

Abstract
Background.
Deprivation of tumor bioenergetics by inhibition of multiple energy pathways has been suggested as an effective therapeutic approach for various human tumors. However, this idea has not been evaluated in glioblastoma (GBM). We hypothesized that dual inhibition of glycolysis and oxidative phosphorylation could effectively suppress GBM tumorspheres (TS).
Methods.
Effects of 2-deoxyglucose (2DG) and metformin, alone and in combination, on GBM-TS were evaluated. Viability, cellular energy metabolism status, stemness, invasive properties, and GBM-TS transcriptomes were examined. In vivo efficacy was tested in a mouse orthotopic xenograft model.
Results.
GBM-TS viability was decreased by the combination of 2DG and metformin. ATP assay and PET showed that cellular energy metabolism was also decreased by this combination. Sphere formation, expression of stemness-related proteins, and invasive capacity of GBM-TS were also significantly suppressed by combined treatment with 2DG and metformin. A transcriptome analysis showed that the expression levels of stemness- and epithelial mesenchymal transition–related genes were also significantly downregulated by combination of 2DG and metformin. Combination treatment also prolonged survival of tumor-bearing mice and decreased invasiveness of GBM-TS.
Conclusion.
The combination of 2DG and metformin effectively decreased the stemness and invasive properties of GBM-TS and showed a potential survival benefit in a mouse orthotopic xenograft model. Our findings suggest that targeting TS-forming cells by this dual inhibition of cellular bioenergetics warrants expedited clinical evaluation for the treatment of GBM.
Keywords: 2-deoxyglucose, glioblastoma, invasion, metformin, stemness
The outcome of glioblastoma (GBM) is still dismal, despite combination treatment with the best modalities currently available.1,2 One of the reasons for treatment failure is thought to be the presence of refractory cancer cells,3,4 which have features like those of GBM-derived tumorspheres (TS).5,6 Although numerous new drugs based on molecular targeting have been tested against various cancers, some of which are clinically very beneficial, most cancers recur, leading to treatment failure.1 Modulation of cancer cell metabolism, one of the emerging therapeutic targets proposed to overcome this limitation, is considered a promising therapeutic approach.7
2-Deoxyglucose (2DG), which is closely related to cellular energy restriction, has been shown to inhibit cancer cell proliferation in pancreatic cancer8 and breast cancer cell models.9 Moreover, it has been reported that 2DG becomes a more effective anticancer agent when combined with biguanide, which inhibits mitochondrial oxidative phosphorylation.10 It has also been reported that metformin, a well-known biguanide, inhibits cancer cell migration and proliferation,11 an effect that is enhanced by combination treatment with other anticancer agents.12
It has been reported that GBM cells preferentially utilize glycolysis for cellular energy production,13 and glycolysis has been targeted in a GBM stem cell model.14 On the other hand, some authors have reported that GBM-TS primarily rely on oxidative phosphorylation,15 and have further shown that GBM-TS survive challenge by a single agent that blocks oxidative phosphorylation by using additional metabolic pathways.16 In addition, there is a report that the anticancer effect of metformin does not mediate the AMP-activated protein kinase (AMPK)/mammalian target of rapamycin (mTOR) pathway in a GBM model, which is a well-established anticancer mechanism of biguanide.16
It is well known that cancer cells gain an advantage by their preferential use of glycolysis for central energy metabolism (Warburg effect),17 but mitochondrial respiration is still a major energy production pathway, even under anaerobic conditions. In particular, refractory cells, characterized by their stem cell–like properties, are more dependent on oxidative phosphorylation.18–20 Many studies have shown that inhibition of either glycolysis or oxidative phosphorylation is effective to decrease proliferation and invasion of cancer cells.21–23 However, exploiting the idea that simultaneous inhibition of multiple metabolic pathways might be a more effective cancer treatment, several researchers have shown that deprivation of tumor bioenergetics through inhibition of multiple energy pathways could be an effective new therapeutic approach for various human tumors8,24—an idea that has not been fully evaluated in a GBM-TS model. Here, we tested the hypothesis that dual inhibition of glycolysis and oxidative phosphorylation effectively and synergistically suppresses GBM-TS in a synergetic manner by evaluating the effect of combined treatment with 2DG and metformin on GBM-TS.
Materials and Methods
GBM-TS Characterization
Five different GBM-TS were used in this study: TS13-20, TS15-88, TS09-03, GSC11, and U87 spheres. The first 3 TS were directly established from fresh GBM tissues as approved by the institutional review board of Severance Hospital, Yonsei University College of Medicine (4-2012-0212). For isolation of TS from GBM specimens, we followed previously published methods for TS isolation from human brain (Supplementary Experimental Procedures). Patient-derived GSC11 cells were provided from another laboratory. U87 spheres were generated from the U87MG cell line under the same culture condition. The capacities of the 5 TS for self-renewal, stemness, and differentiation were verified. In the following experiments, 5 TS were treated with different drugs and doses; 4mM of 2DG, 5mM of metformin alone, their combination, and 15mM of metformin. Detailed description of experimental materials and methods is given in the Supplementary Experimental Procedures.
