

Abstract
Exemestane (EXE) treats estrogen receptor positive (ER+) breast cancer in postmenopausal women by inhibiting the estrogen‐synthesizing cytochrome P450 CYP19A1. Variability in the severity and incidence of side effects as well as overall drug efficacy may be partially explained by genetic factors, including nonsynonymous variation in CYP19A1, also known as aromatase. The present study identified phase I EXE metabolites in human liver microsomes (HLM) and investigated mechanisms that may alter the extent of systemic estrogen deprivation in EXE‐treated women with breast cancer, including whether functional polymorphisms in aromatase cause differential inhibition by EXE and whether EXE metabolites possess anti‐aromatase activity. The potency of EXE and ten of its derivatives was measured with HEK293‐overexpressed wild type aromatase (CYP19A1*1) using a rapid novel UPLC tandem mass spectrometry method. Of the ten compounds assayed, five were poor inhibitors (IC 50 ˃ 50 μmol/L) of wild type aromatase while five others, including the major metabolite, 17β‐dihydroexemestane (17β‐DHE), exhibited moderate potency, with IC 50 values ranging between 1.2 and 7.1 μmol/L. The anti‐aromatase activity of EXE was also tested with two common allozymes, aromataseThr201Met (CYP19A1*3) and aromataseArg264Cys (CYP19A1*4). Differential inhibition of variant aromatase is unlikely to account for variable clinical outcomes as EXE‐mediated inhibition of aromataseThr201Met (IC 50 = 0.86 ± 0.12 μmol/L) and aromataseArg264Cys (IC 50 = 1.7 ± 0.65 μmol/L) did not significantly differ from wild type (IC 50 = 0.92 ± 0.17 μmol/L). Although less potent than the parent drug, these results suggest that active metabolites may contribute to the therapeutic mechanism of EXE.
Keywords: Anti‐aromatase activity, aromatase inhibitor, breast cancer, dihydroexemestane, exemestane, pharmacogenetics, polymorphism
Abbreviations
- 17α‐DHE
17α‐dihydroexemestane
- 17β‐DHE
17β‐dihydroexemestane
- 6‐HME
6‐Hydroxymethylandrosta‐1,4,6‐triene‐3,17‐dione
- AAA
anti‐aromatase activity
- AI
aromatase inhibitor
- AKRs
aldo‐keto reductases
- CBR1
carbonyl reductase 1
- CYP450
cytochrome P450
- ER+
estrogen receptor positive
- EXE
exemestane
- GC‐MS
gas chromatography coupled to mass spectrometry
- HLM
human liver microsomes
- NMR
nuclear magnetic resonance spectroscopy
- UGT
UDP‐glucuronosyltransferase
- UPLC/MS/MS
ultra‐performance liquid chromatography coupled to tandem mass spectrometry
Introduction
Exemestane is a synthetic androgen prescribed to postmenopausal women with ER+ breast cancer (Coombes et al. 2007; Paridaens et al. 2008). As an adjuvant endocrine therapy, EXE irreversibly inhibits the aromatase‐mediated production of estrogens from androgen precursors, a process known as aromatization (Hong et al. 2007). A previous pharmacokinetics study found that the maximum plasma concentration of EXE in postmenopausal women with a prior history of breast cancer ranged from 3.0 to 15.6 ng/mL following 2 weeks of oral dosing (25 mg/day) while the maximum amount of its 17β‐DHE metabolite varied 7‐fold with reported values of 0.22–1.58 ng/mL (Traina et al. 2008).
Prescriptive information states that EXE is extensively metabolized, in part by aldo‐keto reductases (AKRs) (Pfizer, 2016). A key phase I metabolic pathway of EXE is C17 reduction to form a hydroxyl moiety vulnerable to phase II conjugation and excretion (Sun et al. 2010). Recent studies independently confirmed that five purified hepatic cytosolic reductases, AKRs 1C1‐4 and carbonyl reductase 1 (CBR1), reduce EXE to the active metabolite 17β‐DHE (Platt et al. 2016). Formation of 17α‐dihydroexemestane (17α‐DHE), a novel metabolite with unknown anti‐aromatase activity (AAA), was catalyzed by AKR1C4 and CBR1 (Platt et al. 2016). A second metabolic pathway in human liver preparations is C6 exomethylene oxidation by CYP3A4 to form multiple secondary metabolites (Pfizer, 2016). The chemical structures of the C6‐oxidized metabolites, as well as detailed information regarding their capacity to inhibit aromatase are omitted from the product leaflet dispensed with EXE tablets (Pfizer, 2016).
