 0.10 cells/track; F3,37 = 3.03, P = .04, 1-way ANOVA; P < .05, Tukey’s test; figure 1F). No change in the DA population activity in the VTA of animals exposed to RS (n = 11 rats; 82 cells; 1.18 ± 0.10 cells/track; P > .05 vs naïve animals, Tukey’s test) or FS (n = 9 rats; 64 cells; 1.16 ± 0.09 cells/track; P > .05 vs naïve, Tukey’s test; figure 1F) was observed. Furthermore, firing rate and percentage of spikes in bursts of DA cells (supplementary figure S2A and S2C) did not differ significantly across all groups. When we expanded our analysis based on the relative location throughout the mediolateral VTA divisions (supplementary figure S3), we observed that the increased DA population activity in animals exposed to FS + RS was confined exclusively to the lateral VTA (figure 1F). The repeated measures 2-way ANOVA indicated significant effects of stress exposure (F3,37 = 3.66, P = .002). Post hoc analysis indicated that animals exposed to FS+RS showed significantly higher DA population activity in the lateral VTA (P < .05 vs naïve, Tukey’s test; figure 1G). There were no significant changes in either average firing rate or percentage of spikes in burst at any VTA subregion across groups (supplementary figures S2B and S2D).
Given that only the adolescent exposure to the combination of FS + RS induced long-lasting changes in the DA system activity in rats as adults, we decided to evaluate if rats exposed to FS + RS during adulthood would show similar changes to those observed after adolescent stress. The experimental design was similar to that employed with adolescent rats (supplementary figure S4A). Neither behavioral nor electrophysiology changes were observed (supplementary figures S4B–M). These results underscore that adolescence is indeed a developmental period of particular susceptibility to stress.
plPFC Disruption Increases the Vulnerability to Adolescent Stress
Evidence suggests that the plPFC is involved in mitigating the impact of stress.18–20 Thus, we sought to determine if a lesion within this region would increase the vulnerability to FS exposure during adolescence in rats as adults. A representative photomicrograph showing the adolescent plPFC lesion in an adult animal is shown in figure 2B. The extent of damage was estimated by reconstruction from preparations stained with neutral red (supplementary figure S5). All lesions involved the plPFC; although in 6 cases some cingulate cortex was involved, the data from these animals did not differ from those in which the lesion was confined to the plPFC.
Adolescent FS exposure again induced impairment in body weight gain (F1,27 = 5.17, P = .03, repeated measures 3-way ANOVA; P < .05 vs sham + naïve from PD61-76, Tukey’s test; figure 2C), with no effect for plPFC lesion (F1,27 = 0.87, P > .05). As expected, FS by itself also induced anxiety-like responses in the EPM and cognitive impairment in the NOR test. Similar changes were observed in plPFC lesioned animals exposed to FS (figures 2D and 2E). The lesion by itself induced an anxiety-like response but did not induce any change in the NOR test (figures 2D and 2E). In the NOR test, all groups, except plPFC lesioned animals exposed to FS, explored the novel object significantly longer than the familiar object (P < .05, Tukey’s test). Also, the difference in time spent exploring novel vs familiar object was markedly decreased in both sham and plPFC lesioned animals exposed to FS (supplementary figure S6), reflecting in a decreased discrimination index in these 2 groups (F1,27 = 14.79, P = .0007, 2-way ANOVA; P < .05 vs sham + naïve, Tukey’s test; figure 2E).
plPFC lesioned animals exposed to FS also showed DA hyper-responsivity as indicated by an augmented locomotor response to amphetamine (P < .05 vs sham + naïve at 15, 20, and 25 minutes after amphetamine, Tukey’s test; figure 2F) and an increased VTA DA population activity. Two-way ANOVA indicated significant effect of plPFC lesion (F1,26 = 11.33, P = .002), and tendency for an effect of FS exposure (F1,26 = 3.81, P = .06). Post hoc analysis indicated that only the plPFC lesioned animals exposed to FS (n = 8 rats; 85 cells; 1.60 ± 0.09 cells/track) demonstrated significant increases in the number of DA neurons firing spontaneously compared to sham + naive rats (n = 6 rats; 44 cells; 1.06 ± 0.06 cells/track; P < .05, Tukey’s test; figure 2G). No change in the DA population activity in the VTA of sham animals exposed to FS (n = 8 rats; 70 DA cells; 1.11 ± 0.11 DA cells/track; P > .05, Tukey’s test) or plPFC lesion + naïve animals (n = 8 rats; 77 cells; 1.26 ± 0.12 cells/track; P > .05, Tukey’s test; figure 2G) was observed. Furthermore, firing rate and percentage of spikes in bursts (supplementary figure S7A and S7C) did not differ significantly across all 4 groups.
