

ABSTRACT
RhoA and RhoC GTPases are 92% identical but demonstrate unique regulation and function. Phosphorylation of Ser188 has widely been reported to inhibit RhoA activity. RhoC possesses Arg188 in place of Ser188 but retains a canonical upstream PKA recognition sequence. We report here that RhoC-R188S was a PKA substrate in vitro and exhibited less GTP loading compared to wild-type RhoC when expressed in cells. Transiently expressed RhoC was found to be significantly more membrane associated than RhoA. Membrane association of RhoC-R188S and RhoC-R188A were similar to each other and wild-type RhoA, suggesting that Arg188 directly promotes RhoC membrane binding. The positive influence of Arg188 on RhoC membrane association was evident in a constitutively active (Q63L) background. In accordance, RhoA-S188R was significantly more membrane associated than either RhoA or RhoA-S188A. Altogether, these data suggest that swapping residue 188 identity effectively flips the membrane binding profile of wild-type RhoA and RhoC through positive arginine contribution rather than negative phosphoserine regulation.
KEYWORDS: arginine, membrane, RhoA, RhoC, serine
Introduction
RhoA and RhoC are small intracellular GTPases associated with cell proliferation, migration, and transformation.1 RhoC expression has been more extensively linked with advancement of cancer cell metastasis.2,3 These proteins are most active when intercalated into the inner leaflet of plasma membranes and bound to GTP. When active, RhoA and RhoC promote actomyosin contraction and influence cell function by binding to shared and unique effector proteins.1-4
The human RhoC gene is postulated to have originated from duplication of the RhoA gene, with only a small number of subsequent mutations producing the current dominant RhoC allele.5 In accordance, mature RhoA and RhoC are 93% (176/190) identical at the amino acid level, with half of the observed divergences lying within the short 14 to 18 residue hypervariable (HV) domain of their carboxyl termini. The HV domain is a key determinant of isoform-specific subcellular localization and function.6,7
Koo et al. recently analyzed the location of the fluorescent reporter, mEos2, when fused to either full-length RhoA, full-length RhoC, or the HV domain of either RhoA or RhoC.8 The authors found a similar number of diffusion states for mEos2 when attached to either full-length GTPase or its corresponding HV domain, suggesting the HV domain alone dictates much of the diffusibility of RhoA and RhoC within cells, potentially through its intermolecular interactions. Moreover, the HV domain chimeras of RhoA and RhoC were found to be dissimilar. The RhoA HV domain chimera exhibited less diffusibility, providing additional evidence that the Rho HV domains contribute directly to isoform-specific activity. In support of HV-guided regulation, Wang et al. found that fusing the HV of RhoC, but not RhoA, to the carboxyl terminus of p190-RhoGAP produced a chimeric protein capable of reducing anchorage independent growth, invasion, and migration of breast cancer and melanoma cells.9
Within the HV domain of all Rho GTPases is a polybasic region (PBR) whose contribution to GTPase membrane localization is multifaceted.10 SmgGDS (Small G-protein GDP Dissociation Stimulator) is an atypical RhoA and RhoC guanine nucleotide exchange factor (GEF) and chaperone for Rho protein maturation.11,12 SmgGDS binds newly synthesized Rho proteins through their PBR, which in turn fosters a prenylation event required for subsequent membrane intercalation and activity of the given GTPase.12,13 Further, the PBR of Rho GTPases is postulated to directly and positively participate in charge-mediated association with the negatively charged inner leaflet of the plasma membrane or intracellular organelles.14 In support, Welman et al. found that plasma membrane association of K-Ras4B positively correlated with the number of positively charged residues present in its PBR.15
The end of the RhoA PBR (RRGKKKSGC) contains a serine residue (Ser188) that is an established protein kinase A/G substrate.4 Ser188 phosphorylation is inhibitory to RhoA activity within a variety of cell types.16-18 It has been suggested that RhoA Ser188 phosphorylation impedes engagement of the key downstream effector Rho-associated kinase and promotes binding of the intracellular inhibitor RhoGDI concomitant with increased extraction of the GTPase from plasma membranes.4,19 Moreover, it is likely the addition of a negatively charged group to the end of the PBR facilitates RhoA extraction by destabilizing its electrostatic binding to the inner plasma membrane surface.
The PBR of RhoC (RKNKRRRGC) is notably different in that it contains a positively charged arginine residue at position 188, thereby losing phospho-regulation and potentially extending the PBR in promotion of more stable electrostatic association with the inner leaflet of the plasma membrane. Interestingly, Zhang et al. reported RhoC possesses self-stimulatory GTPase-activating protein activity driven by PBR-dependent homooligomerization in vitro.20 Here, Arg188 functions as an “arginine finger” necessary for the GAP activity. In accordance, RhoA displayed self-stimulatory GAP activity only when Ser188 was mutated to Arg188.