Antiproliferation Effect of 2DG and Metformin
Effects of 2DG and metformin on the viability of GBM-TS were determined using assay with MTS (3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium). Each experiment was repeated 3 times in triplicate, and the results were expressed as the percentage of viable cells relative to controls. Cellular energy metabolism status was evaluated with western blotting of AMPK, mTOR, and ATP assay. The uptake of 18F-fluorodeoxyglucose (18F-FDG) PET was measured from GSC11 treated with 2DG, metformin, and their combination. Detailed description of experimental materials and methods is given in the Supplementary Experimental Procedures.
Anti-Stemness Effect of 2DG and Metformin
All 5 TS were cultured for 3 weeks under different conditions. The number of sphere-positive wells was counted, and the proportion of sphere-positive wells of treatment group to that of the control was calculated as a percentage. After sphere formation, lactate dehydrogenase (LDH) assay was performed to check the viability of TS. Protein expression for stemness markers was examined with western blotting. Detailed description of experimental materials and methods is given in the Supplementary Experimental Procedures.
Anti-Invasion Effect of 2DG and Metformin
Green fluorescent protein (GFP)–GBM-TS were generated using GFP-expressing lentiviral supernatants, and cultured in collagen I matrices in different drug conditions. Their viability was determined by assaying green (live) and red (dead) fluorescence. Invasiveness was quantified using the maximal area covered by migrating edges of cells as a parameter, calculated as (invaded area at a certain time/spheroid area at initial time) × 100. Protein expression for epithelial-mesenchymal transition (EMT) markers was examined with western blotting. Detailed description of experimental materials and methods is given in the Supplementary Experimental Procedures.
Gene Expression Microarray and class Comparison
Before and after combination treatment of 2DG and metformin on GSC11, gene expression microarray analysis was performed. Differential expressions of stemness-related, EMT-related, and mitochondrial complex I–related genes were examined. Detailed description of experimental materials and methods is given in the Supplementary Experimental Procedures.
Orthotopic Xenograft Model
Five mice were used for the experiment with different conditions. All experimental procedures were approved by the Yonsei University College of Medicine Institutional Animal Care and Use Committee. Implanted into the right frontal lobe of nude mice were 5×105 GBM-TS (GSC11). Then, 2DG (500mg/kg) and/or metformin (500mg/kg) was administered to mice intraperitoneally every other day. When mice were euthanized, their brains were removed and sectioned with hematoxylin and eosin (H&E) staining and Zeb1 immunostaining. Invading cells, defined as Zeb1-positive cells outside the gross tumor boundary demarcated by H&E staining, were counted. Survival of mice was evaluated using the Kaplan–Meier method. Detailed description of experimental materials and methods is given in the Supplementary Experimental Procedures.
Results
GBM-TS Characterization
Self-renewal capacity was confirmed in all 5 GBM-TS (Fig. 1A). Central necrosis was not observed in the immunofluorescent staining of the TS, which is commonly seen in large-sized TS after long duration of culture. Among 4 tested markers for cell stemness (CD133, nestin, Musashi, and podoplanin [PDPN]), immunocytochemistry revealed that CD133and nestin were positive in all 5 GBM-TS, while positive staining for Musashi and PDPN was observed in GSC11, TS13-20, and TS09-03 but not TS15-88 and U87 spheres (Fig. 1B). Neuroglial differentiation was successfully induced in all TS except U87 sphere, which was confirmed by positive glial fibrillary acidic protein (GFAP), myelin basic protein (MBP), neuronal nuclei (NeuN), and tubulin beta 3 (TUBB3) stains (Fig. 1C). GFAP and MBP were not detectable in U87 sphere. Their molecular characteristics, including molecular subtype, the presence of codeletion of chromosome 1p and 19q, methylation status of O6-DNA methylguanine-methyltransferase promoter, and the presence of isocitrate dehydrogenase mutation are summarized in Supplementary Table S1.
Fig. 1.

Characterization of 5 GBM-TS. (A) TS formation was observed after 3 weeks of culture in GSC11, TS13-20, TS15-88, TS09-03, and U87 spheres. (B) Immunocytochemistry for stemness markers CD133, nestin, Musashi, PDPN. All 4 stem markers were observed in GSC11, TS13-20, and TS09-03. Musashi and PDPN were not detectable in TS15-88 and U87 spheres. (C) Neuroglial differentiation of TS. Two weeks of culture in differentiation medium resulted in successful neuroglial differentiation, as confirmed by positive GFAP, MBP, NeuN, and TUBB3 staining, although GFAP and MBP were not detectable in the U87 sphere. Nuclei were counterstained with 4′,6′-diamidino-2-phenylindole. Images are ×100 original magnification with scale bar = 50 μm for (A) and (B), ×200 original magnification with scale bar = 200 μm for (C).
