

Abstract
Exemestane (EXE) is an endocrine therapy commonly used by postmenopausal women with hormone‐responsive breast cancer due to its potency in inhibiting aromatase‐catalyzed estrogen synthesis. Preliminary in vitro studies sought to identify phase I EXE metabolites and hepatic cytochrome P450s (CYP450s) that participate in EXE biotransformation. Phase I metabolites were identified by incubating EXE with HEK293‐overexpressed CYP450s. CYP450s 1A2, 2C8, 2C9, 2C19, 2D6, 3A4, and 3A5 produce 17β‐dihydroexemestane (17β‐DHE), an active major metabolite, as well as two inactive metabolites. 17β‐DHE formation in pooled human liver microsomes subjected to isoform‐specific CYP450 inhibition was also monitored using tandem mass spectrometry. 17β‐DHE production in human liver microsomes was unaffected by isoform‐specific inhibition of CYP450s 2A6, 2B6, and 2E1 but decreased 12–39% following inhibition of drug‐metabolizing enzymes from CYP450 subfamilies 1A, 2C, 2D, and 3A. These results suggest that redundancy exists in the EXE metabolic pathway with multiple hepatic CYP450s catalyzing 17β‐DHE formation in vitro. To further expand the knowledge of phase I EXE metabolism, the impact of CYP450 genetic variation on 17β‐DHE formation was assessed via enzyme kinetic parameters. Affinity for EXE substrate and enzyme catalytic velocity were calculated for hepatic wild‐type CYP450s and their common nonsynonymous variants by monitoring the reduction of EXE to 17β‐DHE. Several functional polymorphisms in xenobiotic‐metabolizing CYP450s 1A2, 2C8, 2C9, and 2D6 resulted in deviant enzymatic activity relative to wild‐type enzyme. Thus, it is possible that functional polymorphisms in EXE‐metabolizing CYP450s contribute to inter‐individual variability in patient outcomes by mediating overall exposure to the drug and its active metabolite, 17β‐DHE.
Keywords: Aromatase inhibitor, breast cancer, CYP450, dihydroexemestane, exemestane, pharmacogenetics, polymorphism
Abbreviations
- 17α‐DHE
17α‐dihydroexemestane
- 17β‐DHE
17β‐dihydroexemestane
- 6‐HME
6‐Hydroxymethylandrosta‐1,4,6‐triene‐3,17‐dione
- AKR
aldo‐keto reductasecarbonyl reductase 1
- CBR1
CYP450, cytochrome P450
- EXE
exemestane
- HLM
human liver microsomes
- UGT2B17
uridine diphosphate glucose glucuronosyltransferase family 2 member B17 (UGT2B17)
- UPLC/MS/MS
ultra‐performance liquid chromatography coupled to tandem mass spectrometry
Introduction
EXE is a third‐generation steroidal aromatase inhibitor used in the treatment and prevention of breast cancer in postmenopausal women (Geisler et al. 1998; Goss et al. 2011). At present, a lack of consensus exists regarding the exact mechanism by which EXE irreversibly inhibits the enzyme (Brueggemeier 2002; Lombardi 2002; Ghosh et al. 2009, 2012; Viciano and Marti 2016). However, its efficacy in suppressing aromatase‐mediated synthesis of estrogens from adrenal‐supplied androgens (aromatization) is well established. Geisler et al. (1998) have previously reported mean suppression of total body aromatization of approximately 98% in postmenopausal women with advanced breast cancer treated with 25 mg EXE daily for 6–8 weeks. As the principal source of estrogen after menopause, aromatase maintains estrogen excess in hormone receptor‐positive breast cancers (Labrie 2015; To et al. 2015). Deviant estrogen signaling, in turn, promotes tumorigenesis and metastasis of hormone‐responsive mammary tumors (Sommer and Fuqua 2001; Murphy and Watson 2002; Platet et al. 2004). EXE‐induced estrogen suppression is therefore an effective treatment modality used to delay disease progression.
Although several studies have examined EXE metabolic pathways, relatively little is known about its phase I metabolites. 6‐hydroxymethylandrosta‐1,4,6‐triene‐3,17‐dione (6‐HME) has tentatively been identified as an EXE derivative in incubations with human liver microsomes (Kamdem et al. 2011). However, it is unlikely to make meaningful contributions to systemic estrogen deprivation as its potency in inhibiting aromatase is about 21‐67‐fold less potent than its parent drug (Buzzetti et al. 1993; Peterson et al. 2017). Another derivative, 17β‐DHE, is the major metabolite found in the plasma of individuals taking EXE. Aside from its abundance in clinical samples, 17β‐DHE is noteworthy for its ability to inhibit aromatase (IC50 = 2.3 ± 0.83 μmol/L), which by one estimate is equipotent to that of EXE (IC50 = 1.4 ± 0.42 μmol/L) (Sun et al. 2010). The concentration of 17β‐DHE relative to EXE in human plasma may vary fivefold between individuals although the mechanisms underlying this observation have not been fully resolved (Evans et al. 1992).
Several cytosolic reductase enzymes including carbonyl reductase 1 (CBR1) and members of the aldo‐keto reductase 1C (AKR1C) subfamily have been shown to be active in 17β‐DHE formation (Evans et al. 1992; Corona et al. 2009; Platt et al. 2016). Previous studies have suggested that EXE reduction may be augmented by several wild‐type CYP450s, as well (Kamdem et al. 2011). In a small panel (n = 15) of human liver microsomes incubated with EXE, the velocities of 17β‐DHE and 6‐HME formation correlated with the catalytic activity of CYP450s 1A2/4A11 and 2B6/3A, respectively (Kamdem et al. 2011).
Xenobiotic‐metabolizing members of the CYP450 family participate in the hepatic metabolism of hundreds of distinct substrates comprising approximately 75% of all marketed pharmaceuticals and catalyze many diverse reactions as reviewed by Guengerich (2001, 2008), including carbon hydroxylation, dealkylation, and epoxidation. With over 2000 known genetic variants, CYP450s are highly polymorphic enzymes (Preissner et al. 2013). A subset of variant CYP450 isoforms are associated with clinically significant alterations in drug metabolism and disease susceptibility as reviewed by Preissner et al. (2013).
Generally speaking, it is well established that nonsynonymous polymorphisms in genes involved in drug absorption, distribution, metabolism, and excretion (ADME), such as the CYP450s, can lead to genotype‐dependent variability in clinical responses for certain drugs (Kalow et al. 1998). Regarding EXE pharmacogenetics, a recent in vitro enzyme kinetics study showed that the hepatic cytosolic enzymes CBR1 and AKR1C1‐4 reduce EXE with several nonsynonymous variants in AKR1C3 and AKR1C4 significantly altering overall 17β‐DHE metabolism (Platt et al. 2016). While examining phase II EXE metabolism, Sun et al. (2010) found a significant correlation between uridine diphosphate glucose glucuronosyltransferase family 2 member B17 (UGT2B17) genotype in human liver microsomes (HLM) and levels of 17β‐DHE conjugation with glucuronic acid to form a water soluble glucuronide (glucuronidation) for urinary excretion. Overall, glucuronidation (V max/K M) of 17β‐DHE in the HLM panel was 36‐fold lower in UGT2B17‐null homozygotes (*2/*2) compared to wild‐type homozygotes (*1/*1) (Sun et al. 2010). These observations bolster the possibility that genetically determined differences in metabolic capacity contribute to variability in clinical responses in EXE‐treated women by influencing 17β‐DHE production and UGT‐driven clearance. Nonetheless, pharmacogenetic studies describing the impact of variant CYP450s on EXE metabolism are conspicuously absent from the literature. The present study expands upon the currently limited understanding of phase I EXE metabolism by identifying metabolites produced by xenobiotic‐metabolizing hepatic CYP450s. To our knowledge, this is also the first in vitro study to examine the impact of common nonsynonymous variant CYP450s on formation of the active metabolite, 17β‐DHE.
Materials and Methods
Chemicals and materials
Testosterone, boldenone, and 4‐andostene‐3,17‐dione were purchased from Hangzhou DayangChem Co., China. All other reagents used for synthesis of EXE and its phase I metabolites were ACS grade or higher and purchased from Thermo Fisher Scientific (Waltham, MA), Tokyo Chemical Industry Co. (Tokyo, Japan) or Sigma‐Aldrich (St. Louis, MO). Thin‐layer chromatography plates and silica columns used to purify the synthesized steroids were purchased from Bonna‐Agela Technologies Inc. (Wilmington, DE) and Yamazen Corp. (Osaka, Japan), respectively. Variant CYP450 overexpression vectors were made using a QuikChange II Site‐Directed Mutagenesis Kit from Agilent (Santa Clara, CA), as well as oligonucleotides manufactured by Integrated DNA Technologies (Coralville, IA). Negative control baculosomes, SuperScript II First‐Strand Synthesis System for RT‐PCR, cell culture medium, fetal bovine serum, penicillin/streptomycin, and G418 were acquired from Invitrogen (Carlsbad, CA). Choice‐Taq polymerase was purchased from Denville Scientific (Holliston, MA). LC/MS grade organic solvents, tris base, glycine, tetramethylethylenediamine (TEMED), ammonium persulfate (APS), sodium dodecyl sulfate (SDS), SuperSignal West Femto Maximum Sensitivity Substrate, Lipofectamine 2000, oligo(dT) primer, Pierce BCA protein assay kit, PVDF membranes, and monoclonal HRP‐conjugated V5 epitope antibody (catalog # MA5‐15253‐HRP) were procured from Thermo Fisher Scientific (Waltham, MA). Nonfat dry milk used as a blocking agent for Western blotting was prepared by BioRad (Hercules, CA, US). The nicotinamide adenine dinucleotide phosphate (NADPH) regeneration system included in kinetic assays was purchased from Corning (Corning, NY). Ampicillin, dithiothreitol (DTT), Tween 20, and acrylamide/bis‐acrylamide solution were acquired from Sigma‐Aldrich (St. Louis, MO). Pooled mixed gender liver microsomes from 50 human donors were obtained from XenoTech (Lenexa, Kansas, US). The ethnic composition of the pooled hepatic microsomes was 84% Caucasian, 8% Hispanic, 6% African American, and 2% Asian. (‐)‐N‐3‐benzylphenobarbital (NBP) is a product of Cypex Ltd (Dundee, United Kingdom), whereas all other compounds used for CYP450 isoform‐specific inhibition were obtained from Sigma‐Aldrich.