As residue 188 involves RhoA phospho-regulation in vivo, impacts the length of the Rho PBR, and influences self-stimulating GAP activity in vitro, we biochemically analyzed wild-type and residue 188 mutated RhoA and RhoC proteins transiently expressed in cells. We report here that switching residue 188 identity flips RhoA and RhoC membrane binding profiles. Further, Arg188 promotes membrane localization of these GTPases independent of their conformation and with a greater impact than loss or gain of phospho-regulation at this position.
Results
Membranes were isolated from homogenized lysate precleared of unlysed cells, cell debris, and nuclei. GFP localized solely with the cytosolic supernatant and β1-integrin with the isolated membrane pellet fraction (Fig. 1A). As expected, constitutively active Myc-RhoA-Q63L and Myc-RhoC-Q63L were more membrane associated than wild-type counterparts (Fig. 1B and 1C). Transiently expressed Myc-RhoC was found to be at least 2-fold more membrane localized than Myc-RhoA, with the latter largely cytoplasmic in all experiments (Fig. 1D).
Figure 1.
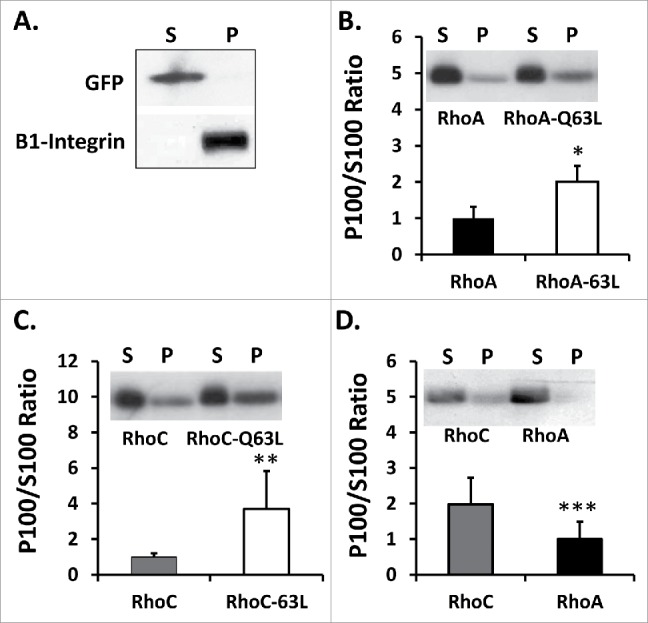
(A) Validation of membrane fractionation approach was achieved by probing the 100,000 × g supernatant (S), which represents the cytosolic fraction, and pellet (P), which represents the membrane fraction, for β1-integrin and exogenously expressed GFP. Constitutively active (B) Myc-RhoA-Q63L and (C) Myc-RhoC-Q63L exhibited significantly more membrane localization than wild-type counterpart qualitatively and by densitometric analysis (*n = 6,6 U = 2 z = −2.482 p=0.013 for Myc-RhoA-Q63L and **n = 9,12 U = 98 z = −3.09 p = 0.002 for Myc-RhoC-Q63L). Data are represented as a normalized signal ratio of the membrane fraction over the cytosolic fraction (P100/S100), with greater values equating with more membrane association. Representative protein gel blot of membrane fractions is inset. (D) Myc-RhoC displays more membrane association than Myc-RhoA (***n = 10,11 U = 15 z = 2.78 p = 0.0054). Representative western blot of membrane fractions is inset.
To begin analyzing the impact of swapping Arg188 in vitroand Ser188 to GTPase function, the ability of PKA to phosphorylate 6xHis-RhoA and 6xHis-RhoC fusion proteins in vitro was assessed. The PKA catalytic domain phosphorylated 6xHis-RhoA and 6xHis-RhoC-R188S, but displayed no detectable activity against either 6xHis-RhoA-S188R or 6xHis-RhoC (Fig. 2), suggesting RhoC-R188S contains the appropriate PKA recognition sequence necessary to be a viable PKA/G substrate in cells.
Figure 2.
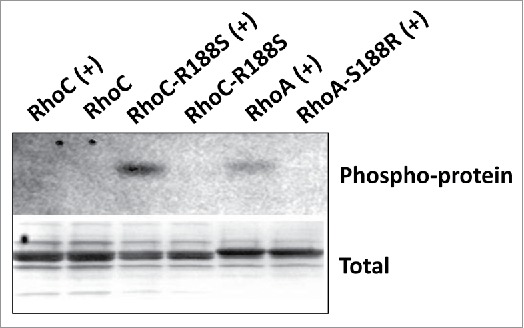
PKA phosphorylates 6xHis-RhoC-R188S. The indicated 6xHis-Rho proteins were incubated with (+) and without (−) the catalytic domain of PKA in kinase buffer. Coomassie brilliant blue was used to confirm total protein and Pro-Q Phosphoprotein stain to visualize phosphorylated protein. 6xHis-RhoA and 6xHis-RhoC-R188S, but not 6xHis-RhoA-S188R and 6xHis-RhoC, are PKA substrates in vitro.