Combination of 2DG and Metformin Inhibits Proliferation and Cellular Energy Metabolism of GBM-TS
GBM-TS cells were treated with 2DG (4mM), metformin (5mM), or both for 3 days, and their viability was evaluated by MTS assay. The combination of 2DG and metformin significantly decreased the proliferation of GBM-TS compared with untreated controls. These effects were consistently observed in all 5 GBM-TS (Fig. 2A). A moderate antiproliferative effect of 2DG was observed in TS13-20, TS15-88, and U87 spheres. Interestingly, metformin alone effectively inhibited proliferation of only TS15-88, and an increase in the viability of TS was observed in GSC11. ATP assay was conducted and revealed significant decrease in ATP level in all TS except TS13-20 when treated with the combination of 2DG and metformin (Fig. 2B). The effects of single treatment with 2DG or metformin on ATP levels were not consistent among the 5 TS. PET images showed significant differences in 18F-FDG uptake in GBM-TS (GSC11) among 4 different treatment groups (Fig. 2C). Notably, 18F-FDG uptake was markedly decreased in GBM-TS treated with the combination of 2DG and metformin (11.0% of controls). Treatment with 2DG alone decreased glucose metabolism to a greater extent (44.1% of controls) than treatment with metformin alone (70.3% of controls).
Fig. 2.

Antiproliferative effects of 2DG and metformin and cellular energy metabolism in GBM-TS. (A) 5mM of metformin (Met) alone did not effectively inhibit proliferation of GBM-TS except TS15-88, and rather increased the viability of TS in GSC11. A moderate antiproliferative effect of 2DG (4mM) was observed only in TS13-20, TS15-88, and U87 spheres. The combination of 2DG and Met, however, constantly exhibited a significant antiproliferative effect in all 5 GBM-TS. (B) ATP assay revealed a significant decrease in ATP levels in 4 out of the 5 TS tested (no decrease observed in TS13-20) when treated with the combination of 2DG and Met. (C) 18F-FDG uptake measured by PET. In GSC11, 2DG and Met alone showed a moderate decrease in 18F-FDG uptake. When GSC11 TS were treated with these 2 simultaneously, PET indicated a much greater decrease in glucose metabolism. (**P < .01, ***P < .001, compared with the control).
The expression of AMPK and mTOR proteins was examined based on the consensus that metformin is a well-known AMPK activator which downregulates mTOR. However, western blot analyses showed no evidence for elevated AMPK expression and its subsequent mTOR inhibition in response to combination treatment as well as single treatment of metformin (Supplementary Fig. S1). In ATP assay, ATP depletion (increased AMP) was observed in 4 out of 5 TS treated by combination except TS13-20; however, it did not lead to AMPK activation in any of 5 tumorspheres. Low-dose metformin (5mM) alone also failed to decrease ATP level or increase AMPK expression. High-dose (15mM) metformin single treatment did not affect cell viability in any of the 5 TS, although a decrease in ATP levels was observed in 4 out of 5 TS (Supplementary Fig. S2).
Combined Treatment with 2DG and Metformin Decreases Stemness of GBM-TS
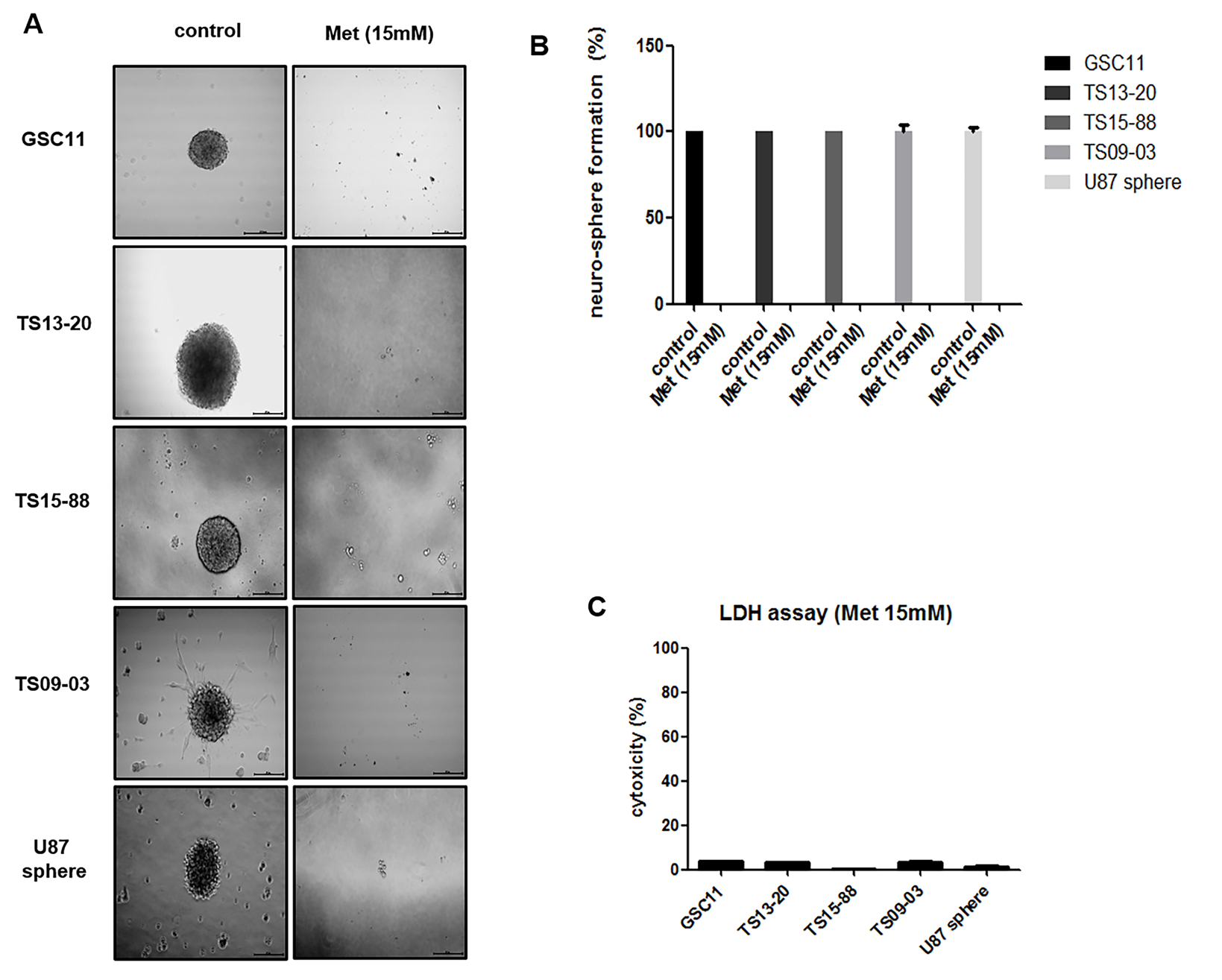