Synthesis of EXE and phase I EXE metabolites
EXE and its phase I metabolites were synthesized at Washington State University using previously published protocols (Buzzetti et al. 1993; Vatèle 2007; Marcos‐Escribano et al. 2009; Varela et al. 2014; Platt et al. 2016). A Yamazen AI‐580s flash chromatography system was utilized in the purification of each compound following synthesis. Purity was estimated for each EXE derivative by photodiode array (PDA) spectrum (210–400 nm) using the Acquity I Class UPLC platform from Waters (Milford, MA). With the kind permission of Gonzaga University's Department of Chemistry (Spokane, WA), the identity of each steroid was authenticated using nuclear magnetic resonance spectra generated by a Bruker AV300 instrument (Billerica, MA). High‐resolution mass spectra were also obtained on a Waters Xevo G2‐S QTof Mass Spectrometer to confirm the parity of the experimental and theoretic mass‐to‐charge values for each compound prior to resuspension in ethanol and storage at −80°C.
Identification of nonsynonymous polymorphisms
Common nonsynonymous polymorphisms for CYP450s 1A2, 2C8, 2C9, 2C19, 2D6, 3A4, and 3A5 were identified using the National Center for Biotechnology Information (NCBI) Variation Viewer (2015). Search filters were set to simultaneously screen dbVar and dbSNP for any missense or nonsense variants caused by single nucleotide variations, deletions, insertions, or frameshifts. Low incidence variants were excluded from the current study and defined as those occurring with a minor allele frequency (MAF) of <1% according to the GO‐ESP dataset or 1000 Genomes Project. Inter‐ethnic differences in the occurrence of common CYP450 variants were examined using NCBI's 1000 Genomes Browser. These results are summarized in Data S1.
Creation of CYP450‐overexpressing HEK293 cell lines
Oligo(dT)20 was used to prime reverse transcription of pooled hepatic total RNA from five human donors (Penn State Cancer Institute Biorepository). The resultant first‐strand cDNA was further amplified using CYP450 isoform‐specific primers and Taq polymerase. cDNA encoding wild‐type CYP450s 1A2, 2C8, 2C9, 2C19, 2D6, 3A4, or 3A5 was introduced into the pcDNA3.1/V5‐His‐TOPO expression vector for stable overexpression in mammalian cells. CYP2D6*2 and CYP2C19*1B cDNA were also amplified from pooled human liver RNA. Additional constitutive overexpression vectors encoding nonsynonymous variants with MAF > 0.01 were produced for CYP450s 1A2, 2C8, 2C9, 2C19, and 3A4 via site‐directed mutagenesis (SDM) using wild‐type plasmid as template. Two common CYP2D6 haplotypes were likewise derived from the CYP2D6*2 overexpression vector through SDM. Oligonucleotide pairs used to amplify wild‐type CYP450 cDNA or prime mutagenesis are detailed in Data S2 and Data S3, respectively.
Each expression vector was transformed into chemically competent BL21. Transformants were then grown overnight on ampicillin selection plates at 37°C. Sanger sequencing was used for sequence confirmation prior to transfecting HEK293 with overexpression plasmids using Lipofectamine 2000 per the manufacturer instructions. Transfected HEK293 were grown for at least 3 weeks under 700 μg/mL G418 selective pressure in DMEM supplemented with 4.5 g/L glucose, l‐glutamine, 110 mg/L sodium pyruvate, 10% FBS, and penicillin/streptomycin. Data S4 describes the quantification of the relative CYP450 content of each overexpressing cell line.
Metabolite identification
CYP450‐derived phase I EXE metabolites were identified in 45‐min incubations performed at 37°C. Each 50‐μL reaction contained 2.5 mmol/L EXE substrate, 3 μL NADPH regeneration system, and 20 μg of microsomal protein from a wild‐type CYP450‐overexpressing HEK293 cell line in phosphate‐buffered saline (PBS), pH 7.4. Similar incubations were conducted to assess background activity of endogenous metabolizing enzymes in non‐transfected HEK293, as well as commercially available negative control baculosomes. These incubations included 100 μM EXE, 50 μg of microsomes, or 50 μg of baculosomes in addition to PBS, pH7.4, and an NADPH regeneration system. Enzymatic incubations were terminated with 50 μL of ice‐cold acetonitrile before centrifugation at 13,200g for 15 min at 4°C. The resulting supernatant was analyzed by ultra‐pressure liquid chromatography paired with tandem mass spectrometry (UPLC/MS/MS) on a Waters ACQUITY platform configured with a 0.2 μm inline filter preceding a 1.7 μm ACQUITY UPLC BEH C18 column (2.1 mm × 100 mm, Ireland). The UPLC gradient used to separate EXE and its phase I metabolites has been described previously (Platt et al. 2016). 800 L/h nitrogen gas was used for drying while desolvation temperature was set at 500°C. A targeted UPLC/multiple reaction monitoring (MRM) method was performed in positive mode using electrospray ionization to monitor mass transitions for EXE (m/z 297.34 → 185.07), 17α‐DHE (m/z 299.14 → 135.07), 17β‐DHE (m/z 299.20 → 135.13), and 6‐HME (m/z 312.89 → 158.98). 0.01 sec dwell time was optimal for all four compounds, whereas 25 V of collision energy was used for EXE and 6‐HME. 20 V of collision energy was used for 17α‐ and 17β‐DHE. An additional non‐targeted screening for phase I EXE metabolites was performed using UPLC/MS to detect positively charged molecular ions with m/z ranging from 100 to 450. The identity of all metabolites observed was verified by comparing the observed retention times and m/z with those of purified standards.
Enzyme kinetic assays
Wild‐type and variant CYP450‐mediated 17β‐DHE production was measured in 45‐min incubations at 37°C in PBS, pH 7.4 with varying concentrations of EXE (25–2500 μmol/L). Each reaction included 20 μg of microsomal protein from CYP450‐overexpressing HEK293, as well as an NADPH regeneration system. The chosen incubation length and protein concentration fell within the linear range of EXE reduction velocity curves for the seven wild‐type CYP450s assayed (data not shown). Cold acetonitrile (50 μl) was used to terminate each reaction. After centrifugation for 15 min at 4°C at 13,200g, 17β‐DHE formation was monitored in the supernatant according to a previously established UPLC/MS/MS method and quantitated against a standard curve constructed of known 17β‐DHE concentrations (Platt et al. 2016).
Isoform‐specific CYP450 inhibition
Reduction of EXE to 17β‐DHE in the presence of isoform‐specific CYP450 inhibitors was monitored in pooled human liver microsomes (HLM). Furafylline (1 μmol/L) was used to inhibit CYP1A2, whereas 0.5 μmol/L tranylcypromine (TCP) and 10 μmol/L thioTEPA inhibited CYPs 2A6 and 2B6, respectively. Other compounds used to systematically inhibit hepatic CYP450s included 0.5 μmol/L montelukast (CYP2C8 inhibitor), 1 μmol/L sulfaphenazole (CYP2C9 inhibitor), 0.5 μmol/L NBP (CYP2C19), 1 μmol/L quinidine (CYP2D6 inhibitor), 5 μmol/L clomethiazole (CYP2E1 inhibitor), and 1 μmol/L ketoconazole (CYP3A inhibitor). Initial dosages were selected from the literature and then further titrated to determine the lowest dose producing maximum isoform‐specific inhibition (Bourrie et al. 1996; Administration USFaD, 2006; Khojasteh et al. 2011). Each 50‐μL inhibition reaction contained 12.5 μg of pooled HLM, one isoform‐specific CYP450 inhibitor dissolved in ethanol, and 10 μmol/L EXE in PBS, pH 7.4. Following a 15‐min pre‐incubation at 37°C, the reactions were initiated by the addition of NADPH regeneration system and incubated for an additional 15 min before termination with 50 μL of cold acetonitrile. After refrigerated centrifugation for 15 min at 13,200g, supernatants were collected and dried at ambient temperature in a Jouan RC10.22 vacuum concentrator. Samples were resuspended in 20 μL of water/acetonitrile (1:1) and analyzed by UPLC/MS/MS using the aforementioned method. A negative control reaction was run in parallel and received ethanol vehicle rather than inhibitor. Organic solvent constituted less than 1% of each incubation.
Statistical analyses
K M and relative V max values were calculated in GraphPad Prism 6 according to the Michaelis–Menten equation (GraphPad Software, Inc., San Diego, California). V max is expressed as pmol min−1 mg−1 and was normalized to total protein content to account for differences in CYP450 expression between cell lines as determined by Western blot analysis. Two‐sided unpaired t‐tests were used to compare the wild‐type catalytic activity of CYP450s 1A2 and 3A4 to their respective variants. Wild‐type CYP450s 2C8, 2C9, 2C19, and 2D6 were compared to their nonsynonymous variants using one‐way ANOVA supplemented with Dunnett's multiple comparison test. In all instances, the threshold for statistical significance was set at a two‐tailed P < 0.05. The percent change in 17β‐DHE formation associated with each inhibitor in pooled HLM was likewise calculated in GraphPad Prism 6. An uninhibited reaction receiving vehicle was considered maximum catalytic activity for the purposes of comparison. All experimental results represent triplicate assays performed independently.
Results
Identification of phase I EXE metabolites
As shown in a representative Western blot analysis, significant levels of expression of individual CYPs were obtained in all HEK293 overexpressing cell lines (Fig. 1, panel A). Ponceau staining was used as a loading control to normalize the CYP450 content of each cell line to the amount of total protein loaded per lane (Fig. 1, panel B). The major metabolite formed in individual 45‐min incubations of EXE with CYP2C9 (Fig. 2, panel E) as well as overexpressed wild‐type CYP450s 1A2, 2C8, 2C19, 2D6, 3A4, and 3A5 (results not shown) was 17β‐DHE. Low levels of 6‐HME and 17α‐DHE formation were observed for all CYPs studied. In all cases, formation of the 17α‐dihydroexemestane (17α‐DHE) and 6‐hydroxymethylandrosta‐1,4,6‐triene‐3,17‐dione (6‐HME) metabolites exceeded the minimum threshold of detection. No EXE metabolite formation was observed in incubations using microsomes from the parent HEK293 cell line (Fig. 2, panel F), but interestingly, significant 17β‐DHE formation was observed using negative control baculosomes (Fig. 2, panel G). Secondary metabolite formation was not observed when overexpressing CYP450 microsomes were presented with 6‐HME or either stereoisomer of 17‐DHE as substrate (results not shown).
Figure 1.
