

Abstract
Functional ex vivo assays that predict a patient’s clinical response to anticancer drugs for guiding cancer treatment have long been a goal, but few have yet proved to be reliable. To address this, we have developed an automated flow cytometry platform for drug screening that evaluates multiple endpoints with a robust data analysis system that can capture the complex mechanisms of action across different compounds. This system, called PharmaFlow, is used to test peripheral blood or bone marrow samples from patients diagnosed with hematological malignancies. Functional assays that use the whole sample, retaining all the microenvironmental components contained in the sample, offer an approach to ex vivo testing that may give results that are clinically relevant. This new approach can help to predict the patients’ response to existing treatments or to drugs under development, for hematological malignancies or other tumors. In addition, relevant biomarkers can be identified that determine the patient’s sensitivity, resistance, or toxicity to a given treatment. We propose that this approach, which better recapitulates the human microenvironment, constitutes a more predictive assay for personalized medicine and preclinical drug discovery.
Keywords: ex vivo, whole patient sample, flow cytometry, precision medicine, drug discovery
Introduction
Ex vivo testing of the activity of cancer drugs with different mechanisms of action (MOAs) has been done for many years, and the measurement of drug activity has become easier because of current technological innovations. Although the ability to find predictive assays prior to clinical treatment has long been sought after to guide or help treatment decisions, few assays meet the standards to be of functional use by health care professionals. Only a limited number of genetic tests against specific DNA alterations have been approved, and the results of these tests are not routinely available prior to the start of treatment.1–3 In addition, these genomics tests also fail to capture the factors that ultimately determine how tumor cells will behave inside the body. Currently, there are multiple approaches to estimate drug efficacy that include, in addition to the above mentioned genetic tests, exposure of isolated tumor cells or tumor-derived cell lines to a panel of drugs to assess the impact on their cell survival or growth.4,5 However, this type of testing has historically had only limited value because of the lack of a correlation between the results of these tests and the clinical outcome of the patient.
Flow cytometry (FCM) is a widely validated technique that is used in all aspects of preclinical development ranging from target selection and validation to MOA identification. Standard chemosensitivity assays, such as the semiautomated colorimetric MTT or bioluminescence assays, report drug activity for an entire population of cells as one result. In contrast, FCM uses single-cell analysis and has the ability to selectively detect the effect of drugs on multiple populations of cells, including cancer cells, normal residual cells, and even the presence of multiples clones. However, FCM applicability for drug screening has been very limited because of the reduced number of assays that can be performed per patient sample and the lack of automation. These limitations have been addressed with the inclusion of FCM platforms that read multiwell plates. We are pioneers in the development of a robust automated FCM platform that incorporates novel innovations to leverage 30 years of FCM experience. We call this PharmaFlow, for pharmacological FCM, and here show how these key innovations supply sound pharmacology using patient samples.
The PharmaFlow platform is a cell-based multicolor screening FCM platform that incorporates both automated sample preparation and automated FCM, in conjunction with proprietary analytical software and a database structure that is geared for rapid data acquisition, analysis, and reporting of results. The automated PharmaFlow platform has been designed to test peripheral blood (PB) or bone marrow (BM) samples, is capable of measuring up to 2000 points per sample, and can evaluate drug effects selectively in the cancer cell population. Our group previously demonstrated the use of the PharmaFlow platform (formerly called ExviTech) to measure the pharmacological activity of drugs and drug combinations as well as model their pharmacological behavior in hematological patient samples.6 This screening has allowed us to accumulate dose-response data in thousands of patient samples for hematological diseases and create a drug reference database to extrapolate differences between patients for personalized medicine purposes or to compare new compounds versus standard of care drugs.
As the MOA of different compounds or antibodies produces different cellular effects, appropriate assays are necessary to cover the different activities. Specific assays have been designed to capture the effects that produce apoptosis, necrosis, cell cycle arrest, proliferation, immune cell modulation, and antibody-dependent cellular cytotoxicity for monoclonal antibodies. FCM is a versatile technique with universal standard protocols that capture the effects produced by a wide range of compounds and, in many cases, can evaluate several effects simultaneously. In addition, the PharmaFlow platform has the ability to count the absolute number of live cells for each well. When a well with a drug is compared with control wells without drug, the percentage of cell depletion can be measured based on live cell counts, and this may be a better estimation of the real drug effect.