Several studies imply that EXE hepatic metabolism may be more complex than previously believed with possibly undiscovered metabolites and coaction by additional cytochrome P450s (CYP450s) (Kamdem et al. 2011; Platt et al. 2016). Comprehensively identifying phase I EXE metabolites is warranted, because EXE derivatives may contribute to systemic estrogen blockade through aromatase inhibition. The presence of 17β‐DHE as a major metabolite in human plasma has been unequivocally confirmed in studies of postmenopausal women taking EXE (Evans et al. 1992; Traina et al. 2008). However, past attempts to identify less‐studied metabolites have been speculative due to the lack of standard reference compounds. Using GC‐MS, three peaks likely corresponding to C6‐oxidized metabolites were detected in the urine of healthy male volunteers (Cavalcanti Gde et al. 2011). Another study found six metabolites, including 17β‐DHE, in human urine following administration of radiolabeled EXE (Cocchiari et al. 1994). However, both studies of urinary EXE metabolites were hampered by a lack of comparison of physiochemical properties between the suspected metabolites and known standards. Six possible metabolite peaks were observed in human liver microsomes presented with EXE substrate (Kamdem et al. 2011). One peak was confirmed to be 17β‐DHE and another was tentatively designated as 6‐hydroxymethylandrosta‐1,4,6‐triene‐3,17‐dione (6‐HME) (Kamdem et al. 2011). The identities of the remaining four peaks could not be established (Kamdem et al. 2011).
The current study addresses methodological issues that have historically undermined phase I EXE metabolite identification. First, a reference library of C6 and C17‐modified EXE analogs was synthesized to confirm the identity of suspected metabolites observed in incubations of EXE with human liver microsomes. Secondly, a newly developed UPLC/MS/MS method eliminates the need for organic extraction to remove residual substrate prior to analysis unlike previous scintillation‐based studies of AAA (Thompson and Siiteri 1974). Instead, low levels of estrone formation are quantitated directly rather than extrapolated from tritiated water release during the aromatization of radiolabeled androstenedione. Interestingly, aromatase from human placental microsomes is used in traditional AAA screenings (Thompson and Siiteri 1974). CYP1A1 is well‐expressed in human placenta and extensively metabolized EXE in an in vitro assay using recombinant baculosome‐expressed CYP450s (Kamdem et al. 2011; Uhlén et al. 2015). Therefore, background phase I metabolism in human placental microsomes may complicate the analysis of AAA assays. However, expression analysis has shown that HEK293 are CYP450 and UDP‐glucuronosyltransferase (UGT)‐null (data not shown). To circumvent potential confounding from endogenous enzymes in placental preparations, aromatase‐overexpressing HEK293 were created in the present study to evaluate the potency of EXE analogs in impeding estrogen biosynthesis.
While it is well‐accepted that genetic differences may influence an individual's drug disposition for many pharmaceuticals, the extent to which polymorphisms in aromatase explain interindividual variation in EXE potency is unclear. Interestingly, aromatase has several common nonsynonymous variants which might contribute to variability in drug disposition by altering its affinity for EXE (Ma et al. 2005), potentially affecting EXE efficacy or toxicity risk. Consequently, we also compared the efficacy of EXE in inhibiting two allozymes, aromataseThr201Met and aromataseArg264Cys relative to the wild type enzyme.