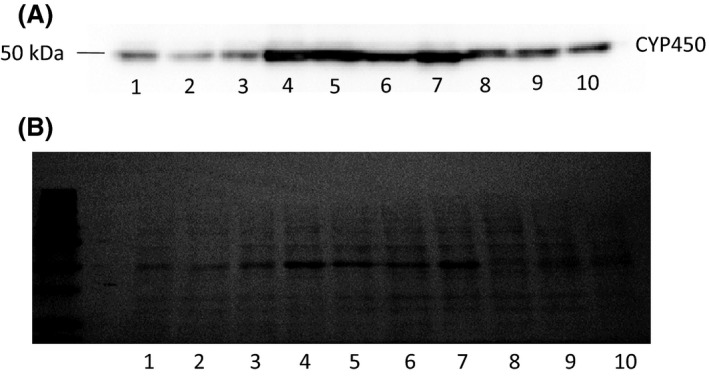
Relative quantification of overexpressed CYP450s in HEK293 microsomes. Panel (A), Detection of V5‐tagged CYP450s by Western blotting. Panel (B), Ponceau total protein staining for CYP450 normalization. Lane 1, CYP2C19; lane 2, CYP2C19Glu92Asp; lane 3, CYP2C19Ile331Val; lane 4, CYP2D6; lane 5, CYP2D6Arg296Cys, Ser486Thr; lane 6, CYP2D6Pro34Ser, Ser486Thr; lane 7, CYP2D6Thr107Ile, Arg296Cys, Ser486Thr; lane 8, CYP3A4; lane 9, CYP3A4Arg162Gln; lane 10, CYP3A5.
Figure 2.
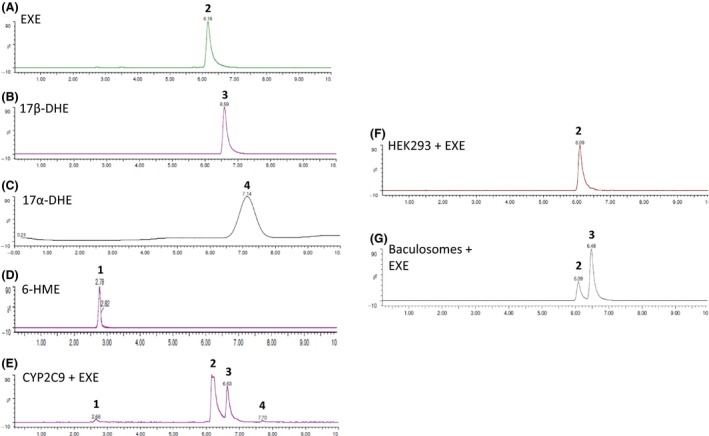
Identification of EXE metabolites. Panel (A), EXE (6‐methyleneandrosta‐1,4‐diene‐3,17‐dione) standard; panel (B), 17β‐DHE (17β‐hydroxy‐6‐methyleneandrosta‐1,4‐dien‐3‐one) standard; panel (C), 17α‐DHE (17α‐hydroxy‐6‐methyleneandrosta‐1,4‐dien‐3‐one) standard; panel (D), 6‐HME (6‐hydroxymethylandrosta‐1,4,6‐triene‐3,17‐dione) standard; panel (E), EXE metabolite profile after incubating CYP2C9‐overexpressing HEK293 microsomes with EXE; panel (F), EXE metabolite profile after incubating microsomes from the parent HEK293 cell line (no CYP450 overexpression) with EXE; panel (G), EXE metabolite profile after incubating negative control commercial baculosomes (no CYP450 overexpression) with EXE. Incubations were performed for 45 min at 37°C with 100 μM exemestane and 20 μg of CYP450‐overexpressing HEK293 microsomes, 50 μg of non‐CYP450‐overexpressing HEK293 microsomes or 50 μg of baculosomes. Peak 1, 6‐HME; peak 2, exemestane; peak 3, 17β‐DHE; peak 4, 17α‐DHE.
Kinetic analysis of 17β‐DHE formation
Representative kinetic curves of 17β‐DHE formation for each wild‐type hepatic CYP450 are shown in Figure 3. Kinetic assays using overexpressed CYP450 protein suggest that CYP2D6 exhibited the highest affinity for EXE (K M = 0.57 ± 0.03 mmol/L) followed by CYP2C8 (K M = 0.66 ± 0.10 mmol/L), CYP3A5 (K M = 0.69 ± 0.18 mmol/L), CYP1A2 (K M = 0.74 ± 0.5 mmol/L), and CYP3A4 (K M = 0.83 ± 0.16 mmol/L) (Table 1). CYP450s 2C9 and 2C19 had approximately the same affinity for EXE with K M values of 0.96 ± 0.22 mmol/L and 0.92 ± 0.18 mmol/L, respectively. CYP2C9 exhibited the slowest rate of EXE reduction, whereas CYP2C8 reduced EXE approximately fivefold faster (V max = 26 ± 0.6 vs. 128 ± 4 pmol min−1 mg −1). CYP1A2 catalyzed 17β‐DHE production at a rate of 61 ± 17 pmol min−1 mg −1 followed by CYP2C19 (51 ± 8 pmol min−1 mg −1) and CYP2D6 (49 ± 3 pmol min−1 mg −1). Catalytic velocities for CYP450s 3A4 and 3A5 were not significantly different (39 ± 7 and 36 ± 4 pmol min−1 mg −1). Intrinsic clearance (V max/K M) was calculated for each wild‐type enzyme as an indication of its overall catalytic activity against EXE. CYP2C8 exhibited the highest intrinsic clearance value (194 nL min−1 mg−1) followed by CYP2D6 (86 nL min−1 mg−1), CYP1A2 (82 nL min−1 mg−1), CYP2C19 (55 nL min−1 mg−1), CYP3A5 (52 nL min−1 mg−1), and CYP3A4 (47 nL min−1 mg−1). In addition to having the slowest rate of EXE reduction, CYP2C9 also exhibited the lowest overall catalytic activity against EXE substrate (27 nL min−1 mg−1).
Figure 3.
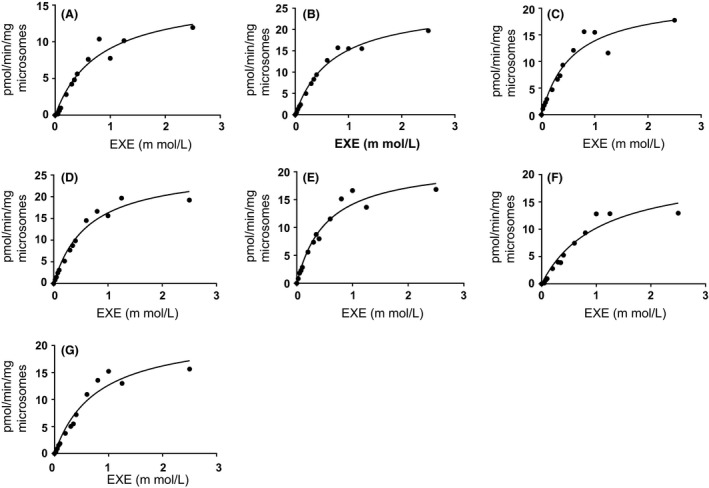
Representative kinetics curves for the reduction of exemestane to 17β‐DHE. Panel (A), CYP1A2; panel (B), CYP2C8; panel (C), CYP2C9; panel (D), CYP2C19; panel (E), CYP2D6; panel (F), CYP3A4; and panel (G), CYP3A5.
Table 1.
Kinetic analysis of wild‐type and variant CYP450s active against exemestane
| Wild‐type CYP450 or variant | NCBI dbSNP identifier | Allele | K M (mmol/L) | V max (pmol min−1 mg−1)a | CLINT (nl min−1 mg−1) |
|---|---|---|---|---|---|
| CYP1A2 | CYP1A2*1A | 0.74 ± 0.5 | 61 ± 17 | 82 | |
| CYP1A2Ser298Arg | rs17861157 | 0.59 ± 0.2b | 80 ± 7 | 136 | |
| CYP2C8 | CYP2C8*1A | 0.66 ± 0.10 | 128 ± 4 | 194 | |
| CYP2C8Ile269Phe | rs11572103 | CYP2C8*2 | 0.86 ± 0.8 | 280 ± 17b | 326 |
| CYP2C8Arg139Lys, Lys399Arg | rs11572080, rs10509681 | CYP2C8*3 | 0.77 ± 0.13 | 144 ± 7 | 187 |
| CYP2C8Ile264Met | rs1058930 | CYP2C8*4 | 0.65 ± 0.3 | 218 ± 11b | 335 |
| CYP2C9 | CYP2C9*1A | 0.96 ± 0.22 | 26 ± 0.6 | 27 | |
| CYP2C9Arg144Cys | rs1799853 | CYP2C9*2 | 0.95 ± 0.4 | 58 ± 4 | 61 |
| CYP2C9Ile359Leu | rs1057910 | CYP2C9*3 | 1.32 ± 0.16 | 36 ± 4 | 27 |
| CYP2C9Arg150His | rs7900194 | CYP2C9*8 | 1.14 ± 0.25 | 73 ± 13b | 64 |
| CYP2C9His251Arg | rs2256871 | CYP2C9*9 | 1.07 ± 0.14 | 116 ± 13b | 108 |
| CYP2C19 | CYP2C19*1A | 0.92 ± 0.18 | 51 ± 8 | 55 | |
| CYP2C19Ile331Val | rs3758581 | CYP2C19*1B | 0.92 ± 0.13 | 59 ± 16 | 64 |
| CYP2C19Glu92Asp | rs17878459 | 0.70 ± 0.10 | 59 ± 7 | 84 | |
| CYP2D6 | CYP2D6*1A | 0.57 ± 0.03 | 49 ± 3 | 86 | |
| CYP2D6Arg296Cys, Ser486Thr | rs16947, rs1135840 | CYP2D6*2 | 0.95 ± 0.10b | 23 ± 0.6b | 24 |
| CYP2D6Pro34Ser, Ser486Thr | rs1065852, rs1135840 | CYP2D6*10 | 1.04 ± 0.01b | 39 ± 6 | 38 |
| CYP2D6Thr107Ile, Arg296Cys, Ser486Thr | rs28371706, rs16947, rs1135840 | CYP2D6*17 | 0.92 ± 0.15 | 40 ± 2 | 43 |
| CYP3A4 | CYP3A4*1A | 0.83 ± 0.16 | 39 ± 7 | 47 | |
| CYP3A4Arg162Gln | rs4986907 | CYP3A4*15A | 1.04 ± 0.09 | 46 ± 6 | 44 |
| CYP3A5 | CYP3A5*1A | 0.69 ± 0.18 | 36 ± 4 | 52 |
All V max values were normalized to reflect the relative CYP450 content of microsomes assayed.
Denotes that a variant exhibited statistically significant deviations (P < 0.05) from the activity of its respective wild‐type CYP450.