Interestingly, different from the changes induced by the combination of FS + RS described above, the increased DA population activity in plPFC lesioned animals exposed to FS was observed throughout the VTA with significant differences in both medial and lateral VTA (P < .05 vs sham + naïve, Tukey’s test; figure 2H). There were no significant changes in either average firing rate or burst activity at any VTA subregion across the groups (supplementary figure S7B and S7D).
Discussion
Numerous studies have shown that stressful events occurring early in life can lead to long-term consequences that extend into adulthood.25,26 Similarly, we showed that stressors applied during adolescence (through PD31–40) induced persistent changes. This period, corresponding to mid-to-late adolescence in humans, is a critical period for both PFC and hippocampal development and inhibitory circuit functional maturation,27–29 both of which are sensitive to stress30,31 and are involved in the pathophysiology of schizophrenia.16,32,33 Although all adolescent stressors (RS, FS, and FS + RS) induced a persistent weight gain impairment and anxiety-like responses in the EPM at adulthood, cognitive deficits in the NOR test were observed only in animals exposed to FS or to FS + RS. Additionally, only the exposure to the combination of FS + RS induced a DA hyper-responsivity in terms of amphetamine hyperlocomotion and increased VTA DA population activity resembling that observed in animal models of schizophrenia.34 These findings suggest that the impact on the DA system seems to depend on intense multiple stressors. Our data also provide evidence that a dysfunction of the plPFC can increase the vulnerability to stress since, unlike intact rats, plPFC lesioned animals exposed only to the FS during adolescence showed a DA hyper-responsivity.
Stress is known to be a risk factor for schizophrenia. Exposure to several socio-environmental factors are associated with risk for psychosis (eg, childhood trauma, urbanicity, ethnic minority status, social disadvantage), and are known to increase the emotional reactivity of individuals and the intensity of psychotic experiences.4,35,36 Stress sensitivity is an important psychological process in the development of psychotic experiences in early stages of schizophrenia.37 Reflecting heightened sensitivity to stress, altered activity of the hypothalamic-pituitary-adrenal axis, as indicated by higher cortisol levels, is common in individuals at high risk and suggestive of conversion to psychosis.38 Furthermore, increased cortisol levels are linked to hippocampal volume loss and to excessive release of DA in the associative striatum in high risk individuals.38,39 Additionally, it has been suggested that exposure to social adversity sensitizes the DA system.40
Although the DA system is hyper-responsive in schizophrenia, there is little evidence for dysfunction within the DA system itself.34 The pathology is likely to originate in the afferent structures that lead to dysregulation of the DA system. The anterior hippocampus, which is functionally equivalent to the ventral hippocampus in rodents, seems to be hyperactive in schizophrenia and this hyperactivity correlates with psychosis.16,41,42 Furthermore, in the methylazoxymethanol acetate (MAM) developmental disruption model of schizophrenia, a DA hyper-responsivity also appears to be a consequence of excessive hippocampal activity.14 Together, this evidence suggests that the hyperdopaminergia states of schizophrenia may be driven by abnormally heightened hippocampal activity, possibly driven by a functional loss of parvalbumin-GABAergic interneurons in MAM rats as well as in schizophrenia patients.32,43 Additionally, given that there is substantial variability among schizophrenia patients in their etiology and functional brain abnormalities, diverse neural pathways can potentially converge on DA hyper-responsiveness.