Sphere-formation assays revealed different inhibitory effects of treatment regimens on the stemness of GBM-TS. With varying degrees of anti-stemness effect, the proportion of sphere-positive wells decreased following treatment of GBM-TS with 4mM 2DG alone (Fig. 3A). In contrast, treatment with metformin alone had a less prominent anti-stemness effect compared with 2DG; this effect was observed in only TS13-20, TS09-03, and U87 spheres. Strikingly, combined treatment of GBM-TS with 2DG and metformin demonstrated a strong synergistic, anti-stemness effect, almost completely inhibiting sphere formation. LDH assay revealed that the majority of sphere cells were viable, which implies that the decrease in sphere formation was not mainly mediated by cell death (Fig. 3B). Western blot analyses further indicated that the combination of 2DG and metformin decreased the expression of stemness-related proteins (Fig. 3C and Supplementary Fig. S3). In samples treated with a combination of 2DG and metformin, sex determining region Y–box 2 (Sox-2) and Notch2 were consistently downregulated in all 5 TS, which is consistent with results of sphere formation assays. Combination treatment also resulted in downregulation of nestin in GSC11, TS13-20, and TS09-03. On the contrary, downregulation of CD133 was observed in only TS15-88 and U87 spheres, and no consistent change in octamer-binding transcription factor 3 or 4 was observed. At a higher concentration (15mM), metformin alone was capable of nearly completely inhibiting sphere formation in all 5 TS (Supplementary Fig. S4). LDH assay also revealed most TS were still viable after high-dose metformin treatment.
Fig. 3.

Combined treatment with 2DG and metformin decreases stemness of GBM-TS (A) Sphere formation was tested under different conditions. 2DG (4mM) alone inhibited sphere formation with varying degrees of anti-stemness effect in all 5 TS. Treatment with 5mM of metformin (Met) alone showed less prominent anti-stemness effect compared with 2DG only in TS13-20, TS09-03, and U87 spheres. Sphere formation was nearly completely inhibited by treatment with the combination of 2DG and metformin. All images are ×50 original magnification with scale bar = 200 μm. (***P < .001, compared with the control). (B) The viability of spheres treated in different conditions was tested using the LDH assay, which revealed that the majority of sphere cells were viable, implying that decreased sphere formation was not mainly mediated by cell death. (C) Downregulation of stemness-related genes by combination treatment was confirmed by western blot analyses. Sox-2 and Notch2 were the most consistent markers when compared with the results of sphere formation assays.
Combined Treatment with 2DG and Metformin Inhibits Invasiveness of GBM-TS
For 3D invasion assays, we implanted GFP-GBM-TS in a collagen type I matrix. The implanted GBM-TS migrated radially into the collagen matrix, a response that is physiologically relevant to in vivo tumor behaviors. When evaluated at 72 hours, the combination of 2DG (4mM) and metformin (5mM) significantly inhibited GBM-TS invasion in all 5 TS compared with untreated GBM-TS (Fig. 4A-E, Supplementary Videos S1 and S2). Treatment with 2DG alone also inhibited invasion in TS15-88, TS09-03, and U87—however, its anti-invasion effect was not comparable to that of combination treatment. At a concentration of 5mM, metformin alone did not affect the invasiveness of any GBM-TS. To exclude the possibility that the anti-invasion effects of drugs were mediated by their cytotoxicity, we checked for the presence of dead cells (red fluorescence) using a Live/Dead assay. The distribution of dead cells was limited to the core of the spheroid and was not different from that in controls. Even at a higher concentration (15mM), metformin alone did not show a remarkable anti-invasion effect on GBM-TS (Supplementary Fig. S6). Western blot analyses also showed that the expression of EMT-related markers Zeb1, β-catenin, and N-cadherin were markedly decreased after combined treatment with 2DG and metformin in all 5 TS (Fig. 4F and Supplementary Fig. S5). Decreased expression of EMT-related genes, to levels comparable to combination treatment, was observed in all TS following treatment with 2DG alone.
Fig. 4.