Impact of functional polymorphisms on EXE reduction
In all, 23 nonsynonymous polymorphisms with MAF > 0.01 were detected using the NCBI Variation Viewer to search for common variants in hepatic CYP450s involved in xenobiotic metabolism. Four polymorphisms of interest were identified for CYP2C9. Two variants were gleaned for CYP2C19, whereas a single variant was reported for both CYP450s 1A2 and 3A4. No variants matching the search criteria were listed for CYP3A5. Variation Viewer search filters returned four common single nucleotide polymorphisms (SNPs) in CYP2C8. In the present study, two of the CYP2C8 polymorphisms were investigated as a single haplotype (CYP2C8*3). In all, 11 nonsynonymous polymorphisms occur in CYP2D6 at > 1%. Although CYP2D6 is notoriously polymorphic, only a handful of common haplotypes are associated with clinically relevant alterations in drug metabolism (Zhou 2009). For this reason, only four of the SNPs identified in Variation Viewer were included in kinetic assays as the 2D6*2, 2D6*10, and 2D6*17 haplotypes.
Although V max was comparable between wild‐type CYP1A2 and CYP1A2Ser298Arg, the variant enzyme exhibited significantly increased affinity for EXE with K M values of 0.74 ± 0.5 and 0.59 ± 0.2 mmol/L, respectively. No notable differences were observed in affinity between wild‐type CYP450s 2C8, 2C9, 2C19, or 3A4 and their respective variants (Table 1). However, CYP2D6*2 (K M = 0.95 ±0.10 mmol/L) and CYP2D6*10 (K M = 1.04 ± 0.01 mmol/L) were both associated with decreased affinity relative to wild‐type CYP2D6 (K M = 0.57 ± 0.03 mmol/L). The CYP2C8*2 and CYP2C8*4 polymorphisms were associated with 2.2 and 1.7‐fold increases in velocity of EXE reduction compared to wild‐type. CYP2C9*8 and CYP2C9*9 variants were likewise associated with increased rates of 17β‐DHE formation (V max = 73 ± 13 and 116 ± 13 versus 26 ± 0.6 pmol min−1 mg−1 for wild‐type). The V max for EXE reduction by CYP2D6*2 (23 ± 0.6 pmol min−1 mg −1) is 53% lower than that of wild‐type CYP2D6 (49 ± 3 pmol min−1 mg −1). V max values were comparable between CYP2C19 and its variants CYP2C19Ile331Val and CYP2C19Glu92Asp. No significant difference in V max was observed between wild‐type CYP3A4 and CYP3A4Arg162Gln (V max = 39 ± 7 and 46 ± 6 pmol min−1 mg −1, respectively).
Isoform‐specific CYP450 inhibition
In incubations of EXE with pooled mixed gender HLMs, inhibition of CYP3A isoforms with ketoconazole resulted in a 39% decrease in 17β‐DHE production (Fig. 4). Isoform‐specific inhibition of CYP450s 2C8, 2C9, and 2C19 resulted in 27%, 12%, and 22% decreases in EXE reduction, respectively. Inclusion of TCP to inhibit CYP2A6 did not impact 17β‐DHE production to any appreciable extent (100.5 ± 5.7% control activity). Similar results were obtained when either thioTEPA or clomethiazole were included to chemically inhibit CYP2B6 (112.5 ± 8.3% control activity) or CYP2E1 (97.3 ± 5.3% control activity), respectively. Inhibition of CYP2D6 decreased EXE reduction by 27%, whereas furafylline‐induced CYP1A2 inhibition reduced formation of the active 17β‐DHE metabolite by 19%.
Figure 4.
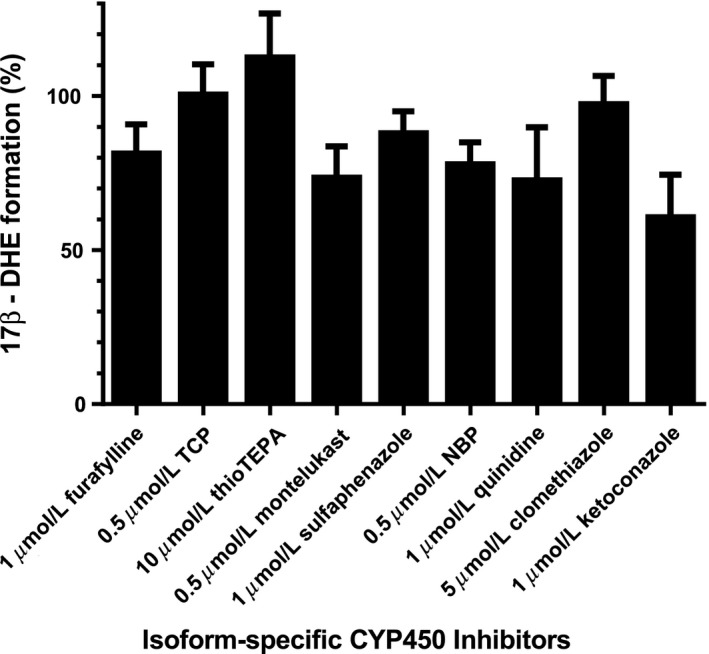
Isoform‐specific chemical inhibition of CYP450‐mediated exemestane metabolism in pooled HLM (see Materials and Methods for details). Data represent means of triplicate independent assays monitoring 17β‐DHE formation.
Discussion
Previous studies examining phase I exemestane metabolism strongly suggest that reduction of EXE at the C17 carbonyl moiety to produce 17β‐DHE represents a major metabolic pathway for this commonly prescribed aromatase inhibitor (Evans et al. 1992; Lonning 1998; Mareck et al. 2006; Traina et al. 2008). Manufacturer‐supplied information regarding EXE metabolism is minimal and attributes metabolism in human liver preparations to CYP3A4 and members of the aldo‐keto reductase superfamily (Pfizer, 2016). CYP3A4 is further identified as the principal enzyme catalyzing EXE oxidative metabolism with subsequent formation of multiple metabolites (Pfizer, 2016). At present, a comprehensive roster of all metabolites observed in these studies has not been disclosed. A recent study has clarified the role of five cytosolic reductases in hepatic EXE metabolism in vitro (Platt et al. 2016), and multiple hepatic CYP450s may contribute to EXE metabolism as work by Kamdem et al. (2011) suggests. The present study examines the involvement of CYP450s in EXE metabolism in vitro and highlights the potential impact of common nonsynonymous CYP450 polymorphisms on 17β‐DHE formation.
EXE reduction to form 17β‐DHE was detected in incubations with HEK293‐overexpressed CYP450s 1A2, 2C8, 2C9, 2C19, 2D6, 3A4, and 3A5. These results are in agreement with previous studies indicating that EXE reduction at C17 is a major metabolic pathway and confirm that CYP3A4 catalyzes the production of multiple phase I metabolites (Evans et al. 1992; Lonning 1998; Mareck et al. 2006; Traina et al. 2008; Pfizer, 2016). Due to overlapping substrate specificity, many currently marketed pharmaceuticals are metabolized by multiple CYP450s (Preissner et al. 2013). The data presented herein bolster previous observations regarding the capacity of multiple hepatic CYP450s to engage in in vitro oxidative metabolism of EXE (Kamdem et al. 2011; Platt et al. 2016).
The relative contribution of each CYP450 to EXE metabolism in vivo is likely dependent upon differential expression in human liver, as well as their overall catalytic activity against EXE (V max/K M). CYPs 3A4 and 3A5 exhibited intermediate intrinsic clearance values in the present study (47 and 52 nL min−1 mg−1, respectively), but the literature indicates that CYP3A isoforms are well expressed in the liver comprising 30% of total CYP450 content and accounting for nearly 55% of xenobiotic metabolism (Chang and Kam 1999). The overall catalytic activity against EXE for CYP2D6 (86 nL min−1 mg−1) was second only to that of CYP2C8 (194 nL min−1 mg−1) in kinetic assays. CYP2D6 is estimated to account for 30% of drug metabolism despite constituting only 2% of total hepatic CYP450 content (Chang and Kam 1999). CYP1A2, while moderately active against EXE in vitro (CLINT = 82 nL min−1 mg−1), accounts for only 2% of overall CYP450‐mediated xenobiotic metabolism in human liver (Chang and Kam 1999). CLINT values for EXE ranged from 27 to 194 nL min−1 mg−1 for CYP450 2C isoforms, which make up 20% of total hepatic CYP450 proteins but contribute only 10% of total hepatic drug metabolism (Chang and Kam 1999). Therefore, CYP3A isoforms and CYP2D6 are potentially key enzymes in phase I EXE metabolism in vivo as they are well expressed hepatically, making significant contributions to overall xenobiotic metabolism, and display high activity against EXE in vitro.
To our knowledge, only one study to date had previously examined EXE metabolism by CYP450s. In incubations with HLMs, Kamdem et al. (2011) reported two primary EXE metabolites, 17‐DHE and 6‐HME. In incubations of EXE with CYP450‐overexpressing HEK293 microsomes, we observed the formation of three EXE metabolites including 17α‐ and 17β‐DHE, as well as 6‐HME (Fig. 2). Formation of several metabolites is unsurprising as CYP450s catalyze diverse reactions (Guengerich 2001). Significantly decreased 17β‐DHE formation (39% reduction) in the presence of the isoform‐specific inhibitor ketoconazole suggests that CYP3As are the major hepatic CYP450s responsible for EXE reduction in human liver microsomes (Fig. 4). This result is in accordance with the manufacturer's limited description of the drug's phase I metabolism (Pfizer, 2016). Isoform‐specific inhibition of major hepatic CYP450s 1A2, 2C8, 2C9, 2C19, and 2D6 also decreased 17β‐DHE formation by 12–27%, which suggests that multiple CYP isoforms in addition to CYP3As may be relevant to EXE metabolism. In contrast, a previously published CYP450 inhibition experiment found that treating pooled HLM with ketoconazole strongly inhibits 6‐HME formation but had no significant effect on EXE reduction to 17β‐DHE (Kamdem et al. 2011). Furthermore, no effect on 17β‐DHE formation was observed in HLM treated with any other CYP inhibitor (Kamdem et al. 2011). The same in vitro study attributed 6‐HME formation predominantly to CYP3As and CYP2B6 (Kamdem et al. 2011). The present study cannot comment on the relative contribution of specific CYP450s to EXE oxidation to form 6‐HME. Kinetic parameters were not collected for 6‐HME formation in the present study since it exhibits low anti‐aromatase activity in vitro (Buzzetti et al. 1993; Peterson et al. 2017). In addition, while 6‐HME was a major metabolite in a previous study (Kamdem et al. 2011), it was a minor metabolite formed by the seven hepatic CYP450s tested in the present study. Discrepancies between the two studies may reflect experimental differences. It is also feasible that significant decreases in 17β‐DHE formation were undetectable in HLM subject to isoform‐specific inhibition in the prior study due to redundancy in EXE clearance by multiple hepatic CYP450s. Of interest, Kamdem et al. (2011) found a significant correlation between 17β‐DHE formation and the rate of activity by CYP450s 1A2 and 4A11 in a panel of HLM. CYP1A2 inhibition with furafylline decreased 17β‐DHE formation by approximately 19% in pooled HLM in the present study and may indeed contribute to hepatic EXE metabolism to a previously unappreciated extent.