Materials and Methods
Materials
Hangzhou DayangChem Co. (Hanzhou City, China) supplied the androgens boldenone, testosterone, and 4‐andostene‐3,17‐dione for the synthesis of EXE and its analogs. Tokyo Chemical Industry Co. (Tokyo, Japan), Thermo Fisher Scientific (Waltham, MA), and Sigma‐Aldrich (St. Louis, MO) produced all other reagents (ACS grade or higher) needed for synthesis. Steroid purification required silica columns (Yamazen Corp., Osaka, Japan) and thin‐layer chromatography plates (Bonna‐Agela Technologies Inc., Wilmington, DE). LC/MS grade methanol, acetonitrile, and formic acid was purchased from Thermo Fisher Scientific. XenoTech (Lenexa, KS) supplied pooled mixed gender human liver microsomes (Cat no. H0610, n = 50). Corning (Corning, NY) and Integrated DNA Technologies (Coralville, IA) manufactured the NADPH regeneration system and oligonucleotide primers, respectively. A QuikChange II Site‐Directed Mutagenesis Kit was purchased from Agilent (Santa Clara, CA) to produce aromatase variant overexpression vectors. The HEK293 cell line was procured from ATCC (Manassas, VA). G418, penicillin/streptomycin, fetal bovine serum, Opti‐MEM, and DMEM supplemented with 4.5 g/L glucose, 110 mg/L sodium pyruvate, and L‐glutamine was purchased from Invitrogen (Carlsbad, CA) along with an XCell electrophoresis system. Lipofectamine 2000, PVDF membranes, Pierce BCA protein assay kit, SuperSignal West Femto Maximum Sensitivity Substrate, sodium dodecyl sulfate (SDS), glycine, tris base, ammonium persulfate (APS), goat anti‐rabbit HRP‐conjugated antibody (cat. No. 31466), and tetramethylethylenediamine (TEMED) were also purchased from Thermo Fisher Scientific. Nonfat dry milk was prepared by BioRad (Hercules, CA). Sigma‐Aldrich (St. Louis, MO) supplied Ponceau staining solution, Tween 20, acrylamide/bis‐acrylamide solution, 2‐mercaptoethanol, estrone, androstenedione substrate, and estrone‐2,3,4‐13C3. Rabbit monoclonal anti‐aromatase antibody (cat. no. ab124776) was purchased from Abcam (Cambridge, MA).
Reference library synthesis
EXE and ten C6‐oxidized or C17‐reduced EXE analogs were resuspended in ethanol and stored at −80°C following synthesis at Washington State University (Spokane, WA). Previous studies provide detailed descriptions of the synthesis, purification, and NMR‐based identity verification of each compound (Buzzetti et al. 1993; Marcos‐Escribano et al. 2009; Platt et al. 2016; Vatèle 2007).
Creation of aromatase‐overexpressing HEK293
Stable overexpression of wild type aromatase in HEK293 was driven by a pcDNA3.1/V5‐His‐TOPO mammalian expression vector as previously described (Sun et al. 2010). Constitutive overexpression vectors encoding common aromatase variants Thr201Met and Arg264Cys were produced via site‐directed mutagenesis using the wild‐type plasmid as template. Variant expression vectors were amplified in BL21 grown under ampicillin selection for 16 h at 37°C. Sanger sequencing was used to confirm successful mutagenesis. Lipofectamine 2000 was used to transfect HEK293 with variant overexpression plasmids. Transfected HEK293 were grown in high‐glucose DMEM containing 700 μg/mL G418, 10% FBS, and penicillin/streptomycin for at least 3 weeks. The cells were then harvested by resuspension in PBS, lysed via 4 freeze‐thaw cycles, and centrifuged for 15 min at 13,200g at 4°C. Microsomes for each cell line were prepared from the supernatant through differential centrifugation (1 h, 34000g) in a chilled Beckman L7‐65 ultracentrifuge (Brea, CA), resuspended in PBS, and stored at −80°C. The relative expression of aromatase was quantitated in triplicate by subjecting 20 μg of protein from each overexpressing cell line to SDS‐PAGE in a 10% tris‐glycine polyacrylamide gel. Following transfer to PVDF for 90 min at 30 V, the membrane was blocked overnight at 4°C in 5% nonfat dry milk, washed for 30 min in 0.1% Tween, and probed overnight with anti‐aromatase primary antibody (1:2500). The next day, the membrane was again washed for 30 min, and probed with HRP‐conjugated goat anti‐rabbit antibody (1:7500) for 1 h at ambient temperature. Following another 30 min wash, the blot was incubated with SuperSignal West Femto Maximum Sensitivity Substrate per the manufacturer instructions and imaged on a ChemiDoc Imager (BioRad, Hercules, CA). Image J software (NIH, Bethesda, MD) was used to measure band density while Ponceau staining was used to validate even loading between lanes.