Transient expression of either Myc-RhoA or Myc-RhoC containing Arg188 drove a hypercontractile phenotype in OVCA429 cells, with rounded cells evident in all fields of observation after 18-24 hours of transient expression (Fig. 3). Conversely, transient expression of Myc-RhoA or Myc-RhoC containing Ser188 produced subpopulations of OVCA429 cells that retained a normal phenotype. These data suggest Arg188 has a positive influence on GTPase signaling compared to Ser188.
Figure 3.
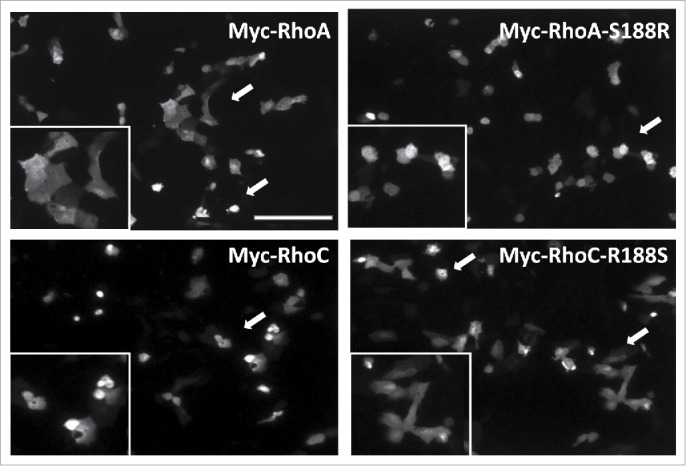
Arg188 promotes a more active Rho phenotype than Ser188. OVCA429 cells transfected with the indicated Myc-Rho expression plasmid were immunostained for the c-Myc epitope and qualitatively examined for actin-driven cell rounding. Myc-RhoA and Myc-RhoC-R188S expressing cells displayed a mixture of both normal and rounded phenotypes independent of fluorescence intensity (arrows). On the other hand, Myc-RhoA-S188R and Myc-RhoC expressing cells were nearly exclusively the rounded cellular phenotype (arrow), suggesting greater ho-driven actin contractility activity when Arg188 is present. Inset panels provide a magnified view of arrow directed fields. ar = 205 μm.
In support, fractionation demonstrated less Myc-RhoC-R188S associated with membranes compared to Myc-RhoC (Fig. 4A and 4B). Pretreatment with forskolin, an adenylyl cyclase agonist, had no measurable impact on wild-type Myc-RhoC or the already low membrane association of Myc-RhoC-R188S. A loss of membrane localization was also observed when Ser188 was placed in a constitutively active background (Fig. 4C), demonstrating membrane destabilization was independent of GTP-binding status. Thus, the negative impact of Ser188 operates above or below GEF activation.
Figure 4.
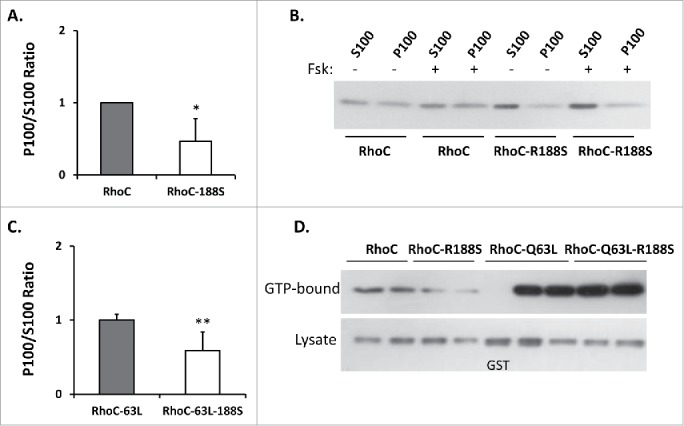
(A) Mutation of Arg188 to a serine reduces Myc-RhoC membrane association (*n = 12,12 U = 12 z = 3.44 p = 0.0006). Data are represented as a normalized signal ratio of the membrane fraction over the cytosolic fraction (P100/S100). (B) Representative protein gel blot of wild-type Myc-RhoC and Myc-RhoC-R188S membrane fractionation. The decreased membrane association of Myc-RhoC-R188S compared to wild-type Myc-RhoC is evident. The already low membrane association of Myc-RhoC-R188S was not obviously impacted by a 15 minute forskolin (+ Fsk) pretreatment. (C) Membrane association of Myc-RhoC-Q63L-R188S was also less than Myc-RhoC-Q63L, indicating that membrane destabilization is GTP-binding independent (**n = 7,8 U = 52 z = −2.72 p = 0.0065). (D) Transiently expressed Myc-RhoC-R188S displayed less GTP-binding than Myc-RhoC as evidenced by a GST-RBD effector pulldown assay, suggesting that GTP-loading of Myc-RhoC may depend on stable membrane association. Constitutively active (Q63L) Myc-RhoC proteins are included as a positive control, while GST bait is used as a negative affinity control. Normalized signal ratio of GTP-Bound/Lysate from 3 independent experiments were found to be 1.00 ± 0.24 for Myc-RhoC and 0.44 ± 0.20 for Myc-RhoC-R188S (n = 6,6 U = 1 z = 2.64 p = 0.0083).