Unfortunately, the enzyme kinetics results presented herein are not comparable to those of the previous in vitro enzyme kinetics study of EXE metabolism, which concluded that baculosome‐expressed CYP1A1, CYP2A6, and CYP4A11 are most active in 17‐DHE production (Kamdem et al. 2011). In lieu of using a commercially available baculosome system, HEK293 cell lines constitutively overexpressing CYP450s were created for kinetics assays because HEK293 are devoid of many drug‐metabolizing enzymes, including the CYP450s. In addition, significant reduction of EXE to 17β‐DHE was observed in negative control baculosomes (see Fig. 2). These results indicate that endogenous CYPs or active reductases are present in baculosome preparations, which is likely to confound the interpretation of kinetic assays. In contrast, background reduction in HEK293 microsomal fractions was undetectable by UPLC/MS/MS in the present study (see Fig. 2). Although overexpressed CYP2A6 was not directly tested in overexpressed HEK293 cells for activity against EXE in the present study, the involvement of CYP2A6 is questionable as it has been shown to preferentially metabolize small substrates (Zhou 2016). Furthermore, its inhibition did not appreciably decrease 17β‐DHE generated by HLM incubated with EXE (100.5 ± 5.7% control activity) in the present study. CYP1A1 expression, in turn, is low in human liver diminishing the likelihood that it is a key participant in phase I EXE metabolism (Stiborova et al. 2005).
Xenobiotic‐metabolizing CYP450s are highly polymorphic (Preissner et al. 2013). Multiple nonsynonymous variants often exist within a single isoform at varying levels of penetrance in the human population. CYP2D6, for instance, can harbor > 100 distinct polymorphisms several of which are associated with altered drug metabolism and increased risk for life‐threatening adverse reactions (Dalen et al. 1997; Wan et al. 2001; Gasche et al. 2004; Raimundo et al. 2004; Preissner et al. 2013). In our enzyme kinetics assays, three variant CYP450s exhibited altered affinity for EXE, whereas five deviated appreciably from their wild‐type counterparts with respect to maximum velocity of EXE reduction.
Maximum velocity of EXE reduction to form 17β‐DHE is similar between wild‐type CYP1A2 and CYP1A2Ser298Arg (see Table 1). However, CYP1A2Ser298Arg had approximately 25% higher affinity, which contributed to a 66% increase in intrinsic clearance of EXE compared to the wild‐type isoform (CLINT = 136 vs. 82 nL min−1 mg−1). Despite its high incidence in African populations (MAF = 0.0893), a three‐dimensional structure examining conformational changes in CYP1A2 arising from arginine substitution is currently unavailable (1000 Genomes Browser, 2016; Watanabe et al. 2016). It is known, however, that amino acid residue 298 is located within a loop distal to the CYP1A2 active site (Watanabe et al. 2016). In silico analyses by Watanabe et al. (2016) predict that when the adjacent glycine at residue 299 is mutated to serine, flexibility increases near Val487 in the C‐terminal loop. Although this particular mutation does not impact overall CYP1A2 enzymatic activity as assessed by 7‐ethoxyresorufin O‐deethylation, it is indicative that polymorphisms can alter structural flexibility in loop regions (Ito et al. 2015; Watanabe et al. 2016). Structural flexibility, in turn, may strongly impact ligand‐binding induced conformational changes, substrate recognition, and overall CYP450 catalytic activity as several studies suggest (Skopalik et al. 2008; Zhang et al. 2011; Kobayashi et al. 2014; de Waal et al. 2014). Similar structural analyses are needed to determine whether CYP1A2Ser298Arg exhibits deviant flexibility, possibly underlying differences observed in kinetic parameters against EXE.
CYP2C8Ile269Phe (*2), CYP2C8Arg139Lys,Lys399Arg (*3), and CYP2C8Ile264Met (*4) are comparable to wild‐type CYP2C8 in affinity for EXE substrate in vitro. Kaspera et al. (2010) likewise found only minor differences in apparent K M values between recombinant wild‐type CYP2C8 and its *2, *3, and *4 variants while monitoring oxidation of cerivastatin to form its O‐desmethyl‐ (M‐1) and 6‐hydroxyl‐ (M‐23) cerivastatin metabolites. Although E. coli‐expressed CYP2C8*2 yielded individual V max values similar to wild‐type CYP2C8 for M‐1 and M‐23 formation, a 53% increase in the sum of cerivastatin metabolite clearance was noted (Kaspera et al. 2010). In the present study, CYP2C8*2 produced 17β‐DHE 2.2‐fold faster than wild‐type contributing to a 68% increase in overall EXE clearance. CYP2C8*2 carriers reported abdominal pain more frequently than wild‐type homozygotes in a small study of West African malaria patients taking amodiaquine, suggesting possible clinical relevance for the polymorphism (Parikh et al. 2007). The CYP2C8*3 genotype is likewise of considerable interest and has been examined extensively with regard to NSAIDS, antidiabetic agents, and 3‐hydroxy‐3‐methyl‐glutaryl‐coenzyme A (HMG‐CoA) reductase inhibitors (Kirchheiner et al. 2003; Martinez et al. 2005; Kirchheiner et al. 2006; Lopez‐Rodriguez et al. 2008; Aquilante et al. 2008; Garcia‐Martin et al. 2004). In clinical studies, CYP2C8*3 is associated with increased drug metabolism for several substrates as evidenced by significantly decreased plasma concentrations of rosiglitazone, pioglitazone, and repaglinide (Niemi et al. 2003, 2005; Kirchheiner et al. 2006; Aquilante et al. 2008; Tornio et al. 2008). In contrast, decreased ibuprofen metabolism has also been noted, implying that the kinetic properties of CYP2C8*3 may be substrate‐dependent (Garcia‐Martin et al. 2004; Martinez et al. 2005). With regard to EXE, our in vitro results indicate that the CYP2C8*3 polymorphism does not alter the drug's overall intrinsic clearance. CYP2C8*4, on the other hand, was associated with a 1.7‐fold increase in EXE clearance due to elevated V max relative to wild‐type (218 ± 11 vs. 128 ± 4 nL min−1 mg−1). Kaspera et al. (2010) estimated that recombinant CYP2C8*4 increased the combined clearance of the M‐1 and M‐23 cerivastatin metabolites by approximately 2.5‐fold compared to wild‐type. Although CYP2C8*4 has demonstrated increased catalytic activity against both cerivastatin and EXE in vitro, human livers with the *4 genotype express lower levels of CYP2C8 protein (Kaspera et al. 2010; Naraharisetti et al. 2010). Differences in hepatic expression of variant CYPs may negate or exacerbate the overall metabolic effect of deviant catalytic activity observed in the present study. Thus, additional in vivo studies are needed to gauge what impact, if any, CYP2C8 polymorphisms have on clinical outcomes in postmenopausal breast cancer patients taking EXE.
Kinetic parameters for EXE metabolism by CYP2C9Arg144Cys (*2) and CYP2C9Ile359Leu (*3) were similar to those of wild‐type CYP2C9. The CYP2C9*2 and *3 polymorphisms are relatively common (MAF > 0.01) in South Asian, European, and Hispanic populations (see Data S1) and are associated with impaired warfarin metabolism (Aithal et al. 1999; Lindh et al. 2009). Although the *2 variant is associated with a poor metabolizer phenotype for certain substrates, its impact on catalytic activity is not entirely clear (Aithal et al. 1999; van der Weide et al. 2001; Rosemary et al. 2006; Lindh et al. 2009; Mosher et al. 2009). Several previous studies of the CYP2C9*2 variant are conflicting with fluvastatin and celecoxib metabolism unaffected, whereas losartan and phenytoin clearance significantly decreased relative to wild type (Aynacioglu et al. 1999; Sandberg et al. 2002; Yasar et al. 2002; Kirchheiner et al. 2003). Impaired warfarin metabolism is likewise associated with CYP2C9*3 necessitating the need for genotype‐based dose reductions in clinical settings (Aithal et al. 1999; Lindh et al. 2009). Work by Wei et al. (2007) suggests that CYP2C9*2 and *3 exhibit altered metabolism due to decreased coupling efficiency in the P450 catalytic cycle. Another study posits that the substrate binding pocket of CYP2C9*3 is enlarged relative to wild type, resulting in reduced enzymatic activity against warfarin (Sano et al. 2010). CYP2C9Arg150His (*8) and CYP2C9His251Arg (*9) polymorphic protein reduced EXE to 17β‐DHE 2.8‐ and 4.5‐fold faster than wild‐type CYP2C9, respectively. The CYP2C9*8 allele also increased clearance of the antidiabetic tolbutamide in vitro (Blaisdell et al. 2004). In vivo, the CYP2C9*8 allele is associated with decreased phenytoin metabolism due to strong linkage disequilibrium with SNPs in the gene promoter that downregulates the expression (Allabi et al. 2005; Cavallari et al. 2013). In silico analyses by Matimba et al. (2009) predicted reduced activity of CYP2C9*9 compared to wild‐type. However, a significant correlation between the CYP2C9*9 allele and phenytoin metabolism was not detected in African epilepsy patients (Matimba et al. 2009). Published discrepancies in variant CYP2C9 activity are common and likely arise from variations in experimental procedures between laboratories (Jarrar and Lee 2014).