Anti‐aromatase Activity Assays
Per 50‐μL reaction in PBS (pH 7.4), 5 μmol/L androstenedione, a NADPH regeneration system (1.55 mmol/L NADP+, 3.3 mmol/L glucose‐6‐phosphate, 3.3 mmol/L MgCl2, 0.5 μL of 40 U/mL glucose‐6‐phosphate dehydrogenase), and 15 μg of microsomes from HEK293 overexpressing wild type or variant aromatase were individually incubated with varying concentrations of each steroid. The steroid concentrations used for anti‐aromatase activity assays are as follows: EXE (0.05‐15 μmol/L), 17β‐DHE (0.05–30 μmol/L), 17α‐DHE (100 μmol/L), 6α‐spirooxiranandrosta‐1,4‐diene‐3,17‐dione (0.05–30 μmol/L), 6β‐spirooxiranandrosta‐1,4‐diene‐3,17‐dione (0.05–40 μmol/L), 6α‐methylandrosta‐1,4,6‐triene‐3,17‐dione (0.5–30 μmol/L), 6α‐methylandrosta‐1,4‐diene‐3,17‐dione (0.2–30 μmol/L), 6‐hydroxymethylandrosta‐1,4,6‐triene‐3,17‐dione (2.5–300 μmol/L), 17β‐hydroxy‐6‐hydroxymethylandrosta‐1,4,6‐triene‐3‐one (100 μmol/L), 6α/β‐hydroxy‐6α/β‐hydroxymethylandrosta‐1,4‐diene‐3,17‐dione (100 μmol/L), and 6α/β,17β‐Dihydroxy‐6α/β‐hydroxymethylandrosta‐1,4‐diene‐3‐one (100 μmol/L). Organic solvent comprised <1% of the total volume of each enzymatic incubation, which proceeded at 37°C for 2 h. Reactions were terminated with 50 μl of ice cold acetonitrile and centrifuged at 4°C for 15 min at 13,200g. Supernatants were collected and spiked with 50 ng of estrone‐2,3,4‐13C3 as an internal standard. An incubation with microsomes derived from non‐transfected HEK293 was also performed to serve as a negative control. Aromatization catalyzed by wild‐type or variant aromatase was likewise monitored in the presence of vehicle rather than EXE or compounds from the reference library to reflect maximal uninhibited estrone formation. Estrone was measured using a novel 6‐min direct detection UPLC/MS/MS method on the Waters Acquity platform using m/z transitions 271.17→133.09 as a marker for estrone and 274.15→162 for estrone‐2,3,4‐13C3. Mobile phase (57% methanol in 0.1% formic acid) was infused isocratically from 0 to 4 min at a flow rate of 0.4 mL/min. The column was then washed with methanol for 1 min followed by 1 min of re‐equilibration with mobile phase. Cone and collision voltages were set at 35 V and 20 V respectively. Dwell time for both compounds was 0.1 sec. IC50 values from incubations with wild‐type aromatase were calculated for each compound in GraphPad Prism 6 (La Jolla, CA). One‐way ANOVA was used to compare the IC50 value for EXE incubated with wild type aromatase with IC50 values for EXE incubated with overexpressed aromatase allozymes.