Such Ser188-driven destabilization of the protein from cell membranes could impact Myc-RhoC activation through loss of GEF accessibility. On the other hand, potential elimination of the putative self-stimulatory GAP activity could instead promote GTP stability.20 Rhotekin-binding domain (RBD) pulldown analysis demonstrated that Myc-RhoC-R188S displayed significantly less GTP-binding (Fig. 4D), supporting immunofluorescence evidence that Myc-RhoC-R188S is a less active protein within cells (Fig. 3).
Comparably, Myc-RhoA-S188R was more membrane associated than Myc-RhoA (Fig. 5A and 5B). Pre-treatment with forskolin had a modest impact (27 ± 12% reduction) on Myc-RhoA membrane association, but had no influence on Myc-RhoA-S188R. The positive impact of Arg188 on Myc-RhoA membrane stability was also observed in the constitutively active background (Fig. 5C), further supporting that residue 188 identity influences above or below the level of GEF activation. Interestingly, although Ser188 had a negative impact on GTP-loading of Myc-RhoC-R188S, the converse was not observed for Myc-RhoA-S188R as its GTP loading status was not increased. At the same time, Myc-RhoA-S188A, which has been reported to drive membrane association, displayed a very modest, but reproducible, increase in GTP loading.
Figure 5.
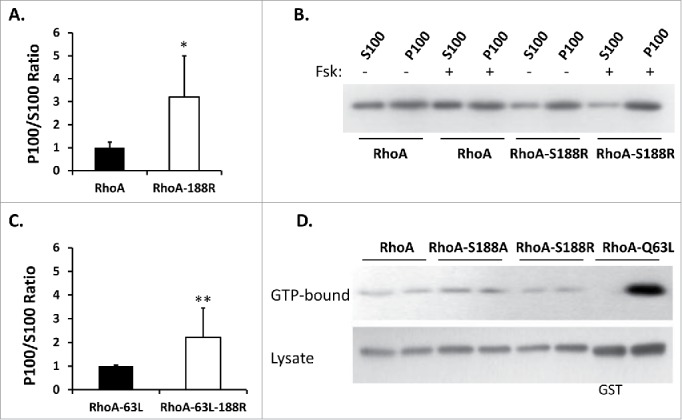
(A) Mutation of Ser188 to an arginine promotes Myc-RhoA membrane association (*n = 8,8 U = 64 z = −3.31 p = 0.001). Data represented as a normalized signal ratio of the membrane fraction over the cytosolic fraction (P100/S100). (B) Representative western blot of wild-type Myc-RhoA and Myc-RhoA-S188R membrane fractionation. The increased membrane association of Myc-RhoA-S188R compared to wild-type Myc-RhoA is evident. Only a very slight increase in Myc-RhoA S100 signal was observed after a 15 minute forskolin (+ Fsk) pretreatment. Myc-RhoA-S188R was not impacted by FSK pretreatment. (C) Myc-RhoA-Q63L-S188R is more membrane associated than Myc-RhoA-Q63L, indicating that Arg188 contributes to membrane stability independent of GTP-binding status (**n = 8,8 U = 64 z = −3.31 p = 0.001). (D) At the same time, GTP binding was relatively insensitive to residue 188 identity (serine, alanine, or arginine), with a modest increase observed with Myc-RhoA-S188A. Normalized signal ratio of GTP-Bound/Lysate from 3 independent experiments were found to be 1.00 ± 0.07 for Myc-RhoA, 1.46 ± 0.51 for Myc-RhoA-S188A, and 0.91 ± 0.34 for Myc-RhoA-S188R (n = 6,7 U = 17 z = 0.5 p = 0.61; Myc-RhoA vs. Myc-RhoA-S188R).
To evaluate whether Arg188 directly promotes membrane association or simply reflects loss of phospho-Ser188 negative regulation, the impact of a neutral Ala residue was analyzed (Fig. 6). Ser188 and Ala188 were found to destabilize Myc-RhoC membrane association to the same extent. Further, Myc-RhoA-S188R was significantly more membrane associated than Myc-RhoA-S188A. In total, these data suggest that swapping residue 188 identity effectively flips the membrane association profile of wild-type RhoA and RhoC through positive arginine contribution rather than negative phosphoserine regulation.
Figure 6.