CYP2D6 is believed to be the most polymorphic of the major drug‐metabolizing hepatic CYP450s (Preissner et al. 2013). A 72% decrease in EXE clearance was observed for CYP2D6Arg296Cys, Ser486Thr (*2) relative to wild‐type CYP2D6 due to significant decreases in affinity and 17β‐DHE formation rate. Sakuyama et al. (2008) reported a similar two‐fold decrease in substrate affinity in transiently expressed CYP2D6*2 while monitoring bufuralol 1′‐hydroxylation. Although analysis of the crystal structure of CYP2D6 suggests that residue 296 may be involved in substrate recognition, a study of CYP2D6*2A in Europeans found no association with altered drug metabolism (Marez et al. 1997; Rowland et al. 2006). In the present study, HEK293‐expressed CYP2D6Pro34Ser, Ser486Thr (*10) increased the K M value for EXE substrate by 82% causing a 56% decrease in clearance compared to CYP2D6*1. CYP2D6*10 is highly prevalent in Asians and has previously been associated with decreased catalytic activity in vitro (Johansson et al. 1994; Bradford 2002; Ji et al. 2002; Ishiguro et al. 2004; Shen et al. 2007; Sakuyama et al. 2008). Shen et al. (2007) estimated that CYP2D6*10 protein decreases catalytic activity in a substrate‐dependent manner with intrinsic clearance of probe substrates reduced to 4–28% that of wild‐type protein. Diminished catalytic activity by the *10 variant is attributed to increased enzyme instability as a result of serine substitution in a proline‐rich region near the N‐terminus (Yokota et al. 1993; Sakuyama et al. 2008). CYP2D6Thr107Ile, Arg296Cys, Ser486Thr (*17) is common in individuals of African heritage and is likewise considered a reduced function allele (Aklillu et al. 1996; Masimirembwa et al. 1996; Griese et al. 1999; Wennerholm et al. 1999; Zhou 2009). CYP2D6*17 exhibits considerable variability in catalytic activity against various substrates (Zhou 2009). Studies using recombinant CYP2D6*17 protein have reported increased metabolism of certain substrates, such as haloperidol, but decreased metabolism of others, including codeine (Oscarson et al. 1997; Bogni et al. 2005). The Thr107Ile substitution (rs28371706) is believed to alter a highly conserved region of CYP2D6 involved in substrate recognition, perhaps explaining the substrate‐dependent effects previously reported (Gotoh 1992; Koymans et al. 1993; Hasler et al. 1994). It is interesting to note that recombinant CYP2D7*17 protein yielded a 61% decrease in EXE affinity relative to wild type although this observation did not reach statistical significance.
Kinetic parameters measuring substrate affinity (K M) and production of the major active metabolite 17β‐DHE (V max) indicate that EXE metabolism by common allelic variants of CYP2C19 and CYP3A4 is comparable to that of their respective wild‐type CYP450s. The neutral effect of the CYP2C19Ile331Val (*1B) polymorphism on EXE reduction confirms prior observations of equivalent catalytic activity for the CYP2C19*1A and *1B alleles (Blaisdell et al. 2002). Although the CYP2C19Glu92Asp SNP (rs17878459) did not directly impact 17β‐DHE formation, it cosegregates with approximately 20% of CYP2C19*2 poor metabolizing alleles in Caucasians (Ibeanu et al. 1998). Found predominantly in Africans, the CYP3A4Arg162Gln (*15A) polymorphism does not appreciably impact EXE metabolism. However, kinetic studies of the variant enzyme with additional substrates are needed to confirm its overall effect on CYP3A4 activity.
The present in vitro study augments existing pharmaceutical knowledge by examining the role of hepatic CYP450s in the metabolism of EXE, a widely used endocrine therapy for hormone‐responsive breast cancer. Qualitative enzymatic incubations with EXE confirm that multiple hepatic monoxygenases from CYP450 families 1, 2, and 3 catalyze the production of 6‐HME, 17α‐DHE, as well as the active metabolite 17β‐DHE. Earlier studies suggested that 17β‐DHE may partially determine overall drug exposure by acting as an androgen agonist, contributing to estrogen blockade through aromatase inhibition, and by serving as a gateway to phase II conjugation and excretion (Ariazi et al. 2007; Sun et al. 2010). Thus, any genetic factors influencing 17β‐DHE formation or clearance may contribute to inter‐individual variation in the overall therapeutic efficacy of EXE by altering a major metabolic pathway. This possibility is bolstered by the observation that three of the variant CYP450s included in this study had altered affinity for EXE substrate leading to differential 17β‐DHE production while five variants had deviant catalytic rates of EXE reduction. To our knowledge, this is the first study to report the impact of common nonsynonymous polymorphisms in CYP450s on EXE reduction to its 17β‐DHE metabolite. A previous in vitro study demonstrated the capacity of genetic variation to alter EXE metabolism by the cytosolic ketosteroid reductases CBR1 and AKR1C1‐4 (Platt et al. 2016). Although differences in experimental procedures preclude a direct comparison of kinetic parameters between the two studies, it appears that both cytosolic and microsomal phase I enzymes contribute to in vitro EXE metabolism. An important limitation of the overexpression model used in this study was the inability to assess the metabolic impact of copy number variations or polymorphisms in noncoding promoter regions. Additional studies are needed to determine whether multiple hepatic CYP450s contribute to overall EXE metabolism in vivo and whether common genetic variants in phase I enzymes are associated with differential metabolite production or clinical outcomes in EXE‐treated breast cancer patients.
Disclosure
There are no conflicts of interest.
Supporting information
Data S1. Digital Content 1.doc.
Data S2. Digital Content 2.doc.
Data S3. Digital Content 3.doc.
Data S4. Digital Content 4.doc.
Acknowledgements
The authors wish to thank the Mass Spectrometry Core facility at the Washington State University Spokane campus for their help in verifying the identity of the androgens synthesized for this study.
Peterson A. , Xia Z. , Chen G. , Lazarus P.. In vitro metabolism of exemestane by hepatic cytochrome P450s: impact of nonsynonymous polymorphisms on formation of the active metabolite 17β‐dihydroexemestane. Pharma Res Per, 5(3), 2017, e00314, doi: 10.1002/prp2.314
References
- Aithal GP, Day CP, Kesteven PJ, Daly AK (1999). Association of polymorphisms in the cytochrome P450 CYP2C9 with warfarin dose requirement and risk of bleeding complications. Lancet 353: 717–719. [DOI] [PubMed] [Google Scholar]
- Aklillu E, Persson I, Bertilsson L, Johansson I, Rodrigues F, Ingelman‐Sundberg M (1996). Frequent distribution of ultrarapid metabolizers of debrisoquine in an ethiopian population carrying duplicated and multiduplicated functional CYP2D6 alleles. J Pharmacol Exp Ther 278: 441–446. [PubMed] [Google Scholar]
- Allabi AC, Gala JL, Horsmans Y (2005). CYP2C9, CYP2C19, ABCB1 (MDR1) genetic polymorphisms and phenytoin metabolism in a Black Beninese population. Pharmacogenet Genomics 15: 779–786. [DOI] [PubMed] [Google Scholar]
- Aquilante CL, Bushman LR, Knutsen SD, Burt LE, Rome LC, Kosmiski LA (2008). Influence of SLCO1B1 and CYP2C8 gene polymorphisms on rosiglitazone pharmacokinetics in healthy volunteers. Hum Genomics 3: 7–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ariazi EA, Leitao A, Oprea TI, Chen B, Louis T, Bertucci AM, et al. (2007). Exemestane's 17‐hydroxylated metabolite exerts biological effects as an androgen. Mol Cancer Ther 6: 2817–2827. [DOI] [PubMed] [Google Scholar]
- Aynacioglu AS, Brockmoller J, Bauer S, Sachse C, Guzelbey P, Ongen Z, et al. (1999). Frequency of cytochrome P450 CYP2C9 variants in a Turkish population and functional relevance for phenytoin. Br J Clin Pharmacol 48: 409–415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blaisdell J, Mohrenweiser H, Jackson J, Ferguson S, Coulter S, Chanas B, et al. (2002). Identification and functional characterization of new potentially defective alleles of human CYP2C19. Pharmacogenetics 12: 703–711. [DOI] [PubMed] [Google Scholar]
- Blaisdell J, Jorge‐Nebert LF, Coulter S, Ferguson SS, Lee SJ, Chanas B, et al. (2004). Discovery of new potentially defective alleles of human CYP2C9. Pharmacogenetics 14: 527–537. [DOI] [PubMed] [Google Scholar]
- Bogni A, Monshouwer M, Moscone A, Hidestrand M, Ingelman‐Sundberg M, Hartung T, et al. (2005). Substrate specific metabolism by polymorphic cytochrome P450 2D6 alleles. Toxicol In Vitro 19: 621–629. [DOI] [PubMed] [Google Scholar]
- Bourrie M, Meunier V, Berger Y, Fabre G (1996). Cytochrome P450 isoform inhibitors as a tool for the investigation of metabolic reactions catalyzed by human liver microsomes. J Pharmacol Exp Ther 277: 321–332. [PubMed] [Google Scholar]
- Bradford LD (2002). CYP2D6 allele frequency in European Caucasians, Asians. Africans and their descendants. Pharmacogenomics 3: 229–243. [DOI] [PubMed] [Google Scholar]
- Brueggemeier RW (2002). Overview of the pharmacology of the aromatase inactivator exemestane. Breast Cancer Res Treat 74: 177–185. [DOI] [PubMed] [Google Scholar]
- Buzzetti F, Di Salle E, Longo A, Briatico G (1993). Synthesis and aromatase inhibition by potential metabolites of exemestane (6‐methylenandrosta‐1,4‐diene‐3,17‐dione). Steroids 58: 527–532. [DOI] [PubMed] [Google Scholar]
- Cavallari LH, Vaynshteyn D, Freeman KM, Wang D, Perera MA, Takahashi H, et al. (2013). CYP2C9 promoter region single‐nucleotide polymorphisms linked to the R150H polymorphism are functional suggesting their role in CYP2C9*8‐mediated effects. Pharmacogenet Genomics 23: 228–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang GW, Kam PC (1999). The physiological and pharmacological roles of cytochrome P450 isoenzymes. Anaesthesia 54: 42–50. [DOI] [PubMed] [Google Scholar]
- Corona G, Elia C, Casetta B, Diana C, Rosalen S, Bari M, et al. (2009). A liquid chromatography‐tandem mass spectrometry method for the simultaneous determination of exemestane and its metabolite 17‐dihydroexemestane in human plasma. J Mass Spectrom 44: 920–928. [DOI] [PubMed] [Google Scholar]
- Dalen P, Frengell C, Dahl ML, Sjoqvist F (1997). Quick onset of severe abdominal pain after codeine in an ultrarapid metabolizer of debrisoquine. Ther Drug Monit 19: 543–544. [DOI] [PubMed] [Google Scholar]
- Evans TR, Di Salle E, Ornati G, Lassus M, Benedetti MS, Pianezzola E, et al. (1992). Phase I and endocrine study of exemestane (FCE 24304), a new aromatase inhibitor, in postmenopausal women. Cancer Res 52: 5933–5939. [PubMed] [Google Scholar]
- Garcia‐Martin E, Martinez C, Tabares B, Frias J, Agundez JA (2004). Interindividual variability in ibuprofen pharmacokinetics is related to interaction of cytochrome P450 2C8 and 2C9 amino acid polymorphisms. Clin Pharmacol Ther 76: 119–127. [DOI] [PubMed] [Google Scholar]
- Gasche Y, Daali Y, Fathi M, Chiappe A, Cottini S, Dayer P, et al. (2004). Codeine intoxication associated with ultrarapid CYP2D6 metabolism. N Engl J Med 351: 2827–2831. [DOI] [PubMed] [Google Scholar]
- Geisler J, King N, Anker G, Ornati G, Di Salle E, Lonning PE, et al. (1998). In vivo inhibition of aromatization by exemestane, a novel irreversible aromatase inhibitor, in postmenopausal breast cancer patients. Clin Cancer Res 4: 2089–2093. [PubMed] [Google Scholar]
- 1000 Genomes Browser . 2016[Internet]. Available at: http://www.ncbi.nlm.nih.gov/variation/tools/1000genomes/(accessed 25 August 2016).