It is well known that the tumor microenvironment (TME) affects tumor cell survival, proliferation, and drug resistance.7,8 Its role has become even more important with the discovery and development of cancer drugs that specifically target its components.8–10 TME contains key factors such as stromal cells, plasma, platelets, and red blood cells (RBCs) that will influence the clinical efficacy of many drugs. In this sense, keeping all these components in the in vitro assays seems to be relevant to improve the predictive value. Assays using a patient’s isolated tumor cells, referred to as individualized tumor response testing (ITRT), have been in use for almost 40 years to try and predict the patient’s response to treatment.11,12 We propose that isolating the tumor cells prior to testing, thus removing the TME components, affects the observed potency and efficacy of the drugs, which has contributed to the lack of clinical predictability for ITRT. Maintaining the TME is critical for preventing artifacts in ITRT ex vivo drug testing.6 We have previously reported that whole-sample incubation, maintaining the TME, results in higher predictive accuracy regarding clinical responses for patients diagnosed with acute myeloid leukemia (AML), with overall correlation and predictive values of 83%13 (Montesinos et al., unpublished manuscript). We refer to these whole-sample experiments as native environment (NE) assays.
We report here a novel method for testing samples from patients diagnosed with hematological malignancies that may overcome the limitations of current procedures and offer clinically significant results. This approach incorporates three main advantages compared with standard methods: 1) the use of whole PB or BM samples, 2) screening with an automated FCM-based system that counts live cells, and 3) data analysis that uses pharmacokinetic and pharmacodynamic (PKPD) models along with a drug reference database developed by screening thousands of patient samples.
In summary, the integration of NE with automated FCM and robust data analysis allows the PharmaFlow platform to serve as a useful tool for drug screening of individual compounds and/or combinations. Here, we demonstrate the robustness of this system and how it can be used to test patient samples with cell depletion, proliferation, and immunomodulation assays using a variety of drugs. This approach evaluates multiple cell populations, and it is a useful tool for personalized treatment strategies for clinical applications and for new drug development and validation.
Materials and Methods
Analytical Performance of the PharmaFlow Platform
The study conducted for the evaluation of analytical performance was done using Perfect-Count Microspheres (Cytognos, Salamanca, Spain) per the manufacturer’s recommendations. To test the precision of the PharmaFlow platform, reproducibility was assessed in five measurements analyzed in six different volumes of Perfect-Count Microspheres in the range of expected number of cells in samples. Volumes were adjusted to 60 µL per well in phosphate-buffered saline (PBS), except for the highest concentration, which was 90 µL. To prevent the injection of air into the cytometer, the final volume acquired was 55 µL or 80 µL, whereby the relevant calculations were made to estimate the observed number of beads recovered for that volume as recommended by the manufacturer.
To evaluate the homogeneity and accuracy in terms of yield in a cell population, an acute lymphoblastic leukemia (ALL) human cell line, TOM-1, was seeded at a concentration of 500 cells/µL in culture medium. The mixture was dispensed with a Multidrop Combi into 96-well plates at a final volume of 60 µL/well. After a wash step, the pellet was resuspended in 80 µL of binding buffer (BB; 10 mL 4-[2-hydroxyethyl]-1-piperazineethanesulfonic acid [HEPES], 8.19 g NaCl, 0.37 g Cl2Ca, H2O to 1 L, pH 7.4) for analysis in the PharmaFlow platform.
Patients and Sample Validation
BM and PB samples were collected in heparinized tubes (Vacuette; Greiner Bio One, Kremsmünster, Austria) from patients with hematological malignancies from 67 Spanish and 7 European hospitals. Most samples were received at the laboratory within 24 h from extraction, although an extended time of up to 72 h was also acceptable for some samples after checking the viability. All patients gave informed consent for study participation, and studies were approved by the ethical committees of the corresponding hospitals. All samples received an internal alphanumeric code that was used thereafter for all the sample processes and data generation, which afforded further security to patients while preserving traceability.