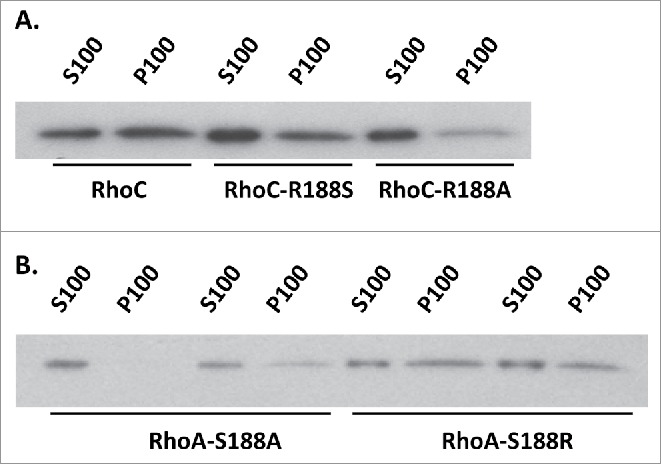
(A) To examine whether presence of a serine or loss of the arginine has the greatest impact on Myc-RhoC membrane association, membrane association of Myc-RhoC-R188S was compared to Myc-RhoC-R188A. Western blot analysis of membrane fractionations revealed both mutations (R188S and 188A) negatively impacted membrane stability to the same extent (n = 6,6 U = 14 z = 0.56 p = 0.5755 for Myc-RhoC-R188S vs. Myc-RhoC-R188A). By densitometric analysis, P100/S100 ratio for Myc-RhoC-R188S was 0.29 ± 0.27 and Myc-RhoC-R188A 0.20 ± 0.15 when compared to Myc-RhoC. (B) At the same time, Myc-RhoA-S188R was more membrane associated than Myc-RhoA-S188A. P100/S100 ratio was 2.05 ± 0.78 higher for Myc-RhoA-S188R when compared to Myc-RhoA-S188A (n = 9,9 U = 78 z = −3.31 p = 0.0009).
Discussion
Ser188 phosphorylation has been reported to negatively impact RhoA binding of Rho-associated kinase while promoting engagement of RhoGDI-α.4,19 We found that 6xHis-RhoC-R188S is a PKA substrate in vitro, therefore a potential mechanism behind its diminished cell contractility phenotype, membrane association, and GTP-loading could involve acquisition of phospho-Ser188 inhibition of ROCK binding and/or increased RhoGDI-α inhibition and extraction from membranes.
At the same time, it has recently been demonstrated that PKA phosphorylation of Ser179/180 negatively influences Rab1B binding of SmgGDS-607, thereby suppressing Rab1b prenylation.21 Williams has extended this finding to hypothesize that prenylation of RhoA might be similarly negatively regulated by phosphorylation of Ser188.22 It follows that reduction of Myc-RhoC-R188S membrane association might also be due to acquisition of phospho-Ser188 regulation of prenylation that obstructs processing events needed for effective membrane intercalation of the GTPase. As both constitutively active Myc-RhoA and Myc-RhoC retain sensitivity to residue 188 identity, such post-translational regulation could explain the influence of residue 188 identity above the level of GEF activation.
Gain or loss of Ser188 phospho-regulation does not adequately explain our results, as we found the presence of Arg188 was largely responsible for swapping the membrane association profile of Myc-RhoA and Myc-RhoC (Fig. 5). One mechanism for Arg188 driving Rho membrane association involves direct lengthening of the PBR, thereby providing a positive tract for stable electrostatic engagement between the GTPase and the inner plasma membrane leaflet.15 Additionally, substitution of a positively charged Arg to the PBR could enhance binding between the nascent GTPase and a complementary acidic patch of SmgGDS isozymes,11 which in turn could foster RhoC or Myc-RhoA-S188R prenylation, processing, and subsequent membrane binding.
Increased Myc-RhoA-S188R membrane association did not completely correlate with increased GTP-binding. Further, Myc-RhoA-S188A exhibited slightly more GTP binding than Myc-RhoA-S188R (Fig. 4), even though the opposite was true in terms of membrane association. One explanation is that isolated preparations of RhoA-S188R exhibited enhanced self-stimulatory GAP activity in vitro compared to wild-type enzyme, though it is unclear as to the extent of homooligizermation of the GTPase within cells.
In conclusion, our work offers evidence that Arg188 directly promotes RhoC membrane association independent of its GTP-binding conformation. Assessing the potential contribution of both Arg188 and (phospho)Ser188 to SmgGDS binding and post-translational processing of these 2 GTPases is an attractive area of future work. Interestingly, RhoG and Rac1 share a similar PBR evolutionary divergence as do RhoA and RhoC. Here, the PBR of RhoG (IKRGRSC) is shorter and terminates in a serine residue, while the PBR of Rac1 (KKRKRKC) lacks this potential serine regulation site in favor of a positive lysine residue. Moreover, RhoG has previously been shown to be phosphorylated by PKA at Ser187 in vitro,16 suggesting phospho-regulation of RhoG post-translational processing and membrane stability might also be an important area of future investigation.