Maintained stress can lead to dendritic shrinkage and neuronal loss in the hippocampus.10 Given that the NOR test is a hippocampus-dependent task,44 deleterious effects of stress in this brain region can be associated with the cognitive deficits observed in animals exposed to FS or to the combination of FS + RS. Additionally, stressful events can lead to loss of parvalbumin interneuron function, which is suggested to result in permanent consequences in adulthood.45,46 Thus, the enhanced locomotor response to amphetamine and increased VTA DA population activity observed in animals submitted to the combination of FS + RS during adolescence could result from stress-induced hippocampal damage and/or loss of parvalbumin interneuron function in the ventral hippocampus, given that these changes drive a hyper-responsive DA system.14,43
In addition to the hippocampus, the mPFC also plays a role in modulating stress and emotional responses. Interestingly, we observed that plPFC lesioned animals exposed to FS alone showed an increased locomotor response to amphetamine and augmented VTA DA population activity. These changes were not present in sham animals exposed to the same stressor. Additionally, nonstressed animals with a lesion of the plPFC showed anxiety-like responses in the EPM. Given that the plPFC and infralimbic PFC have opposite roles modulating stress responses, reducing and facilitating, respectively,47–49 we avoided lesion of the ilPFC. Thus, most damage was confined to the plPFC, albeit in some cases the cingulate cortex may have been affected. However, all results were consistent independent of cingulate involvement.
The mPFC has also been linked to the pathophysiology of schizophrenia,33 and deficits in this region have been observed in the prodromal state of schizophrenia and subjects at ultra-high risk during tasks involving emotion regulation.50,51 Moreover, the plPFC plays a regulatory role over the stress consequences by regulating amygdala responsivity to stress in rats and in humans.19,20,52 Therefore, a failure of the plPFC to regulate the impact of stress in individuals at high risk for schizophrenia could contribute to pathological changes observed in this disorder. It has been shown that activation of the amygdala, which occurs during stress, causes a loss of hippocampal parvalbumin interneurons.53 This, in turn, is proposed to lead to hippocampal hyperactivity and DA system hyper-responsivity.43 Therefore, heightened stress sensitivity secondary to disruption of plPFC functional regulation of amygdala reactivity to stress during adolescence could initiate a cascade of events to lead to hippocampal dysfunction and the emergence of DA system hyper-responsivity.34 Thus, efforts to reduce stress sensitivity in the risk period could reduce transition to psychosis. Cognitive behavioral therapy has shown some efficacy in preventing psychosis onset among at-risk individuals, likely through reducing stress sensitivity.54 Additionally, the idea of alleviating stress to control transition to psychosis was recently supported by studies showing that attenuating the peripubertal heightened stress response in MAM rats21 by administrating the anti-anxiety drug diazepam during adolescence (PD31–40) reduced heightened anxiety and amygdala hyperactivity and prevented the parvalbumin loss in the ventral hippocampus and the hyperdopaminergic state when tested as adults.55–57 While in these studies diazepam was used as a pharmacological tool in the animal model, it is likely that nonpharmacological interventions to control stress reactivity may be effective in humans.
In the rat, VTA DA populations can be anatomically parcellated in a mediolateral manner based on an overall topography in the efferent projections to the forebrain.58 DA neurons in the medial and central parts of the VTA project to the mPFC, amygdala, and reward-related nucleus accumbens shell, whereas DA neurons in the lateral VTA project selectively to the nucleus accumbens core and associative striatum.58,59 Interestingly, we observed that the increased DA population activity observed in animals exposed to FS + RS during adolescence was confined exclusively to the lateral VTA which projects to associative regions of the striatum homologous to those found to be hyper-responsive in schizophrenia patients.60 Similar to the MAM rat,61 plPFC lesioned animals exposed to FS displayed a more widespread increase in DA neuron activity, with significant differences in both medial and lateral VTA regions. Given that the medial and central parts of the VTA send projections to the mPFC and amygdala and these projections play a role in emotional states,59 an increased DA activity in these VTA subregions may reflect a mechanism related to a disruption of the plPFC control of amygdala reactivity to stress.
Altogether, our findings indicate that extreme stress during adolescence may lead to the emergence of psychosis at adulthood. Furthermore, a failure of the plPFC to regulate the impact of stress, which may be present in at-risk individuals, increases the vulnerability to stress consequences. Thus, predisposition to stress hyper-responsivity, or exposure to substantial stressors, during adolescence can initiate a cascade of events that result in a schizophrenia-like profile in adults. This data can provide essential information with respect to identifying markers for schizophrenia vulnerability early in life and, by mitigating the impact of stressors, prevent the transition to psychosis in susceptible individuals.
Supplementary Material
Supplementary material is available at http://schizophreniabulletin.oxfordjournals.org.
Funding
This study was funded by US National Institutes of Health (MH57440 to A.A.G.).