- Ghosh D, Griswold J, Erman M, Pangborn W (2009). Structural basis for androgen specificity and oestrogen synthesis in human aromatase. Nature 457: 219–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh D, Lo J, Morton D, Valette D, Xi J, Griswold J, et al. (2012). Novel aromatase inhibitors by structure‐guided design. J Med Chem 55: 8464–8476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goss PE, Ingle JN, Ales‐Martinez JE, Cheung AM, Chlebowski RT, Wactawski‐Wende J, et al. (2011). Exemestane for breast‐cancer prevention in postmenopausal women. N Engl J Med 364: 2381–2391. [DOI] [PubMed] [Google Scholar]
- Gotoh O (1992). Substrate recognition sites in cytochrome P450 family 2 (CYP2) proteins inferred from comparative analyses of amino acid and coding nucleotide sequences. J Biol Chem 267: 83–90. [PubMed] [Google Scholar]
- Griese EU, Asante‐Poku S, Ofori‐Adjei D, Mikus G, Eichelbaum M (1999). Analysis of the CYP2D6 gene mutations and their consequences for enzyme function in a West African population. Pharmacogenetics 9: 715–723. [PubMed] [Google Scholar]
- Guengerich FP (2001). Common and uncommon cytochrome P450 reactions related to metabolism and chemical toxicity. Chem Res Toxicol 14: 611–650. [DOI] [PubMed] [Google Scholar]
- Guengerich FP (2008). Cytochrome p450 and chemical toxicology. Chem Res Toxicol 21: 70–83. [DOI] [PubMed] [Google Scholar]
- Hasler JA, Harlow GR, Szklarz GD, John GH, Kedzie KM, Burnett VL, et al. (1994). Site‐directed mutagenesis of putative substrate recognition sites in cytochrome P450 2B11: importance of amino acid residues 114, 290, and 363 for substrate specificity. Mol Pharmacol 46: 338–345. [PubMed] [Google Scholar]
- Ibeanu GC, Blaisdell J, Ghanayem BI, Beyeler C, Benhamou S, Bouchardy C, et al. (1998). An additional defective allele, CYP2C19*5, contributes to the S‐mephenytoin poor metabolizer phenotype in Caucasians. Pharmacogenetics 8: 129–135. [DOI] [PubMed] [Google Scholar]
- Ishiguro A, Kubota T, Sasaki H, Iga T (2004). A long PCR assay to distinguish CYP2D6*5 and a novel CYP2D6 mutant allele associated with an 11‐kb EcoRI haplotype. Clin Chim Acta 347(1–2): 217–221. [DOI] [PubMed] [Google Scholar]
- Ito M, Katono Y, Oda A, Hirasawa N, Hiratsuka M (2015). Functional characterization of 20 allelic variants of CYP1A2. Drug Metab Pharmacokinet 30: 247–252. [DOI] [PubMed] [Google Scholar]
- Jarrar YB, Lee SJ (2014). Molecular functionality of CYP2C9 polymorphisms and their influence on drug therapy. Drug Metabol Drug Interact 29: 211–220. [DOI] [PubMed] [Google Scholar]
- Ji L, Pan S, Marti‐Jaun J, Hanseler E, Rentsch K, Hersberger M (2002). Single‐step assays to analyze CYP2D6 gene polymorphisms in Asians: allele frequencies and a novel *14B allele in mainland Chinese. Clin Chem 48: 983–988. [PubMed] [Google Scholar]
- Johansson I, Oscarson M, Yue QY, Bertilsson L, Sjoqvist F, Ingelman‐Sundberg M (1994). Genetic analysis of the Chinese cytochrome P4502D locus: characterization of variant CYP2D6 genes present in subjects with diminished capacity for debrisoquine hydroxylation. Mol Pharmacol 46: 452–459. [PubMed] [Google Scholar]
- Kalow W, Tang BK, Endrenyi L (1998). Hypothesis: comparisons of inter‐ and intra‐individual variations can substitute for twin studies in drug research. Pharmacogenetics 8: 283–289. [DOI] [PubMed] [Google Scholar]
- Kamdem LK, Flockhart DA, Desta Z (2011). In vitro cytochrome P450‐mediated metabolism of exemestane. Drug Metab Dispos 39: 98–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaspera R, Naraharisetti SB, Tamraz B, Sahele T, Cheesman MJ, Kwok PY, et al. (2010). Cerivastatin in vitro metabolism by CYP2C8 variants found in patients experiencing rhabdomyolysis. Pharmacogenet Genomics 20: 619–629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khojasteh SC, Prabhu S, Kenny JR, Halladay JS, Lu AY (2011). Chemical inhibitors of cytochrome P450 isoforms in human liver microsomes: a re‐evaluation of P450 isoform selectivity. Eur J Drug Metab Pharmacokinet 36: 1–16. [DOI] [PubMed] [Google Scholar]
- Kirchheiner J, Kudlicz D, Meisel C, Bauer S, Meineke I, Roots I, et al. (2003). Influence of CYP2C9 polymorphisms on the pharmacokinetics and cholesterol‐lowering activity of (‐)‐3S,5R‐fluvastatin and (+)‐3R,5S‐fluvastatin in healthy volunteers. Clin Pharmacol Ther 74: 186–194. [DOI] [PubMed] [Google Scholar]
- Kirchheiner J, Thomas S, Bauer S, Tomalik‐Scharte D, Hering U, Doroshyenko O, et al. (2006). Pharmacokinetics and pharmacodynamics of rosiglitazone in relation to CYP2C8 genotype. Clin Pharmacol Ther 80: 657–667. [DOI] [PubMed] [Google Scholar]
- Kobayashi K, Takahashi O, Hiratsuka M, Yamaotsu N, Hirono S, Watanabe Y, et al. (2014). Evaluation of influence of single nucleotide polymorphisms in cytochrome P450 2B6 on substrate recognition using computational docking and molecular dynamics simulation. PLoS ONE 9: e96789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koymans LM, Vermeulen NP, Baarslag A (1993). Donne‐Op den Kelder GM. A preliminary 3D model for cytochrome P450 2D6 constructed by homology model building. J Comput Aided Mol Des 7: 281–289. [DOI] [PubMed] [Google Scholar]
- Labrie F (2015). All sex steroids are made intracellularly in peripheral tissues by the mechanisms of intracrinology after menopause. J Steroid Biochem Mol Biol 145: 133–138. [DOI] [PubMed] [Google Scholar]
- Lindh JD, Holm L, Andersson ML, Rane A (2009). Influence of CYP2C9 genotype on warfarin dose requirements–a systematic review and meta‐analysis. Eur J Clin Pharmacol 65: 365–375. [DOI] [PubMed] [Google Scholar]
- Lombardi P (2002). Exemestane, a new steroidal aromatase inhibitor of clinical relevance. Biochim Biophys Acta 1587(2–3): 326–337. [DOI] [PubMed] [Google Scholar]
- Lonning PE (1998). Pharmacological profiles of exemestane and formestane, steroidal aromatase inhibitors used for treatment of postmenopausal breast cancer. Breast Cancer Res Treat 49(Suppl 1): S45–S52; discussion S73‐7. [DOI] [PubMed] [Google Scholar]
- Lopez‐Rodriguez R, Novalbos J, Gallego‐Sandin S, Roman‐Martinez M, Torrado J, Gisbert JP, et al. (2008). Influence of CYP2C8 and CYP2C9 polymorphisms on pharmacokinetic and pharmacodynamic parameters of racemic and enantiomeric forms of ibuprofen in healthy volunteers. Pharmacol Res 58: 77–84. [DOI] [PubMed] [Google Scholar]
- Marcos‐Escribano A, Bermejo FA, Bonde‐Larsen AL, Retuerto JI, Sierra IH (2009). 1,2‐Dehydrogenation of steroidal 6‐methylen derivatives. Tetrahedron. 65:7587–7590. [Google Scholar]
- Mareck U, Geyer H, Guddat S, Haenelt N, Koch A, Kohler M, et al. (2006). Identification of the aromatase inhibitors anastrozole and exemestane in human urine using liquid chromatography/tandem mass spectrometry. Rapid Commun Mass Spectrom 20: 1954–1962. [DOI] [PubMed] [Google Scholar]
- Marez D, Legrand M, Sabbagh N, Lo Guidice JM, Spire C, Lafitte JJ, et al. (1997). Polymorphism of the cytochrome P450 CYP2D6 gene in a European population: characterization of 48 mutations and 53 alleles, their frequencies and evolution. Pharmacogenetics 7: 193–202. [DOI] [PubMed] [Google Scholar]
- Martinez C, Garcia‐Martin E, Blanco G, Gamito FJ, Ladero JM, Agundez JA (2005). The effect of the cytochrome P450 CYP2C8 polymorphism on the disposition of (R)‐ibuprofen enantiomer in healthy subjects. Br J Clin Pharmacol 59: 62–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masimirembwa C, Persson I, Bertilsson L, Hasler J, Ingelman‐Sundberg M (1996). A novel mutant variant of the CYP2D6 gene (CYP2D6*17) common in a black African population: association with diminished debrisoquine hydroxylase activity. Br J Clin Pharmacol 42: 713–719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matimba A, Del‐Favero J, Van Broeckhoven C, Masimirembwa C (2009). Novel variants of major drug‐metabolising enzyme genes in diverse African populations and their predicted functional effects. Hum Genomics 3: 169–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mosher CM, Tai G, Rettie AE (2009). CYP2C9 amino acid residues influencing phenytoin turnover and metabolite regio‐ and stereochemistry. J Pharmacol Exp Ther 329: 938–944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy LC, Watson P (2002). Steroid receptors in human breast tumorigenesis and breast cancer progression. Biomed Pharmacother 56: 65–77. [DOI] [PubMed] [Google Scholar]
- Naraharisetti SB, Lin YS, Rieder MJ, Marciante KD, Psaty BM, Thummel KE, et al. (2010). Human liver expression of CYP2C8: gender, age, and genotype effects. Drug Metab Dispos 38: 889–893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niemi M, Leathart JB, Neuvonen M, Backman JT, Daly AK, Neuvonen PJ (2003). Polymorphism in CYP2C8 is associated with reduced plasma concentrations of repaglinide. Clin Pharmacol Ther 74: 380–387. [DOI] [PubMed] [Google Scholar]