EXE Metabolite Identification
A 50‐μl incubation containing 50 μg of HLM in PBS (pH 7.4), 400 μmol/L EXE, and an NADPH regeneration system was placed in a 37°C water bath for 4 h before termination with 50 μL of cold acetonitrile. After a 15‐min refrigerated centrifugation at 13,200g, the supernatant was examined for phase I EXE metabolites. A 10‐min UPLC method was used to separate and detect EXE and the ten other reference compounds through multiple reaction monitoring with positive mode electrospray ionization on a Waters ACQUITY UPLC/MS/MS system (Milford, MA). The 1.7 μm ACQUITY UPLC BEH C18 column (2.1 mm × 50 mm, Ireland) used for these analyses was protected by a 0.2 μm in‐line filter. The UPLC gradient conditions used have previously been described (Platt et al. 2016). The fragmentation characteristics and retention time of suspected metabolite peaks were compared to compounds from the reference library.
Results and Discussion
Wild‐type aromatase inhibition by EXE and its metabolites
A reference library of purified androgens was assayed for in vitro inhibition of wild type aromatase by monitoring estrone formation (Fig. 1). In the present study, EXE (IC50 = 0.92 ± 0.17 μmol/L) and its major metabolite 17β‐DHE (IC50 = 4.3 ± 0.56 μmol/L) were potent and moderate inhibitors of aromatase respectively. These results agree with an earlier study which found that 17β‐DHE was approximately 2.6‐fold less potent than EXE (Buzzetti et al. 1993). Manufacturer data also references the diminished potency of C6‐oxidized or C17‐reduced EXE derivatives (Pfizer, 2016). Interestingly, the epoxide 6α‐spirooxiranandrosta‐1,4‐diene‐3,17‐dione was the most potent EXE analog assayed (IC50 = 1.2 ± 0.19 μmol/L), exhibiting nearly 5‐fold more potency than its 6β stereoisomer (IC50 = 5.7 ± 1.6 μmol/L). 17α‐DHE and three additional compounds exhibited negligible aromatase inhibition with IC50 values exceeding 100 μmol/L (Fig. 1B). 6‐HME was determined to be 67‐fold less potent (IC50 = 61 ± 20 μmol/L) than EXE in the present study, a large difference in potency also observed by Buzzetti et al. (1993). The remaining androgens assayed were 4–8‐fold less potent than EXE (IC50 = 3.3–7.1 μmol/L). In keeping with the observations of Buzzetti et al. (1993), non‐epoxide C6‐oxidized metabolites exhibited minimal AAA.
Figure 1.
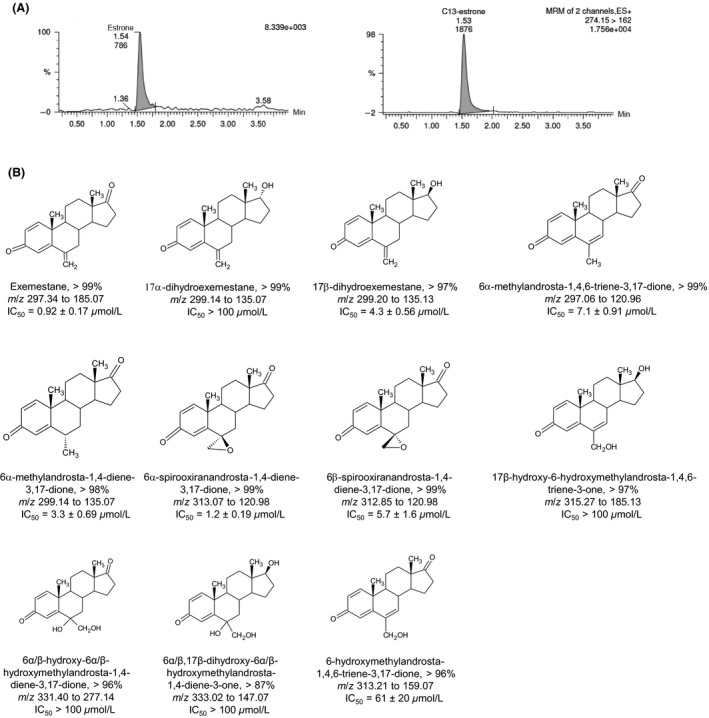
Results of anti‐aromatase activity assays using synthetic androgen inhibitors. (A) Chromatograms showing estrogen detection. Left, estrone; right, estrone‐2,3,4‐13C3 internal standard. (B) Chemical structures of species included in the synthesized reference library of EXE analogs. Percent purity is provided for each compound. Mass transitions used for UPLC/MS/MS‐based detection are listed, as well as IC 50 values for wild‐type aromatase as determined by anti‐aromatase activity assay.