Materials and methods
Materials
Bovine serum albumin and buffer reagents were acquired from Sigma-Aldrich. Polyvinylidene fluoride (PVDF) membranes were purchased from Millipore. OVCA 429 ovarian epithelial cancer cells were maintained in minimum essential medium (Invitrogen) supplemented with 10% fetal bovine serum (Biowhittaker).
Expression constructs
All cDNAs used in this study were Homo sapiens. Creation of pGEX4T-1-RhoA, pCMV-Myc-RhoA, pCMV-Myc-RhoA-S188A, pCMV-Myc-RhoC, pCMV-Myc-RhoC-Q63L and pCMV-Myc-RhoA-Q63L expression plasmids was previously described.16,23,24 Rho mutations (RhoA-S188R, RhoA-Q63L-S188R, RhoC-Q63L, RhoC-R188S, RhoC-R188A, RhoC-Q63L-R188S) were created through PCR mutagenesis using the Quickchange mutagenesis kit (Stratagene). Mutations were confirmed by DNA sequencing, and cDNAs subcloned into either pGEX4T-1 (Amersham Biosciences), pProEx-HTa (Invitrogen), or pCMV-Myc (Clontech) expression plasmids.
Fusion proteins
GST-RBD (GST fusion protein containing the Rho-binding domain [RBD], amino acids 7-89 of Rhotekin) were purified from BL21 E. coli cells (Stratagene) using glutathione-Sepharose 4B (Amersham Biosciences). GST-RBD sepharose was maintained in TBS (50 mM Tris, pH 7.0, 150 mM NaCl, 1 mM DTT) at 4 degrees Celsius and used within 3 days of initial isolation. 6xHis-Rho fusion proteins were purified from BL21 E. coli cells (Stratagene) using NiNTa Agarase (Qiagen) with a gradient imidazole elution. Free imidazole was cleared with a PD10 desalting column (Amersham Biosciences) prior to protein storage in TBS containing 30% glycerol.
Protein phosphorylation and detection
10 µg of 6xHis-Rho fusion protein was incubated with or without 3,750 units of purified protein kinase A catalytic subunit (New England BioLabs) for 3 hours at 37 degrees Celsius in kinase buffer (50 mM Tris-HCL, pH 7.5, 10 mM MgCl2, 200 µM ATP). Proteins were subsequently resolved by SDS-PAGE using 4-20% gradient polyacrylamide Tris-Glycine gels (Invitrogen) and stained with either Coomassie Blue R-250 (Sigma-Aldrich) or Pro-Q Diamond Phosphoprotein Gel Stain (Invitrogen) according to manufacturer's protocol.
Transfections
OVCA 429 ovarian cancer cells grown on either 22 mm glass coverslips or 10 cm tissue-culture dishes were transfected with the indicated expression vectors according to the manufacturer's protocol using LipofectAMINE 2000 (Invitrogen). To avoid aberrant clonal or overexpression effects, exogenous Myc-Rho fusion protein expression was performed transiently and vigorously monitored throughout the study. Cells were maintained in the presence of serum for the duration of the exogenous Myc-protein expression period.
Membrane fractionation
Fifteen minutes prior to collection, some OVCA cells received either 25 µM of forskolin or the appropriate DMSO vehicle control. Cells were washed with Phosphate Buffer Saline (PBS) and scraped into cold homogenization buffer (10 mM Hepes, pH 7.4, 125 mM Sucrose, 5 mM KCl and 2 mM MgCl2) containing fresh DTT (1 mM) and mammalian protease inhibitor cocktail (1:1,000, Sigma-Aldrich). Resulting cell suspensions were kept on ice and passed 20 times through a 26 gauge needle. The homogenized material was incubated on ice for 15 minutes and then clarified at 13,000 × g to remove unlysed cells, cell debris, and nuclei. Supernatants were immediately centrifuged for one hour at 100,000 × g to isolate membrane pellets. The supernatant was removed, stored, and analyzed as the cytosolic (S100) fraction. Membrane pellets were then resuspended in 1 mL of cold homogenization buffer with DTT (1:1000), passed 10 times through a 26 gauge needle to encourage resuspension, and centrifuged for 15 minutes at 100,000 × g. After supernatants were removed, membrane pellets were briefly air-dried on ice, resuspended in 2X Laemmli buffer, boiled, and analyzed as the membrane (P100) fraction.
RhoA and RhoC GTP profile assays
The amount of GTP-bound RhoA or RhoC protein was examined using a technique similar to the method described by Ren and colleagues.25 Transfected cells were lysed in 300 µL of 50 mM Tris, pH 7.4, 10 mM MgCl2, 500 mM NaCl, 1% Triton X-100, 0.1% SDS, 0.5% deoxycholate, and protease inhibitors. 500-750 µg of lysates were cleared at 16,000 × g for 5 minutes, and the supernatant rotated for 30 minutes with 30 µg GST-RBD bound to glutathione-sepharose beads. Samples were washed in 50 mM Tris, pH 7.4, 10 mM MgCl2, 150 mM NaCl, 1% Triton X-100, and protease inhibitors. GST-RBD pulldowns and lysates were then western blotted with anti-c-Myc antibodies.