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgments
We thank Niki MacMurdo and Christy Smolak for technical assistance. A.A.G. has received funds from Johnson & Johnson, Lundbeck, Pfizer, GSK, Merck, Takeda, Dainippon Sumitomo, Otsuka, Lilly, Roche, Asubio, Abbott, Autofony, and Janssen. F.V.G. declares no conflict of interest.
References
- 1. Spear LP. The adolescent brain and age-related behavioral manifestations. Neurosci Biobehav Rev. 2000;24:417–463. [DOI] [PubMed] [Google Scholar]
- 2. Sisk CL, Foster DL. The neural basis of puberty and adolescence. Nat Neurosci. 2004;7:1040–1047. [DOI] [PubMed] [Google Scholar]
- 3. Thompson JL, Pogue-Geile MF, Grace AA. Developmental pathology, dopamine, and stress: a model for the age of onset of schizophrenia symptoms. Schizophr Bull. 2004;30:875–900. [DOI] [PubMed] [Google Scholar]
- 4. van Os J, Kenis G, Rutten BP. The environment and schizophrenia. Nature. 2010;468:203–212. [DOI] [PubMed] [Google Scholar]
- 5. Tsuang M. Schizophrenia: genes and environment. Biol Psychiatry. 2000;47:210–220. [DOI] [PubMed] [Google Scholar]
- 6. Yung AR, Cotter J, Wood SJ, et al. Childhood maltreatment and transition to psychotic disorder independently predict long-term functioning in young people at ultra-high risk for psychosis. Psychol Med. 2015;45:3453–3465. [DOI] [PubMed] [Google Scholar]
- 7. Corcoran CM, Smith C, McLaughlin D, Auther A, Malaspina D, Cornblatt B. HPA axis function and symptoms in adolescents at clinical high risk for schizophrenia. Schizophr Res. 2012;135:170–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Sugranyes G, Thompson JL, Corcoran CM. HPA-axis function, symptoms, and medication exposure in youths at clinical high risk for psychosis. J Psychiatr Res. 2012;46:1389–1393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Walker EF, Trotman HD, Pearce BD, et al. Cortisol levels and risk for psychosis: initial findings from the North American prodrome longitudinal study. Biol Psychiatry. 2013;74:410–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. McEwen BS, Nasca C, Gray JD. Stress effects on neuronal structure: hippocampus, amygdala, and prefrontal cortex. Neuropsychopharmacology. 2016;41:3–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Belujon P, Grace AA. Regulation of dopamine system responsivity and its adaptive and pathological response to stress. Proc Biol Sci. 2015;282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Mondelli V, Cattaneo A, Belvederi Murri M, et al. Stress and inflammation reduce brain-derived neurotrophic factor expression in first-episode psychosis: a pathway to smaller hippocampal volume. J Clin Psychiatry. 2011;72:1677–1684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Heckers S, Konradi C. Hippocampal pathology in schizophrenia. Curr Top Behav Neurosci. 2010;4:529–553. [DOI] [PubMed] [Google Scholar]
- 14. Lodge DJ, Grace AA. Aberrant hippocampal activity underlies the dopamine dysregulation in an animal model of schizophrenia. J Neurosci. 2007;27:11424–11430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Modinos G, Allen P, Grace AA, McGuire P. Translating the MAM model of psychosis to humans. Trends Neurosci. 2015;38:129–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Schobel SA, Chaudhury NH, Khan UA, et al. Imaging patients with psychosis and a mouse model establishes a spreading pattern of hippocampal dysfunction and implicates glutamate as a driver. Neuron. 2013;78:81–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. McEwen BS, Morrison JH. The brain on stress: vulnerability and plasticity of the prefrontal cortex over the life course. Neuron. 2013;79:16–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Quirk GJ, Beer JS. Prefrontal involvement in the regulation of emotion: convergence of rat and human studies. Curr Opin Neurobiol. 2006;16:723–727. [DOI] [PubMed] [Google Scholar]