Impact of nonsynonymous polymorphisms on EXE potency
IC50 values describing EXE‐mediated aromatase inhibition did not significantly differ (P = 0.71) between wild type enzyme (0.92 ± 0.17 μmol/L), aromataseThr201Met (0.86 ± 0.12 μmol/L), and aromataseArg264Cys (0.97 ± 0.09 μmol/L) in AAA assays normalized for relative aromatase expression (Fig. 2). Many aromatase polymorphisms exist, but data regarding the functional significance of variant alleles on human health is inconsistent (Baxter et al. 2001; Ma et al. 2005; Miyoshi et al. 2003). The prevalence of the Thr201Met allele is estimated as 5% in Caucasians and African Americans while the frequency of the Arg264Cys allele is 2.5% and 22.5% in Caucasian and African Americans respectively (Ma et al. 2005). One study of variant aromatase found that enzyme activity strongly correlated with expression levels in transiently transfected COS‐1 and further concluded that any differences from wild‐type in the overall activity of the Thr201Met and Arg264Cys allozymes are likely mediated by differential expression (Ma et al. 2005).
Figure 2.
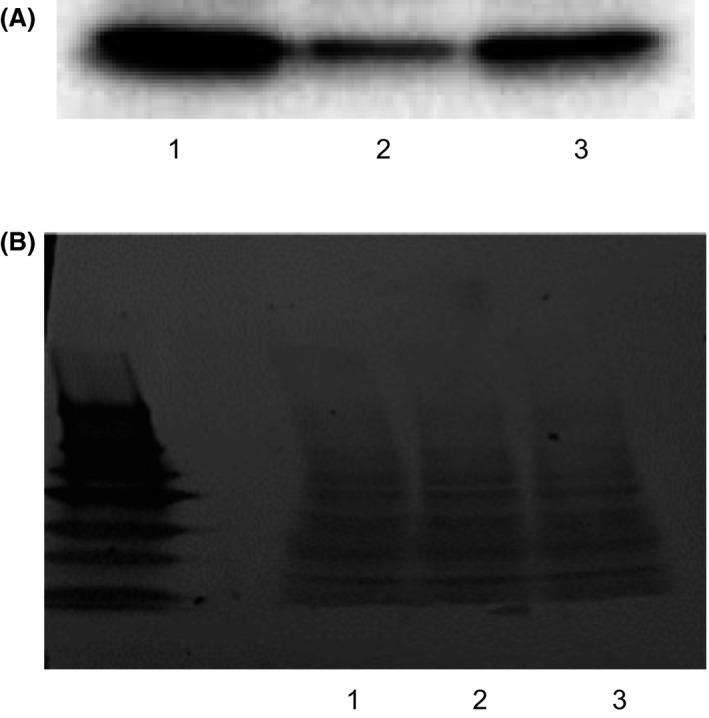
Relative quantification of overexpressed wild type and variant aromatase in HEK293 microsomes by Western blotting. (A) Lane 1, wild type aromatase; lane 2, aromataseThr201Met; lane 3, aromataseArg264Cys. Aromatase was detected using a monoclonal anti‐aromatase antibody (Abcam). (B) Ponceau total protein staining for aromatase normalization. Lanes correspond to the same three lanes described in panel A.