Western blot
Samples were resolved on 4-20% gradient polyacrylamide Tris-Glycine gels (Invitrogen), transferred on to PVDF membrane, probed using either anti-c-Myc mouse monoclonal antibody (clone 9E10, Sigma-Aldrich), anti-GFP mouse monoclonal antibody (Roche), or anti-β1-Integrin mouse monoclonal antibody (clone EP1041Y, Millipore), and visualized using HRP-conjugated goat anti-mouse antibody (ImmunoResearch Laboratories) and enhanced chemiluminescence reagent (Pierce). Results from multiple experiments were combined through densitometric analysis and presented as a normalized signal ratio of either membrane/cytosolic fraction (P100/S100) or GTP-Bound/Lysate input, with greater values equating with more membrane association or GTP-Rho protein, respectively. Mann Whitney U rank analysis was performed to assess statistical significance.
Immunofluorescence
OVCA 429 cells grown on glass coverslips and transfected with the indicated Myc-Rho expression plasmid were incubated for 24 hours before being fixed (3.7% formaldehyde/PBS) and permeabilized (0.5% Triton-X100/PBS). Myc-Rho protein expressing cells were visualized through a combination of anti-c-Myc mouse monoclonal (1 µg mL−1) and Alexa Fluor-488 secondary antibodies. Coverslips were mounted on glass slides using ProLong Gold antifade reagent (Invitrogen) and visualized using an Olympus spinning disc confocal microscope using a CoolSNAP E2 CCD camera (Photometrics) and Metamorph Image software (Universal Imaging Corp.).
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Funding
This work was supported by a research grant from the McElroy Trust Foundation and the Otto Endowed Professorship Research Fund.
References
- [1].Ellenbroek SIJ, Collard JG. Rho GTPases: functions and associations with cancer. Clin Exp Metastasis 2007; 24:657-672; PMID:18000759; https://doi.org/ 10.1007/s10585-007-9119-1 [DOI] [PubMed] [Google Scholar]
- [2].Ridley AJ. RhoA, RhoB and RhoC have different roles in cancer cell migration. J Microsc 2013; 251:242-249; PMID:23488932; https://doi.org/ 10.1111/jmi.12025 [DOI] [PubMed] [Google Scholar]
- [3].Clark EA, Golub TR, Lander ES, Hynes RO. Genomic analysis of metastasis reveals an essential role for RhoC. Nature 2000; 406(6795):532-535; PMID:10952316; https://doi.org/ 10.1038/35020106 [DOI] [PubMed] [Google Scholar]
- [4].Lang P, Gesbert F, Delespine-Carmagnat M, Stancou R, Pouchelet M, Bertoglio J. Protein kinase A phosphorylation of RhoA mediates the morphological and functional effects of cyclic AMP in cytotoxic lymphocytes. EMBO J 1996; 15(3):510-519; PMID:8599934 [PMC free article] [PubMed] [Google Scholar]
- [5].Wheeler AP, Ridley AJ. Why three Rho proteins? RhoA, RhoB, RhoC, and cell motility. Exp Cell Res 2004; 301:43-49; PMID:15501444; https://doi.org/ 10.1016/j.yexcr.2004.08.012 [DOI] [PubMed] [Google Scholar]
- [6].Adamson P, Paterson HF, Hall A. Intracellular localization of the P21rho proteins. J Cell Biol 1992; 119:617-627; PMID:1383236; https://doi.org/ 10.1083/jcb.119.3.617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Michaelson D, Silletti J, Murphey G, D'Eustachio P, Rush M, Philips MR. Differential localization of Rho GTPases in live cells: regulation by hypervariable regions and RhoGDI binding. J Cell Biol 2011; 152:111-126; https://doi.org/ 10.1083/jcb.152.1.111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Koo PK, Weitzman M, Sabanaygam CR, van Golen KL, Mochrie SG. Extracting diffusive states of Rho GTPase in live cells: towards in vivo biochemistry. Plos Comput Biol 2015; 11:1-26; https://doi.org/ 10.1371/journal.pcbi.1004297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Wang L, Yang L, Luo Y, Zheng Y. Novel strategy for specifically down-regulating individual Rho GTPase activity in tumor cells. J Biol Chem 2003; 278:44617-44625; PMID:12939257; https://doi.org/ 10.1074/jbc.M308929200 [DOI] [PubMed] [Google Scholar]
- [10].Williams CL. The polybasic region of Ras and Rho family small GTPases: a regulator of protein interactions and membrane association and a site of nuclear localization signal sequences. Cell Signal 2003; 15:1071-1080; PMID:14575862; https://doi.org/ 10.1016/S0898-6568(03)00098-6 [DOI] [PubMed] [Google Scholar]