Before each study, the CyAn flow cytometer (Beckman Coulter, Brea, CA) and PharmaFlow platform were calibrated per recommendations given by EuroFlow.14 Initial cell count and validation of each sample were performed as previously described in detail,6 with slight modifications based on the type of sample. Briefly, between 1 µL and 7 µL of whole NE samples were aliquoted in duplicate into a 96-well plate and lysed twice with 180 µL ammonium chloride solution, incubated for 10 min at 4 °C followed by a centrifugation step for 5 min at 1200 rpm. Once the RBCs were lysed, a combination of 20 µL of BB, annexin V, and different monoclonal antibody (MoAb) cocktails recommended by the EuroFlow Consortium,15 specific for detecting the leukemic cells in each hematological disease, were used. The specific MoAb staining cocktails used for AML and chronic lymphoid leukemia (CLL) patient samples were
CLL: CD19-PECy7 (0.2 µL; Beckman Coulter, Indianapolis, IN), CD5-PerCP (0.5 µL; Immunostep), CD20-PB (0.5 µL; Biolegend, London, UK), CD45-PO (0.5 µL; Invitrogen, Carlsbad, CA), CD10-APC (0.5 µL; BD, San Jose, CA), and CD23-PE (0.5 µL; BD)
- AML:
- Immature blasts: CD34-PerCP (0.1 µL; Biolegend), CD45-PO (0.5 µL; Invitrogen), HLADR-PE (0.5 µL; Immunostep), CD117-APC (0.5 µL; Beckman Coulter), CD19-PECy7 (0.2 µL; Beckman Coulter).
- Monocytic lineage: CD34-PerCP (0.1 µL; Biolegend), CD45-PO (0.5 µL; Invitrogen), CD64-PE (0.5 µL; Cytognos), CD14-APC (0.5 µL; Immunostep), and CD33-BV421 (0.1 µL; BD).
- Neutrophylic lineage: CD34-PerCP (0.1 µL; Biolegend), CD45-PO (0.5 µL; Invitrogen), CD13-PE (0.5 µL; Immunostep), and CD11b-APC (0.5 µL; Immunostep).
Plate Setup
All drugs used in this study were dissolved in DMSO, or in PBS when the drugs were not stable or soluble in DMSO, at a stock concentration between 5 mM and 30 mM, or in the case of immune-oncology (I-O) drugs at 0.11 mg/mL. Drug plates were prepared using an Echo 550 Liquid Handler (LabCyte, Sunnyvale, CA). Serial dilutions were performed enabling dose-response studies with final drug concentrations ranging from 150 µM to 0.54 nM or from 3.5 µg/mL to 0.2 ng/mL for I-O drugs. Once the drugs were plated, quality controls were performed to test the expected drug activity on a cell line sensitive to that drug or compound. Other quality controls were performed to ensure the data quality throughout the entire process, including systematic quality controls on all the equipment involved, such as the Echo 550 Liquid Handler and the Multidrop Combi (Thermo Scientific, Waltham, MA).
Assay Setup: NE Assays
NE assays were those in which whole BM or PB fresh samples were incubated retaining the erythrocyte population and serum proteins. After initial cell count, if the sample had enough live pathological cells (≥40%), the whole sample was prepared for the different NE assays described below.
NE Depletion Assay
For depletion assays, the whole AML BM or normal BM (NBM) samples shown here were diluted with culture medium, supplemented with 20% (vol/vol) fetal bovine serum (FBS; Thermo Scientific), 2% HEPES (Sigma-Aldrich, St. Louis, MO), 1% antibiotic (Zell Shield, Labclinics, Barcelona, Spain), and 1% L-Glutamine 200 mM (Lonza, Hopkinton, MA) to a final concentration between 130 and 500 pathological cells/µL. The mixture was dispensed with a Multidrop Combi into 96-well plates containing the drugs at a final volume of 60 µL/well. Plates were incubated 48 h at 37 °C in 5% CO2. At the end of the incubation period, the RBCs were lysed twice with an ammonium chloride solution by incubating for 10 min at 4 °C followed by a centrifugation step for 5 min at 1200 rpm, ensuring removal of all the RBCs, which could have interfered later in the MoAb staining and analysis of leukemic cells. The remaining cells were labeled with a combination of 20 µL of BB with Annexin-V FITC and the two best monoclonal antibodies for unequivocally identifying the pathological cells (this was determined in the initial cell count using the EuroFlow panels15), as described above. After 15 min of incubation at room temperature in the dark, a wash step was performed and the pellet was resuspended in 80 µL of BB for analysis in the PharmaFlow platform. Leukemic cells were identified by their specific MoAb markers and their light scatter properties. The annexin-V staining was used to exclude apoptotic or necrotic cells and debris to determine live cell populations.