Combined treatment with 2DG and metformin inhibits invasiveness in GBM-TS. (A), (B), (C), (D), and (E) GBM TS invasion in collagen type I matrices was significantly inhibited by combined treatment with 4mM of 2DG and 5mM of metformin (Met) in all 5 different TS. The anti-invasion effect of 2DG alone was observed in TS15-88, TS09-03, and U87 spheres. Met alone failed to inhibit invasion of GBM-TS. When compared with the control, red fluorescence indicating dead cells was a little increased in 2DG, Met, and their combination treatment groups; however, the majority of red fluorescing cells were localized in the core of spheroids. Images were obtained after 72 hours. Scale bar: 100 μm. (*P < .05, **P < .01, ***P < .001, compared with the control at 72 hours). (F) Western blotting of EMT-related genes; Zeb1, β-catenin, and N-cadherin. Decreased expression of EMT-related genes was observed in all TS treated by combination of 2DG and Met. The downregulation of EMT-related gene expression by 2DG treatment alone was mostly observed in all TS.
Gene Expression Microarray and class Comparison
We performed gene expression microarray analyses and compared genes encoding adhesion junction proteins, cell adhesion molecules, focal adhesion, regulation of actin cytoskeleton, and the transforming growth factor–β signaling pathway between GSC11 treated by combination of 2DG and metformin and controls (Fig. 5A). Especially, upon treatment, a subset of genes encoding proteins involved in regulating cell stemness and EMT such as NES (nestin), PROM1 (CD133), SNAI2 (TWIST), and ZEB1 were significantly downregulated, validating the results of western blotting (Fig. 5B). It was also observed that many mitochondrial complex I genes were downregulated following combination treatment with 2DG and metformin (Fig. 5C).
Fig. 5.

Gene expression microarray and class comparison. (A) Heatmap of genes differentially expressed before and after 2DG/metformin (Met)-treatment. (B) Genes of interest. Combined treatments of 2DG and Met downregulated gene expression of stemness- and EMT-related genes; NES (nestin), PROM1 (CD133), SNAI2 (TWIST), and ZEB1. (C) Expression levels of mitochondrial complex I genes were evaluated using microarray experiment. The values were z-transformed using whole genes (n = 3, *P < .05, **P < .01, ***P < .001 by Student’s t-test).
Effects of 2DG and Metformin in an Orthotopic Xenograft Model
Following treatments, mice were sacrificed and their brains were removed and examined by H&E staining (Fig. 6A). A comparison of the number of Zeb1-positive cells located outside the gross tumor boundary revealed significant intergroup differences in the number of Zeb1-stained, invading cells (Fig. 6B). Whereas treatment with either agent alone did not exert a significant anti-invasion effect, combined treatment demonstrated a remarkable inhibition of invasion of GBM-TS. A Kaplan–Meier survival analysis showed different anticancer effects of treatment regimens (Fig. 6C). Whereas there was no survival benefit associated with treatment with 2DG or metformin alone, combined treatment with 2DG and metformin showed apparent survival benefits compared with the control, 2DG, and metformin alone.
Fig. 6.

Effects of combined treatment with 2DG and metformin in an orthotopic xenograft model. Sections of mouse brains, obtained from euthanized mice at the end of the experiment, were H&E stained to show the margins of gross tumors (A), and immunostained for Zeb1 to identify invading cells (B). Original magnification, ×12. Combination treatment of 2DG and metformin (Met) markedly inhibited invasion of GBM-TS compared with the control and other single treatments. (C) Kaplan‒Meier survival curve showed increased survival of mice treated with the combination of 2DG and Met compared with the control other single treatments. (***P < .001).
Discussion
Modulation of cancer metabolism is an emerging approach for cancer treatment.7 This concept is based on the idea that inhibition of cellular bioenergetics could possibly override oncogenic signaling pathways. Because cancer cells are more dependent on glycolysis (Warburg effect),25 there have been various attempts to block glycolysis as a strategy for inhibiting cancer cells.8,26 A number of drugs that target mitochondrial oxidative phosphorylation in the tricarboxylic acid cycle, such as AICAR (5-aminoimidazole-4-carboxamide ribonucleotide), oxaloacetate, oligomycin, and metformin, have also been extensively tested.27,28 In our study, rather than using specific GBM cell lines, we chose GBM-TS, as we believe new therapeutic approaches should target the subpopulation of GBM cells responsible for treatment failure.29 Although the metabolic characteristics of TS have not been clearly elucidated, there are a number of reports that TS preferentially utilize glycolysis.18,19 Other research based on the selective toxicity of metformin toward cancer stem cells (CSCs), however, supports a “reverse Warburg effect,” arguing that CSCs are more dependent on oxidative phosphorylation.15,22,23 Viale et al20 demonstrated that the surviving cancer cells that are responsible for tumor relapse have features of CSCs and depend on oxidative phosphorylation for their survival.
Although a number of the studies cited above have presented results supporting the therapeutic potential of single-pathway inhibition, it is apparent that cancer cells can utilize both pathways, allowing one pathway to serve as an alternative when the other is blocked.30 Cheong and colleagues12 suggested that dual inhibition of glycolysis and oxidative phosphorylation would result in more serious cellular energetic deprivation and thus a better anticancer effect. Consistent with this suggestion, many studies have shown that the combination of 2DG and metformin exerts stronger anticancer effects than either agent alone. Kennedy et al31 evaluated the efficacy of dual inhibition of glycolysis and oxidative phosphorylation in GBM using a GBM cell line model, reporting that this combined treatment regimen produced a strong antiproliferative effect.