- 19. Rosenkranz JA, Grace AA. Cellular mechanisms of infralimbic and prelimbic prefrontal cortical inhibition and dopaminergic modulation of basolateral amygdala neurons in vivo. J Neurosci. 2002;22:324–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Rosenkranz JA, Moore H, Grace AA. The prefrontal cortex regulates lateral amygdala neuronal plasticity and responses to previously conditioned stimuli. J Neurosci. 2003;23:11054–11064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Zimmerman EC, Bellaire M, Ewing SG, Grace AA. Abnormal stress responsivity in a rodent developmental disruption model of schizophrenia. Neuropsychopharmacology. 2013;38:2131–2139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Grace AA, Bunney BS. The control of firing pattern in nigral dopamine neurons: burst firing. J Neurosci. 1984;4:2877–2890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Grace AA, Bunney BS. The control of firing pattern in nigral dopamine neurons: single spike firing. J Neurosci. 1984;4:2866–2876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ungless MA, Grace AA. Are you or aren’t you? Challenges associated with physiologically identifying dopamine neurons. Trends Neurosci. 2012;35:422–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Sánchez MM, Ladd CO, Plotsky PM. Early adverse experience as a developmental risk factor for later psychopathology: evidence from rodent and primate models. Dev Psychopathol. 2001;13:419–449. [DOI] [PubMed] [Google Scholar]
- 26. Robbins TW, Jones GH, Wilkinson LS. Behavioural and neurochemical effects of early social deprivation in the rat. J Psychopharmacol. 1996;10:39–47. [DOI] [PubMed] [Google Scholar]
- 27. Caballero A, Diah KC, Tseng KY. Region-specific upregulation of parvalbumin-, but not calretinin-positive cells in the ventral hippocampus during adolescence. Hippocampus. 2013;23:1331–1336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Caballero A, Flores-Barrera E, Cass DK, Tseng KY. Differential regulation of parvalbumin and calretinin interneurons in the prefrontal cortex during adolescence. Brain Struct Funct. 2014;219:395–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Keshavan MS, Giedd J, Lau JY, Lewis DA, Paus T. Changes in the adolescent brain and the pathophysiology of psychotic disorders. Lancet Psychiatry. 2014;1:549–558. [DOI] [PubMed] [Google Scholar]
- 30. Czeh B, Simon M, van der Hart MG, Schmelting B, Hesselink MB, Fuchs E. Chronic stress decreases the number of parvalbumin-immunoreactive interneurons in the hippocampus: prevention by treatment with a substance P receptor (NK1) antagonist. Neuropsychopharmacology. 2005;30:67–79. [DOI] [PubMed] [Google Scholar]
- 31. Zaletel I, Filipović D, Puškaš N. Chronic stress, hippocampus and parvalbumin-positive interneurons: what do we know so far? Rev Neurosci. 2016;27:397–409. [DOI] [PubMed] [Google Scholar]
- 32. Heckers S, Konradi C. GABAergic mechanisms of hippocampal hyperactivity in schizophrenia. Schizophr Res. 2015;167:4–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Lewis DA, Hashimoto T, Volk DW. Cortical inhibitory neurons and schizophrenia. Nat Rev Neurosci. 2005;6:312–324. [DOI] [PubMed] [Google Scholar]
- 34. Grace AA. Dysregulation of the dopamine system in the pathophysiology of schizophrenia and depression. Nat Rev Neurosci. 2016;17:524–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Howes OD, Murray RM. Schizophrenia: an integrated sociodevelopmental-cognitive model. Lancet. 2014;383:1677–1687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Morgan C, Charalambides M, Hutchinson G, Murray RM. Migration, ethnicity, and psychosis: toward a sociodevelopmental model. Schizophr Bull. 2010;36:655–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Reininghaus U, Kempton MJ, Valmaggia L, et al. Stress sensitivity, aberrant salience, and threat anticipation in early psychosis: an experience sampling study. Schizophr Bull. 2016;42:712–722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Holtzman CW, Trotman HD, Goulding SM, et al. Stress and neurodevelopmental processes in the emergence of psychosis. Neuroscience. 2013;249:172–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Mizrahi R, Addington J, Rusjan PM, et al. Increased stress-induced dopamine release in psychosis. Biol Psychiatry. 2012;71:561–567. [DOI] [PubMed] [Google Scholar]
- 40. Gevonden M, Booij J, van den Brink W, Heijtel D, van Os J, Selten JP. Increased release of dopamine in the striata of young adults with hearing impairment and its relevance for the social defeat hypothesis of schizophrenia. JAMA Psychiatry. 2014;71:1364–1372. [DOI] [PubMed] [Google Scholar]
- 41. Silbersweig DA, Stern E, Frith C, et al. A functional neuroanatomy of hallucinations in schizophrenia. Nature. 1995;378:176–179. [DOI] [PubMed] [Google Scholar]