NE Proliferation Assay
To simulate the lymph node microenvironment, the stromal cell line HS-5 was seeded in a 96-well flat-bottom plate at a concentration of 1:100 (HS-5 cells:leukemic cells) 24 h before incubation with leukemic cells to allow stromal cells to adhere.
For all proliferation assays, the Vybrant CFDA SE Cell Tracer Kit (Molecular Probes, Eugene, OR) was used. This reagent covalently attached to cytoplasmatic components of cells, resulting in uniform bright fluorescence, and allowed for analysis of cell proliferation. The CFDA SE (component A) was dissolved in DMSO (component B) at a concentration of 5 mM as stock solution and kept at −20 °C until used. Whole CLL PB sample was diluted in culture medium and adjusted so that the number of CLL cells was 2 × 105 cells/well. CFDA was added to 1 mL cell suspension of these CLL cells, to a final concentration of 5 µM. After addition of CFDA, cells were vortexed and incubated at room temperature for 10 min with continuous shaking and protection from light. At the end of the incubation period, the cells were resuspended in 10 mL of cooled RPMI-1640 (Sigma-Aldrich) with 20% FBS and kept on ice for 5 min. The cells were washed twice with this cooled media, then finally resuspended in AIM-V (Invitrogen) culture medium and maintained at 4 °C until used. This step was performed prior to all other steps in the experimental preparation for proliferation assays. To induce cell proliferation, the culture medium with the CFDA-stained leukemic cells was supplemented with human cytokines, IL-2 (Peprotech, London, UK), CpG (ODN2006; Miltenyi Biotech, Bergisch Gladbach, Germany), and 10% human serum (Sigma-Aldrich). Once the leukemic CFDA-labeled cells were resuspended in this proliferating media, they were dispensed into 96-well plates containing Idelalisib at eight different concentrations ranging from 0.54 nM to 150 µM, with the Multidrop Combi at a final volume of 60 µL per well. CLL leukemic cells were dispensed in the drug plates and then transferred into the previously seeded plate containing the HS5 (1:100) cell line. In all cases, no stimulation was also used as a control of both viability and proliferation.
A control of proliferation was performed at 24 h to fix the CFDA peak, where null or minimum cell division was expected and the green channel fluorescence intensity of CFDA had become stable, showing a well-established CFDA peak. For this purpose, five control wells were lysed at 24 h, stained with 20 µL of a combination of BB with Annexin-V CF Blue (Immunostep, Salamanca, Spain) and the best monoclonal antibodies that identified the pathological cells in the initial cell count using EuroFlow panels,15 as described above, and then washed and resuspended in BB for analysis in the PharmaFlow platform.
Ninety-six hours after incubation at 37 °C in 5% CO2, the plates were washed and processed as described above. Annexin-V staining discerned between apoptotic and live cells to test viability, and CFDA allowed for proliferation to be monitored. The leukemic cells were identified using a gating strategy based on forward scatter and/or side scatter and the expression of different surface markers. Dose-response curves for the drugs were measured for each proliferative subset based on the CFDA peak signal.
NE I-O Assays
Fresh BM samples from AML patients were processed as described in the depletion assay and incubated with the bispecific antibody CD3-CD123 (Creative Biolabs, Shirley, NY) for 72 h, 96 h, and 120 h at eight dose-response concentrations ranging from 0.2 ng/mL to 3.5 µg/mL. After the incubation time, the plates were washed and processed as described above. A specific eight-color FCM staining including Annexin V, a blast marker (CD117, CD64, or CD33) for leukemic cells, and a monoclonal antibody cocktail with CD4 (Biolegend), CD5 (Biolegend), CD3 (BD), CD45, CD8 (BD), and CD25 (BD) were used in a total volume of 20 µL per well in BB for simultaneously identifying the live leukemic cells, the activated T-cells, and the residual normal cells.
Data Analysis
The response effect was measured by counting the number of live cells from the population subject of the study that remained after a given incubation time to increasing concentrations of drug. Whenever possible, estimation of pharmacodynamic parameters was carried out using a population modelling approach and a nonlinear mixed effect regression analysis using NONMEM software version 7.2, as described in our previous publication.6 By using this methodology, dose-response curves from all samples were calculated and processed at the same time. Residual errors and interindividual variability were calculated and taken into account to determine the population standard profile for each drug. The aim of this methodology was to get a characterization of the pharmacodynamic behavior of a drug or combination in a representative sampling population, improve the individual fittings, and reduce the confidence interval associated with each individual estimation.