Here, we first tested our 5 different TS: GSC11, TS13-20, TS15-88, TS09-03, and U87 spheres. After characterizing the capacities of the 5 TS for self-renewal, stemness, and differentiation, we verified that all were appropriate for the following experiments. In the experiment for the cytotoxicity of drugs on GBM-TS, the combination of 2DG and metformin showed strong antiproliferative effects, as we expected. 2-Deoxyglucose alone showed moderate antiproliferative effect on GBM-TS, although it was not observed in all 5 TS. Metformin single treatment did not effectively inhibit proliferation of GBM-TS at either a low concentration (5mM) or a high concentration (15mM). Würth et al23 reported that the antiproliferative effect of metformin on GBM tumor-initiating cells was also not apparent at low concentration. They found that metformin inhibits the viability of GBM tumor-initiating cells in a concentration-dependent manner. Although 5mM of metformin showed mild antiproliferative effect on some GBM-initiating cells, they concluded that low metformin concentrations were mainly cytostatic, while concentrations higher than the calculated half-maximal inhibitory concentration (IC50) resulted in a cytotoxic effect. In their experiment, the IC50 was about 10mM, which was similar to the IC50 in our experiment (14.53mM, unpublished data). However, in our experiments, the antiproliferative effect of metformin was not observed in most tumorspheres, even at the higher dose of metformin (15mM; Fig. 2A and Supplementary Figure S2). Although biguanide, the most widely used antidiabetic drug, is known to exert its anticancer effects through activation of AMPK and consequent inhibition of the mTOR pathway,32,33 this AMPK-dependent mTOR inhibition was not observed in our experiment.
To confirm that these metabolism-modulating agents actually alter cellular energy metabolism in GBM-TS, we measured ATP levels of GBM-TS after drug treatment. Indeed, their ATP levels were also mostly decreased by combination treatment. In addition, glucose uptake of TS was evaluated by GBM-TS using micro-PET in which a decreased 18F-FDG uptake was observed in all conditions and combination treatment produced a more profound decrease in glucose metabolism in GBM-TS compared with treatment with either agent alone. The result of microarray, which showed the downregulated expression of mitochondrial complex I genes, also supports the findings above. Although metformin alone did not show cytotoxicity on GBM-TS regardless of the dose, the decrease in cellular ATP levels was more prominent with treatment with high-dose metformin. Because metformin blocks oxidative phosphorylation in mitochondria, it increases the dependency on anaerobic glycolysis for cell energy production, which is less efficient. Consequently, metformin treatment is expected to lead to increased 18F-FDG uptake under physiological conditions. However, treatment of GBM-TS with metformin alone decreased uptake of 18F-FDG compared with the control. It could be possibly because the concentration of metformin used (5mM) translates to a dose higher than that used medically. However, this definitely needs further clarification, as another group observed no decrease in 18-F-FDG with even 20mM of metformin.23
More importantly, combination treatment resulted in strong inhibition of stemness in a GBM-TS model. Almost no sphere formation was observed following dual treatment with 4mM of 2DG and 5mM of metformin. This finding was further supported by western blotting, which showed that expression of stem cell markers such as nestin, Sox-2, and Notch2 were markedly decreased in the combination treatment groups. The anti-stemness effect of combination treatment was not mainly mediated by cytotoxicity, as confirmed by the observation that the majority of TS cells were still viable (as measured using the LDH assay) following treatment. Metformin alone failed to show anti-stemness effect at a low dose (5mM); however, high-dose metformin (15mM) dramatically decreased GBM-TS formation. This implies that metformin inhibits the stemness property of GBM-TS in a dose-dependent manner. Because stem cells are known to be responsible for treatment resistance, metabolism-modulating interventions, with their inhibitory effects on stemness, could be a promising strategy for treating GBM patients.29
Numerous reports have also suggested that tumor invasion is mediated by the CSC population.34,35 Using a physiologically relevant in vivo–like tumor model, we investigated whether modulation of cancer metabolism could inhibit the invasive properties of GBM-TS by observing their 3D invasion into type I collagen—the most abundant matrix in the human body. After implantation of spheroids in a type I collagen matrix, GBM TS cells in the cell-matrix boundary radially invaded into the matrix, possibly reflecting collagen matrix-mediated changes in their behavior.36 The combination of 2DG (4mM) and metformin (5mM) effectively inhibited the invasion of GBM TS cells at 3 days compared with the control. Although dead cells were observed after drug treatment, they were mainly localized in the hypoxic core of spheroids, and the majority of the cells were viable. As for treatment with metformin alone, we found that GBM TS invasion was not inhibited by treatment with either a low concentration (5mM) or a high concentration (15mM) of metformin.
Transcriptome analyses conducted to compare expression profiles before and after combined treatment of 2DG and metformin also found that a subset of genes related to stemness and EMT were significantly downregulated. This finding supports the result of western blot for stemness- and EMT-related genes.