- 42. Tregellas JR. Neuroimaging biomarkers for early drug development in schizophrenia. Biol Psychiatry. 2014;76:111–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Lodge DJ, Behrens MM, Grace AA. A loss of parvalbumin-containing interneurons is associated with diminished oscillatory activity in an animal model of schizophrenia. J Neurosci. 2009;29:2344–2354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Broadbent NJ, Gaskin S, Squire LR, Clark RE. Object recognition memory and the rodent hippocampus. Learn Mem. 2010;17:5–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Do KQ, Cuenod M, Hensch TK. Targeting oxidative stress and aberrant critical period plasticity in the developmental trajectory to schizophrenia. Schizophr Bull. 2015;41:835–846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Steullet P, Cabungcal JH, Kulak A, et al. Redox dysregulation affects the ventral but not dorsal hippocampus: impairment of parvalbumin neurons, gamma oscillations, and related behaviors. J Neurosci. 2010;30:2547–2558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Lacroix L, Spinelli S, Heidbreder CA, Feldon J. Differential role of the medial and lateral prefrontal cortices in fear and anxiety. Behav Neurosci. 2000;114:1119–1130. [DOI] [PubMed] [Google Scholar]
- 48. Radley JJ, Arias CM, Sawchenko PE. Regional differentiation of the medial prefrontal cortex in regulating adaptive responses to acute emotional stress. J Neurosci. 2006;26:12967–12976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Tavares RF, Corrêa FM, Resstel LB. Opposite role of infralimbic and prelimbic cortex in the tachycardiac response evoked by acute restraint stress in rats. J Neurosci Res. 2009;87:2601–2607. [DOI] [PubMed] [Google Scholar]
- 50. Phillips LK, Seidman LJ. Emotion processing in persons at risk for schizophrenia. Schizophr Bull. 2008;34:888–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. van der Velde J, Opmeer EM, Liemburg EJ, et al. Lower prefrontal activation during emotion regulation in subjects at ultrahigh risk for psychosis: an fMRI-study. NPJ Schizophr. 2015;1:15026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Hariri AR, Mattay VS, Tessitore A, Fera F, Weinberger DR. Neocortical modulation of the amygdala response to fearful stimuli. Biol Psychiatry. 2003;53:494–501. [DOI] [PubMed] [Google Scholar]
- 53. Berretta S, Lange N, Bhattacharyya S, Sebro R, Garces J, Benes FM. Long-term effects of amygdala GABA receptor blockade on specific subpopulations of hippocampal interneurons. Hippocampus. 2004;14:876–894. [DOI] [PubMed] [Google Scholar]
- 54. Hardy A, Emsley R, Freeman D, et al. Psychological mechanisms mediating effects between trauma and psychotic symptoms: the role of affect regulation, intrusive trauma memory, beliefs, and depression. Schizophr Bull. 2016;42(suppl 1):S34—S43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Du Y, Grace AA. Peripubertal diazepam administration prevents the emergence of dopamine system hyperresponsivity in the MAM developmental disruption model of schizophrenia. Neuropsychopharmacology. 2013;38:1881–1888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Du Y, Grace AA. Loss of parvalbumin in the hippocampus of MAM schizophrenia model rats is attenuated by peripubertal diazepam. Int J Neuropsychopharmacol. In press. doi:10.1093/ijnp/pyw065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Du Y, Grace AA. Amygdala hyperactivity in MAM model of schizophrenia is normalized by peripubertal diazepam administration. Neuropsychopharmacology. 2016;41:2455–2462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Ikemoto S. Dopamine reward circuitry: two projection systems from the ventral midbrain to the nucleus accumbens-olfactory tubercle complex. Brain Res Rev. 2007;56:27–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Lammel S, Hetzel A, Häckel O, Jones I, Liss B, Roeper J. Unique properties of mesoprefrontal neurons within a dual mesocorticolimbic dopamine system. Neuron. 2008;57:760–773. [DOI] [PubMed] [Google Scholar]
- 60. Kegeles LS, Abi-Dargham A, Frankle WG, et al. Increased synaptic dopamine function in associative regions of the striatum in schizophrenia. Arch Gen Psychiatry. 2010;67:231–239. [DOI] [PubMed] [Google Scholar]
- 61. Lodge DJ, Grace AA. Divergent activation of ventromedial and ventrolateral dopamine systems in animal models of amphetamine sensitization and schizophrenia. Int J Neuropsychopharmacol. 2012;15:69–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.