EXE metabolite identification
17β‐DHE, 6‐HME, 6α/β‐hydroxy‐6α/β‐hydroxy‐methylandrosta‐1,4‐diene‐3,17‐dione, and 6α/β,17β‐dihydroxy‐6α/β‐hydroxymethyl‐androsta‐1,4‐diene‐3‐one were identified in incubations of EXE with pooled human liver microsomes through comparison to reference compounds (Fig. 3). Although we found four EXE metabolites, an in vitro study of EXE metabolism by Kamdem et al. (2011) detected six peaks corresponding to putative metabolites. Our assay was not designed to identify phase II metabolites suggesting that the two additional peaks observed in the previous study may correspond to conjugated metabolites, such as the 17β‐DHE‐glucuronide produced by UGT2B17 (Sun et al. 2010). Considering their low abundance and limited capacity to inhibit aromatase in our novel AAA assay, the three C6‐oxidized metabolites detected are unlikely to contribute to the overall pharmacology of EXE in vivo. However, these results show that 17β‐DHE is not only the predominant EXE metabolite formed in human liver microsomes, but also capable of inhibiting aromatase with moderate potency suggesting that it may make clinically relevant contributions to the overall response to EXE in women with ER+ breast cancer (Platt et al. 2016).
Figure 3.
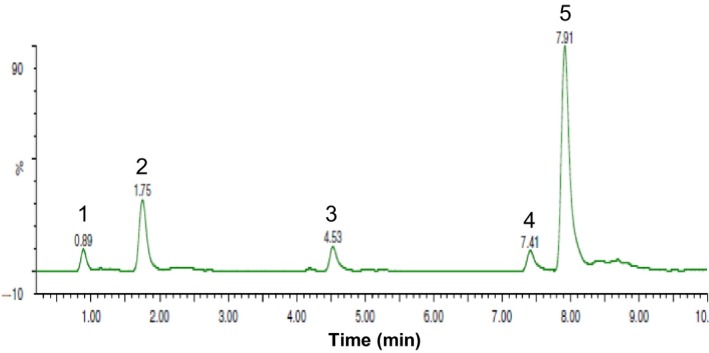
Identification of EXE metabolites in human liver microsomes. EXE metabolite profile was examined by UPLC/MS/MS after a 4 h incubation of pooled (n = 50) mixed gender human liver microsomes with EXE. Peak 1, 6α/β,17β‐dihydroxy‐6α/β‐hydroxymethylandrosta‐1,4‐diene‐3‐one; peak 2, 6α/β‐hydroxy‐6α/β‐hydroxymethylandrosta‐1,4‐diene‐3,17‐dione; peak 3, 6‐HME; peak 4, EXE; peak 5, 17β‐DHE.
Author Contribution
Participated in research design: Peterson and Lazarus. Conducted experiments: Peterson. Contributed new reagents or analytic tools: Xia, Chen, and Peterson. Performed data analysis: Peterson. Wrote or contributed to the writing of the manuscript: Peterson, Chen, Xia, and Lazarus.
Disclosure
None declared.
Acknowledgements
The thank the Washington State University Spokane campus Nuclear Magnetic Resonance Core facility for their help in analyzing newly synthesized EXE derivatives.
Peterson A., Xia Z., Chen G., Lazarus P.. Exemestane potency is unchanged by common nonsynonymous polymorphisms in CYP19A1: results of a novel anti‐aromatase activity assay examining exemestane and its derivatives, Pharma Res Per, 5(3), 2017, e00313, doi: 10.1002/prp2.313
References
- Baxter SW, Choong DY, Eccles DM, Campbell IG (2001). Polymorphic variation in CYP19 and the risk of breast cancer. Carcinogenesis 22: 347–349. [DOI] [PubMed] [Google Scholar]
- Buzzetti F, Di Salle E, Longo A, Briatico G (1993). Synthesis and aromatase inhibition by potential metabolites of exemestane (6‐methylenandrosta‐1,4‐diene‐3,17‐dione). Steroids 58: 527–532. [DOI] [PubMed] [Google Scholar]
- Cavalcanti Gde A, Garrido BC, Leal FD, Padilha MC, de la Torre X, de Aquino Neto FR (2011). Detection of new urinary exemestane metabolites by gas chromatography coupled to mass spectrometry. Steroids 76: 1010–1015. [DOI] [PubMed] [Google Scholar]
- Cocchiari G, Allievi C, Berardi A, Zugnoni P, Strolin Benedetti M, Dostert P (1994). Urinary metabolism of exemestane, a new aromatase inhibitor, in rat, dog, monkey, and human volunteers. J Endocrinol Invest 17 (Suppl. 1 to no. 3): 78. [Google Scholar]