It is known that 2DG and metformin are capable of crossing the blood–brain barrier and can be detected in cerebrospinal fluid.37,38 Indeed, we found that the combination of 2DG and metformin exerted strong anticancer effects in an in vivo model as well. In these experiments using an orthotopic xenograft model, combination treatment with 2DG and metformin strongly inhibited GBM TS invasion, as revealed by Zeb1 staining. Moreover, these strong anticancer actions were also supported by Kaplan–Meier analyses, which showed a statistically significant survival benefit. Sato el al22 observed that metformin treatment alone significantly inhibited tumor growth in an orthotopic mouse model, which was not seen in our experiment. They used the same dose of metformin (500mg/kg) with the same delivery method. However, the drug schedule was different, as they daily administered 500mg/kg of metformin for up to 10 consecutive days. In our experiment, 500mg/kg metformin was given to mice every other day and maintained until mice died or terminated, which makes direct comparison between 2 experiments difficult. In our experiments, we used 500mg/kg of metformin administered by intraperitoneal injection every other day, which is the maximal dose the mice can tolerate. Although metformin clearly crosses the blood–brain barrier, the concentration of metformin in the brain was only 10% of that in serum (data not shown). Increasing the concentration of metformin in the brain will inevitably require administration of higher doses of metformin. A 500mg/kg dose of metformin is apparently quite high for use in humans; however, it should be noted that the maximal safe dose of metformin was determined based on daily long-term use for diabetes control and not for anticancer applications. Pollak also noted the possibility of short-term use of metformin at higher doses.32 Because metformin alone at a dose of 500mg/kg failed to show anticancer effect in an in vivo setting, the dose of metformin required for anticancer effects when used in combination with 2DG needs to be confirmed. The use of other biguanides such as phenformin could be another way to decrease toxicity, as its CSF concentration is much higher than that of metformin.32
In summary, the combination of 2DG and metformin was not cytotoxic toward GBM-TS but did effectively decrease the stemness and invasion capacity of GBM-TS, and showed potential survival benefits in a mouse orthotopic xenograft model. We believe that by targeting cells that give rise to TS, this dual inhibition of bioenergetic pathways could be helpful in the treatment of GBM patients.
Supplementary Material
Supplementary material is available at Neuro-Oncology online.
Funding
This study was supported by the Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education, Science and Technology (NRF-2013R1A1A2006427) and the Korean Health Technology R&D Project, Ministry of Health & Welfare, Republic of Korea (HI14C0042).
Conflict of interest statement. The authors declare no relevant conflicts of interest.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgments
We thank Dr Lang (Department of Neurosurgery, The University of Texas MD Anderson Cancer Center) for providing patient-derived GBM TS (GSC11).
References
- 1. Bai RY, Staedtke V, Riggins GJ. Molecular targeting of glioblastoma: drug discovery and therapies. Trends Mol Med. 2011;17(6):301–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Stupp R, Hegi ME, Mason WP, et al. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol. 2009;10(5):459–466. [DOI] [PubMed] [Google Scholar]
- 3. Liu G, Yuan X, Zeng Z, et al. Analysis of gene expression and chemoresistance of CD133+ cancer stem cells in glioblastoma. Mol Cancer. 2006;5:67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bao S, Wu Q, McLendon RE, et al. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature. 2006;444(7120):756–760. [DOI] [PubMed] [Google Scholar]
- 5. Binda E, Reynolds BA, Vescovi AL. Glioma stem cells: turpis omen in nomen? (The evil in the name?). J Intern Med. 2014;276(1):25–40. [DOI] [PubMed] [Google Scholar]
- 6. Sundar SJ, Hsieh JK, Manjila S, et al. The role of cancer stem cells in glioblastoma. Neurosurg Focus. 2014;37(6):E6. [DOI] [PubMed] [Google Scholar]
- 7. Kim S-Y. Cancer metabolism: strategic diversion from targeting cancer drivers to targeting cancer suppliers. Biomol Ther. 2015;23(2):99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Cheng G, Zielonka J, McAllister D, et al. Profiling and targeting of cellular bioenergetics: inhibition of pancreatic cancer cell proliferation. Br J Cancer. 2014;111(1):85–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ciavardelli D, Rossi C, Barcaroli D, et al. Breast cancer stem cells rely on fermentative glycolysis and are sensitive to 2-deoxyglucose treatment. Cell Death Dis. 2014;5(7):e1336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ben Sahra I, Tanti J-F, Bost F. The combination of metformin and 2 deoxyglucose inhibits autophagy and induces AMPK-dependent apoptosis in prostate cancer cells. Autophagy. 2010;6(5):670–671. [DOI] [PubMed] [Google Scholar]