- Niemi M, Backman JT, Kajosaari LI, Leathart JB, Neuvonen M, Daly AK, et al. (2005). Polymorphic organic anion transporting polypeptide 1B1 is a major determinant of repaglinide pharmacokinetics. Clin Pharmacol Ther 77: 468–478. [DOI] [PubMed] [Google Scholar]
- Oscarson M, Hidestrand M, Johansson I, Ingelman‐Sundberg M (1997). A combination of mutations in the CYP2D6*17 (CYP2D6Z) allele causes alterations in enzyme function. Mol Pharmacol 52: 1034–1040. [DOI] [PubMed] [Google Scholar]
- Parikh S, Ouedraogo JB, Goldstein JA, Rosenthal PJ, Kroetz DL (2007). Amodiaquine metabolism is impaired by common polymorphisms in CYP2C8: implications for malaria treatment in Africa. Clin Pharmacol Ther 82: 197–203. [DOI] [PubMed] [Google Scholar]
- Peterson A, Xia Z, Chen G, Lazarus P (2017). Exemestane potency is unchanged by common nonsynonomous polymorphisms in CYP19A1: results of a novel anti‐aromatase activity assay examining exemestane and its derivatives. Pharmaol Res Perspec. doi:10.1002/prp2.313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfizer (2016). Aromasin Exemestane Tablets 2016 [updated October 2016. Available at: http://www.pfizer.com/files/products/uspi_aromasin.pdf (accessed 14 December 2016)
- Platet N, Cathiard AM, Gleizes M, Garcia M (2004). Estrogens and their receptors in breast cancer progression: a dual role in cancer proliferation and invasion. Crit Rev Oncol Hematol 51: 55–67. [DOI] [PubMed] [Google Scholar]
- Platt A, Xia Z, Liu Y, Chen G, Lazarus P (2016). Impact of nonsynonymous single nucleotide polymorphisms on in‐vitro metabolism of exemestane by hepatic cytosolic reductases. Pharmacogenet Genomics 26: 370–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Preissner SC, Hoffmann MF, Preissner R, Dunkel M, Gewiess A, Preissner S (2013). Polymorphic cytochrome P450 enzymes (CYPs) and their role in personalized therapy. PLoS ONE 8: e82562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raimundo S, Toscano C, Klein K, Fischer J, Griese EU, Eichelbaum M, et al. (2004). A novel intronic mutation, 2988G>A, with high predictivity for impaired function of cytochrome P450 2D6 in white subjects. Clin Pharmacol Ther 76: 128–138. [DOI] [PubMed] [Google Scholar]
- Rosemary J, Surendiran A, Rajan S, Shashindran CH, Adithan C (2006). Influence of the CYP2C9 AND CYP2C19 polymorphisms on phenytoin hydroxylation in healthy individuals from south India. Indian J Med Res 123: 665–670. [PubMed] [Google Scholar]
- Rowland P, Blaney FE, Smyth MG, Jones JJ, Leydon VR, Oxbrow AK, et al. (2006). Crystal structure of human cytochrome P450 2D6. J Biol Chem 281: 7614–7622. [DOI] [PubMed] [Google Scholar]
- Sakuyama K, Sasaki T, Ujiie S, Obata K, Mizugaki M, Ishikawa M, et al. (2008). Functional characterization of 17 CYP2D6 allelic variants (CYP2D6.2, 10, 14A‐B, 18, 27, 36, 39, 47‐51, 53‐55, and 57). Drug Metab Dispos 36: 2460–2467. [DOI] [PubMed] [Google Scholar]
- Sandberg M, Yasar U, Stromberg P, Hoog JO, Eliasson E (2002). Oxidation of celecoxib by polymorphic cytochrome P450 2C9 and alcohol dehydrogenase. Br J Clin Pharmacol 54: 423–429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sano E, Li W, Yuki H, Liu X, Furihata T, Kobayashi K, et al. (2010). Mechanism of the decrease in catalytic activity of human cytochrome P450 2C9 polymorphic variants investigated by computational analysis. J Comput Chem 31: 2746–2758. [DOI] [PubMed] [Google Scholar]
- Shen H, He MM, Liu H, Wrighton SA, Wang L, Guo B, et al. (2007). Comparative metabolic capabilities and inhibitory profiles of CYP2D6.1, CYP2D6.10, and CYP2D6.17. Drug Metab Dispos 35: 1292–1300. [DOI] [PubMed] [Google Scholar]
- Skopalik J, Anzenbacher P, Otyepka M (2008). Flexibility of human cytochromes P450: molecular dynamics reveals differences between CYPs 3A4, 2C9, and 2A6, which correlate with their substrate preferences. J Phys Chem B 112: 8165–8173. [DOI] [PubMed] [Google Scholar]
- Sommer S, Fuqua SA (2001). Estrogen receptor and breast cancer. Semin Cancer Biol 11: 339–352. [DOI] [PubMed] [Google Scholar]
- Stiborova M, Martinek V, Rydlova H, Koblas T, Hodek P (2005). Expression of cytochrome P450 1A1 and its contribution to oxidation of a potential human carcinogen 1‐phenylazo‐2‐naphthol (Sudan I) in human livers. Cancer Lett 220: 145–154. [DOI] [PubMed] [Google Scholar]
- Sun D, Chen G, Dellinger RW, Sharma AK, Lazarus P (2010). Characterization of 17‐dihydroexemestane glucuronidation: potential role of the UGT2B17 deletion in exemestane pharmacogenetics. Pharmacogenet Genomics 20: 575–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- To SQ, Knower KC, Cheung V, Simpson ER, Clyne CD (2015). Transcriptional control of local estrogen formation by aromatase in the breast. J Steroid Biochem Mol Biol 145: 179–186. [DOI] [PubMed] [Google Scholar]
- Tornio A, Niemi M, Neuvonen PJ, Backman JT (2008). Trimethoprim and the CYP2C8*3 allele have opposite effects on the pharmacokinetics of pioglitazone. Drug Metab Dispos 36: 73–80. [DOI] [PubMed] [Google Scholar]
- Traina TA, Poggesi I, Robson M, Asnis A, Duncan BA, Heerdt A, et al. (2008). Pharmacokinetics and tolerability of exemestane in combination with raloxifene in postmenopausal women with a history of breast cancer. Breast Cancer Res Treat 111: 377–388. [DOI] [PubMed] [Google Scholar]
- United States Food and Drug Administration (2006). Drug Development and Drug Interactions: Table of Substrate, Inhibitors and Inducers.
- Varela CL, Amaral C, Tavares da Silva E, Lopes A, Correia‐da‐Silva G, Carvalho RA, et al. (2014). Exemestane metabolites: synthesis, stereochemical elucidation, biochemical activity and anti‐proliferative effects in a hormone‐dependent breast cancer cell line. Eur J Med Chem 87: 336–345. [DOI] [PubMed] [Google Scholar]
- Variation Viewer [Internet] (2015). National Center for Biotechnology Information, U.S. National Library of Medicine. Available at: http://www.ncbi.nlm.nih.gov/variation/view/. (accessed 29 December 2015)
- Vatèle J (2007). 2‐(Prenyloxymethyl)benzoyl (POMB) group: a new temporary protecting group removable by intramolecular cyclization. Tetrahedron 63: 10921–10929. [Google Scholar]
- Viciano I, Marti S (2016). Theoretical Study of the Mechanism of Exemestane Hydroxylation Catalyzed by Human Aromatase Enzyme. J Phys Chem B 120: 3331–3343. [DOI] [PubMed] [Google Scholar]
- de Waal PW, Sunden KF, Furge LL (2014). Molecular dynamics of CYP2D6 polymorphisms in the absence and presence of a mechanism‐based inactivator reveals changes in local flexibility and dominant substrate access channels. PLoS ONE 9: e108607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan YJ, Poland RE, Han G, Konishi T, Zheng YP, Berman N, et al. (2001). Analysis of the CYP2D6 gene polymorphism and enzyme activity in African‐Americans in southern California. Pharmacogenetics 11: 489–499. [DOI] [PubMed] [Google Scholar]
- Watanabe Y, Fukuyoshi S, Hiratsuka M, Yamaotsu N, Hirono S, Takahashi O, et al. (2016). Prediction of three‐dimensional structures and structural flexibilities of wild‐type and mutant cytochrome P450 1A2 using molecular dynamics simulations. J Mol Graph Model 68: 48–56. [DOI] [PubMed] [Google Scholar]
- Wei L, Locuson CW, Tracy TS (2007). Polymorphic variants of CYP2C9: mechanisms involved in reduced catalytic activity. Mol Pharmacol 72: 1280–1288. [DOI] [PubMed] [Google Scholar]
- van der Weide J, Steijns LS, van Weelden MJ, de Haan K (2001). The effect of genetic polymorphism of cytochrome P450 CYP2C9 on phenytoin dose requirement. Pharmacogenetics 11: 287–291. [DOI] [PubMed] [Google Scholar]
- Wennerholm A, Johansson I, Massele AY, Lande M, Alm C, Aden‐Abdi Y, et al. (1999). Decreased capacity for debrisoquine metabolism among black Tanzanians: analyses of the CYP2D6 genotype and phenotype. Pharmacogenetics 9: 707–714. [PubMed] [Google Scholar]
- Yasar U, Forslund‐Bergengren C, Tybring G, Dorado P, Llerena A, Sjoqvist F, et al. (2002). Pharmacokinetics of losartan and its metabolite E‐3174 in relation to the CYP2C9 genotype. Clin Pharmacol Ther 71: 89–98. [DOI] [PubMed] [Google Scholar]
- Yokota H, Tamura S, Furuya H, Kimura S, Watanabe M, Kanazawa I, et al. (1993). Evidence for a new variant CYP2D6 allele CYP2D6J in a Japanese population associated with lower in vivo rates of sparteine metabolism. Pharmacogenetics 3: 256–263. [DOI] [PubMed] [Google Scholar]
- Zhang T, Liu LA, Lewis DF, Wei DQ (2011). Long‐range effects of a peripheral mutation on the enzymatic activity of cytochrome P450 1A2. J Chem Inf Model 51: 1336–1346. [DOI] [PubMed] [Google Scholar]
- Zhou SF (2009). Polymorphism of human cytochrome P450 2D6 and its clinical significance: part I. Clin Pharmacokinet 48: 689–723. [DOI] [PubMed] [Google Scholar]
- Zhou S (2016). Cytochrome P450 CYP2D6: Structure, Function, Regulation and Polymorphism. Taylor & Francis Group, LLC, Boca Raton, FL. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S1. Digital Content 1.doc.
Data S2. Digital Content 2.doc.
Data S3. Digital Content 3.doc.
Data S4. Digital Content 4.doc.