- [11].Hamel B, Monaghan-Benson E, Rojas RJ, Temple BR, Marston DJ, Burridge K, Sondek J. SmgGDS is a guanine nucleotide exchange factor that specifically activates RhoA and RhoC. J Biol Chem 2011; 286:12141-12148; PMID:21242305; https://doi.org/ 10.1074/jbc.M110.191122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Berg TJ, Gastonguay AJ, Lorimer EL, Kuhnmuench JR, Li R, Fields AP, Williams CL. Splice variants of SmgGDS control small GTPase prenylation and membrane localization. J Biol Chem. 2010; 285:35255-35266; PMID:20709748; https://doi.org/ 10.1074/jbc.M110.129916 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Schuld NJ, Vervacke JS, Lorimer EL, Simon NC, Hauser AD, Barbieri JT, Distefano MD, Williams CL. The chaperone protein SmgGDS interacts with small GTPases entering the prenylation pathway by recognizing the last amino acid in the CAAX motif. J Biol Chem 2014; 289:6862-6876; PMID:24415755; https://doi.org/ 10.1074/jbc.M113.527192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Wright LP, Philips MR. CAAX modification and membrane targeting of Ras. J Lipid Res 2006; 47:883-891; PMID:16543601; https://doi.org/ 10.1194/jlr.R600004-JLR200 [DOI] [PubMed] [Google Scholar]
- [15].Welman A, Burger MM, Hagmann J. Structure and function of the C-terminal hypervariable region of K-Ras4B in plasma membrane targeting and transformation. Oncogene 2000; 19:4582-4591; PMID:11030147; https://doi.org/ 10.1038/sj.onc.1203818 [DOI] [PubMed] [Google Scholar]
- [16].Ellerbroek SM, Wennerberg K, Burridge K. Serine phosphorylation negatively regulates RhoA in vivo. J Biol Chem 2003; 278(21):19023-19031; PMID:12654918; https://doi.org/ 10.1074/jbc.M213066200 [DOI] [PubMed] [Google Scholar]
- [17].Jones SE, Palmer TM. Protein kinase A-mediated phosphorylation of RhoA on serine 188 triggers the rapid induction of a neuroendocrine-like phenotype in prostate cancer epithelial cells. Cell Signal 2012; 24:1504-1514; PMID:22504159; https://doi.org/ 10.1016/j.cellsig.2012.03.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Dovas A, Couchman JR. RhoGDI: multiple functions in the regulation of Rho family GTPase activities. Biochem J 2005; 390:1-9; PMID:16083425; https://doi.org/ 10.1042/BJ20050104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Nusser N, Gosmanova E, Makarova N, Fujiwara Y, Yang L, Guo F, Luo Y, Zheng Y, Tigyi G. Serine phosphorylation differentially affects RhoA binding to effectors: implications to NGF-induced neurite outgrowth. Cell Signal 2006; 18:704-714; PMID:16109481; https://doi.org/ 10.1016/j.cellsig.2005.06.010 [DOI] [PubMed] [Google Scholar]
- [20].Zhang B, Zhang Y, Collinst CC, Johnson DI, Zheng Y. A built-in arginine finger triggers the self-stimulatory GTPase-activating activity of Rho family GTPases. J Biol Chem 2001; 5:2609-2612. [DOI] [PubMed] [Google Scholar]
- [21].Ntantie E, Gonyo P, Lorimer EL, Hauser AD, Schuld N, McAllister D, Kalyanaraman B, Dwinell MB, Auchampach JA, Williams CL. An adenosine-mediated signaling pathway suppresses prenylation of the GTPase Rap1B and promotes cell scattering. Sci Signal 2013; 6:ra39; PMID:23716716; https://doi.org/ 10.1126/scisignal.2003374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Williams CL. A new signaling paradigm to control the prenylation and trafficking of small GTPases. Cell Cycle 2013; 12:2933-2934; PMID:23974087; https://doi.org/ 10.4161/cc.26230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Arthur WT, Ellerbroek SM, Der CJ, Burridge K, Wennerberg K. XPLN, a guanine nucleotide exchange factor for RhoA and RhoB, but not RhoC. J Biol Chem 2002; 277:42964-72; PMID:12221096; https://doi.org/ 10.1074/jbc.M207401200 [DOI] [PubMed] [Google Scholar]
- [24].Sloan CM, Quinn CV, Peters JP, Farley J, Goetzinger C, Wernli M, DeMali KA, Ellerbroek SM. Divergence of Rho residue 43 impacts GEF activity. Small GTPases 2012; 3:15-22; PMID:22673745; https://doi.org/ 10.4161/sgtp.19557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Ren XD, Schwartz MA. Determination of GTP loading on Rho. Methods Enzymol 2000; 325:264-272; PMID:11036609; https://doi.org/ 10.1016/S0076-6879(00)25448-7 [DOI] [PubMed] [Google Scholar]