Pharmacological effects of single drugs were evaluated using models based on the Hill equation following either a population or individual fitting approach. The dependent variable used for the fitting was always the absolute count of the specific cell population of the study, at increasing drug concentrations. To achieve a more comprehensive analysis, a normalization step was done after fitting the data. One hundred percent of the survival index in each case referred to the basal parameter value estimated with the individual model fit. Drug interaction analysis was carried out by calculating the combination index as described by Chou16 or by using a complex interaction surface model.17 As in the case of single drugs, modelling for interaction analysis was usually carried out following a population approach. After the pharmacological modelling was concluded, a set of graphical tools was used to generate figures and graphs to visualize results in a more comprehensive way depending on the type of analysis. We used the following software products: Spotfire (client and server) 7.6, Piraña 2.9.4 for NONMEM, RStudio 1.0.44 and R 3.2.3.
Results and Discussion
In this report, we present the use of an automated FCM screening platform, PharmaFlow, to test whole samples from patients diagnosed with hematological malignancies. Automation is key for high-throughput screening as it provides the consistency and traceability required. Drug plates are prepared with an acoustic liquid handler (Echo 550), samples and reagents are dispensed into the prepared drug plates with a Multidrop Combi, and automated FCM allow for volumetric sample acquisition in a system that has been developed to produce consistent results.
System Development
FCM has long been a robust tool for reliable cell analysis and has many advantages over other techniques. These include ease of use, the ability to rapidly analyze very large cell numbers, analysis of rare populations of cells, and the ability to obtain multiparameter information on individual cells, which is particularly important for heterogeneous cell samples. In the past decade, there has been increased use of platforms incorporated into commercial flow cytometers. With the introduction of multiwell plate-based sampling, FCM ex vivo testing is now a reliable tool for biomolecular screening and drug discovery and development. The PharmaFlow FCM platform has been designed to overcome the main limitations present in other FCM methods. We compare different conditions that are critical for obtaining homogeneity and reproducibility from well to well during acquisition. Experiments performed with the PharmaFlow platform analyze the effect on cell viability of temperature fluctuations and mixing the content of each well, either by a plate shaker or via aspiration, before acquisition. For each condition, the TOM-1 human cell line, derived from the BM of a patient with refractory ALL, is used to evaluate the following variables: 1) homogeneity, defined by the coefficient of variation (CV) of the whole plate and the difference between the well containing the maximum number of cells and the well containing the minimum number of cells, and 2) yield, defined as the actual number of cells recovered as a percentage of the expected number of cells. The summary of these results is shown in Table 1 , reflecting a clear advantage of the PharmaFlow system that incorporates mixing via a temperature-controlled plate shaker. There is a distinct advantage seen by continuously mixing samples during plate acquisition with shaking, over a single mixing step via aspiration just prior to the acquisition of the well contents. Moreover, by using the shaker with temperature control, we have avoided positional effects on the plate, observing no difference between the number of cells acquired from each well depending on its location in the plate.
Table 1.
Cell-Based Assay Using the Human Cell Line TOM-1 to Compare Different Conditions for Obtaining High Reproducibility and Homogeneity of Cell Counts with an Automated Flow cytometry Platform.a
| Condition | Coefficient of Variation (%) | Differences, Max-Min (%) | Yield (%) |
|---|---|---|---|
| PharmaFlow platform | 3.77 | 14.69 | 81.6 |
| Other platforms | |||
| No shaking/no mixing/ no temperature control | 21.81 | 54.53 | 58.4 |
| Mixing via aspiration | 17.33 | 53.30 | 54.4 |
Sample acquisition conditions for the PharmaFlow platform were shaking at 1000 rpm with temperature controlled at 20 °C. An initial step of 5 min of shaking is performed for the three conditions.
To test the accuracy of the platform, we acquired six different volumes of Perfect Count Microspheres. A good correlation (R2 = 0.99) is observed between the volume of beads added and the number of beads recovered ( Fig. 1A ). The reproducibility is assessed using five different measurements of six different volumes of Perfect-Count Microspheres. Figure 1B shows the median of the CV for the five measurements performed for each volume and the number of beads recovered for each of the six different volumes tested. As can be seen, reproducibility is very high, with a mean intra-assay CV of 4.48%. The PharmaFlow platform has been developed incorporating these critical functions (shaking and controlled temperature) to achieve a screening technology with a robust process for the determination of specific cell counts with a low intra-assay variability and high reproducibility.