- 11. Bao B, Wang Z, Ali S, et al. Metformin inhibits cell proliferation, migration and invasion by attenuating CSC function mediated by deregulating miRNAs in pancreatic cancer cells. Cancer Prev Res (Phila). 2012;5(3):355–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Cheong J-H, Park ES, Liang J, et al. Dual inhibition of tumor energy pathway by 2-deoxyglucose and metformin is effective against a broad spectrum of preclinical cancer models. Mol Cancer Ther. 2011;10(12):2350–2362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Beckner ME, Gobbel GT, Abounader R, et al. Glycolytic glioma cells with active glycogen synthase are sensitive to PTEN and inhibitors of PI3K and gluconeogenesis. Lab Invest. 2005;85(12):1457–1470. [DOI] [PubMed] [Google Scholar]
- 14. Pelicano H, Martin D, Xu R, et al. Glycolysis inhibition for anticancer treatment. Oncogene. 2006;25(34):4633–4646. [DOI] [PubMed] [Google Scholar]
- 15. Janiszewska M, Suvà ML, Riggi N, et al. Imp2 controls oxidative phosphorylation and is crucial for preserving glioblastoma cancer stem cells. Genes Dev. 2012;26(17):1926–1944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Vlashi E, Lagadec C, Vergnes L, et al. Metabolic state of glioma stem cells and nontumorigenic cells. Proc Natl Acad Sci U S A. 2011;108(38):16062–16067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. science. 2009;324(5930):1029–1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Bednar F, Simeone DM. Metformin and cancer stem cells: old drug, new targets. Cancer Prev Res (Phila). 2012;5(3):351–354. [DOI] [PubMed] [Google Scholar]
- 19. Rattan R, Ali Fehmi R, Munkarah A. Metformin: an emerging new therapeutic option for targeting cancer stem cells and metastasis. J Oncol. 2012;2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Viale A, Pettazzoni P, Lyssiotis CA, et al. Oncogene ablation-resistant pancreatic cancer cells depend on mitochondrial function. Nature. 2014;514(7524):628–632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Prasanna VK, Venkataramana NK, Dwarakanath B, et al. Differential responses of tumors and normal brain to the combined treatment of 2-DG and radiation in glioablastoma. J Cancer Res Ther. 2009;5(9):44. [DOI] [PubMed] [Google Scholar]
- 22. Sato A, Sunayama J, Okada M, et al. Glioma-initiating cell elimination by metformin activation of FOXO3 via AMPK. Stem Cells Transl Med. 2012;1(11):811–824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Wurth R, Pattarozzi A, Gatti M, et al. Metformin selectively affects human glioblastoma tumor-initiating cell viability: a role for metformin-induced inhibition of Akt. Cell Cycle. 2013;12(1):145–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Sahra IB, Tanti J-F, Bost F. The combination of metformin and 2-deoxyglucose inhibits autophagy and induces AMPK dependent apoptosis in prostate cancer cells. Autophagy. 2010;6(5):670–671. [DOI] [PubMed] [Google Scholar]
- 25. Gatenby RA, Gillies RJ. Why do cancers have high aerobic glycolysis? Nature Reviews Cancer. 2004;4(11):891–899. [DOI] [PubMed] [Google Scholar]
- 26. Zhu Z, Jiang W, McGinley JN, et al. 2-Deoxyglucose as an energy restriction mimetic agent: effects on mammary carcinogenesis and on mammary tumor cell growth in vitro. Cancer research. 2005;65(15):7023–7030. [DOI] [PubMed] [Google Scholar]
- 27. Owen MR, Halestrap AP. The mechanisms by which mild respiratory chain inhibitors inhibit hepatic gluconeogenesis. Biochim Biophys Acta (BBA)-Bioenergetics. 1993;1142(1):11–22. [DOI] [PubMed] [Google Scholar]
- 28. Wallace DC. Mitochondria and cancer. Nat Rev Cancer. 2012;12(10):685–698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kang SG, Cheong JH, Huh YM, et al. Potential use of glioblastoma tumorsphere: clinical credentialing. Arch Pharm Res. 2015;38(3):402–407. [DOI] [PubMed] [Google Scholar]
- 30. Scott DA, Richardson AD, Filipp FV, et al. Comparative metabolic flux profiling of melanoma cell lines: beyond the Warburg effect. J Biol Chem. 2011;286(49):42626–42634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kennedy CR, Tilkens SB, Guan H, et al. Differential sensitivities of glioblastoma cell lines towards metabolic and signaling pathway inhibitions. Cancer Lett. 2013;336(2):299–306. [DOI] [PubMed] [Google Scholar]
- 32. Pollak M. Potential applications for biguanides in oncology. J Clin Invest. 2013;123(9):3693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Sahra IB, Regazzetti C, Robert G, et al. Metformin, independent of AMPK, induces mTOR inhibition and cell-cycle arrest through REDD1. Cancer Res. 2011;71(13):4366–4372. [DOI] [PubMed] [Google Scholar]
- 34. Korkaya H, Paulson A, Iovino F, et al. HER2 regulates the mammary stem/progenitor cell population driving tumorigenesis and invasion. Oncogene. 2008;27(47):6120–6130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Wicha MS, Liu S, Dontu G. Cancer stem cells: an old idea—a paradigm shift. Cancer Res. 2006;66(4):1883–1890. [DOI] [PubMed] [Google Scholar]
- 36. Kaufman L, Brangwynne C, Kasza K, et al. Glioma expansion in collagen I matrices: analyzing collagen concentration-dependent growth and motility patterns. Biophys J. 2005;89(1):635–650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Labuzek K, Suchy D, Gabryel B, et al. Quantification of metformin by the HPLC method in brain regions, cerebrospinal fluid and plasma of rats treated with lipopolysaccharide. Pharmacol Rep. 2010;62(5):956–965. [DOI] [PubMed] [Google Scholar]
- 38. McAllister MS, Krizanac-Bengez L, Macchia F, et al. Mechanisms of glucose transport at the blood–brain barrier: an in vitro study. Brain Res. 2001;904(1):20–30. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.