- Coombes RC, Kilburn LS, Snowdon CF, Paridaens R, Coleman RE, Jones SE, et al. (2007). Survival and safety of exemestane versus tamoxifen after 2‐3 years' tamoxifen treatment (Intergroup Exemestane Study): a randomised controlled trial. Lancet 369: 559–570. [DOI] [PubMed] [Google Scholar]
- Evans TR, Di Salle E, Ornati G, Lassus M, Benedetti MS, Pianezzola E, et al. (1992). Phase I and endocrine study of exemestane (FCE 24304), a new aromatase inhibitor, in postmenopausal women. Cancer Res 52: 5933–5939. [PubMed] [Google Scholar]
- Hong Y, Yu B, Sherman M, Yuan YC, Zhou D, Chen S (2007). Molecular basis for the aromatization reaction and exemestane‐mediated irreversible inhibition of human aromatase. Mol Endocrinol 21: 401–414. [DOI] [PubMed] [Google Scholar]
- Kamdem LK, Flockhart DA, Desta Z (2011). In vitro cytochrome P450‐mediated metabolism of exemestane. Drug Metab Dispos 39: 98–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma CX, Adjei AA, Salavaggione OE, Coronel J, Pelleymounter L, Wang L, et al. (2005). Human aromatase: gene resequencing and functional genomics. Cancer Res 65: 11071–11082. [DOI] [PubMed] [Google Scholar]
- Marcos‐Escribano A, Bermejo FA, Bonde‐Larsen AL, Retuerto JI , Sierra IH (2009). 1,2‐Dehydrogenation of steroidal 6‐methylen derivatives. Tetrahedron 65: 7587–7590. [Google Scholar]
- Miyoshi Y, Ando A, Hasegawa S, Ishitobi M, Yamamura J, Irahara N, et al. (2003). Association of genetic polymorphisms in CYP19 and CYP1A1 with the oestrogen receptor‐positive breast cancer risk. Eur J Cancer 39: 2531–2537. [DOI] [PubMed] [Google Scholar]
- Paridaens RJ, Dirix LY, Beex LV, Nooij M, Cameron DA, Cufer T, et al. (2008). Phase III study comparing exemestane with tamoxifen as first‐line hormonal treatment of metastatic breast cancer in postmenopausal women: the European Organisation for Research and Treatment of Cancer Breast Cancer Cooperative Group. J Clin Oncol 26: 4883–4890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfizer (2016) Aromasin Exemestane Tablets.
- Platt A, Xia Z, Liu Y, Chen G, Lazarus P (2016). Impact of nonsynonymous single nucleotide polymorphisms on in‐vitro metabolism of exemestane by hepatic cytosolic reductases. Pharmacogenet Genomics 26: 370–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun D, Chen G, Dellinger RW, Sharma AK, Lazarus P (2010). Characterization of 17‐dihydroexemestane glucuronidation: potential role of the UGT2B17 deletion in exemestane pharmacogenetics. Pharmacogenet Genomics 20: 575–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson EA Jr, Siiteri PK (1974). Utilization of oxygen and reduced nicotinamide adenine dinucleotide phosphate by human placental microsomes during aromatization of androstenedione. J Biol Chem 249: 5364–5372. [PubMed] [Google Scholar]
- Traina TA, Poggesi I, Robson M, Asnis A, Duncan BA, Heerdt A, et al. (2008). Pharmacokinetics and tolerability of exemestane in combination with raloxifene in postmenopausal women with a history of breast cancer. Breast Cancer Res Treat 111: 377–388. [DOI] [PubMed] [Google Scholar]
- Uhlén M, Fagerberg L, Hallström B, Lindskog C, Oksvold P, Mardinoglu A, et al. (2015). The human protein atlas. http://www.proteinatlas.org (last accessed April 2017).
- Vatèle J (2007). 2‐(Prenyloxymethyl)benzoyl (POMB) group: a new temporary protecting group removable by intramolecular cyclization. Tetrahedron 63: 10921–10929. [Google Scholar]