Figure 1.

Evaluation of the accuracy and reproducibility of the PharmaFlow platform. (A) To evaluate the accuracy, different volumes of Perfect Count Microspheres, which have a reference value per microliter, are acquired in the PharmaFlow platform showing a good correlation (R2 = 0.99) between the volume of beads added (x-axis) and the number of beads recovered (y-axis). (B) Evaluation of the reproducibility using five different measurements of six different volumes of Perfect-Count Microspheres. This graph shows the median of the coefficients of variation (y-axis) for the five measurements performed and the number of beads recovered for each of the six different volumes tested (x-axis).
NE FCM Depletion Assays
The first assay developed with the PharmaFlow technology is a cell depletion assay based on exact counts of live cells. The combination of fluorescently labeled antibodies to identify leukemic cells and Annexin-V staining to exclude dying cells allows for the measurement of specific cell populations in both the presence and absence of a drug. The difference in the number of live leukemic cells in a well with drug compared with the number of live leukemic cells in the control wells is used to determine the percentage of depletion induced by the drug. The robustness of this process depends on automated sample preparation together with the PharmaFlow technology that allows for volumetric sample input into the flow cytometer.
With the NE depletion assay, the PharmaFlow platform represents an attractive method to establish or personalize the therapeutic index for each drug by simultaneously evaluating drug efficacy and toxicity in healthy or pathological cell populations. Figure 2A shows the effect of a standard AML drug, cytarabine, in an AML patient sample and the different behavior of the leukemic and normal mature cell populations. Moreover, with this assay, we can calculate the potential hematotoxicity or myelosuppression of CD34+ progenitors in NBM samples ( Fig. 2B ). For a proof of concept to test this hypothesis, we selected two known and related cytotoxic drugs (cytarabine and clofarabine) and two novel drugs with specific cell targets and low expected cytotoxicity (ruxolitinib and volasertib). A multiple staining (AnnexinV/CD45/CD117/CD34/CD38/CD19) is used to distinguish the most immature population. Drug response is evaluated as a depletion survival index of each cell population relative to the average of six control wells in each plate. In contrast to the nucleoside cytotoxic drugs, a better safety profile in the myeloid precursor cells is observed with the novel drugs ruxolotinib and volasertib. This corroborates the lack of clinical hematotoxicity expected for these novel drugs even at very high concentrations never achieved in vivo.
Figure 2.

Dose-response curves from different assays performed by the PharmaFlow platform. (A) Evaluation of the therapeutic index determining the effect of cytarabine in both leukemic and residual mature lymphoid cells in an acute myeloid leukemia sample with the depletion assay. (B) Evaluation of the cytotoxic effects of compounds on CD34+ progenitor cells in NBM samples (n = 10) with the depletion assay. Each line represents the median for four approved drugs. (C, D) Evaluation of the antiproliferative (C) or cytotoxic (D) activity of idelalisib determining selectively the effects in proliferative (C) and whole-cell population (D) with the native environment proliferation assay. Panels C and D represent the median and the standard deviation (vertical error bars) in 19 chronic lymphoid leukemia samples for idelalisib.
The advantage of using a cell depletion assay over a simple apoptosis assay is that some drugs induce cell death through processes other than apoptosis. In addition, the assay includes wash steps, known to cause cell loss most predominantly in the apoptotic cell population.18 Cell proliferation or cell differentiation has also been known to occur during incubation, and with the depletion assay, this can be identified by comparison with controls. Cell depletion is measured at different drug concentrations to evaluate the dose-response effect of the tested drug. Parameters associated with the dose-response pharmacodynamics can be calculated. Cell depletion indicates the highest efficacy yielded by a drug on a particular cell subpopulation from a patient sample. Appropriate choice of the time point to evaluate each individual drug is selected to capture its activity as well as to identify the time that best differentiates between samples. In this way, the PharmaFlow platform can quantify the activity of drugs, operating through different MOAs.
NE FCM Proliferation Assays
The NE proliferation assays are performed for those compounds in which cell cycle arrest is suspected to be more relevant than depletion, although both effects are simultaneously captured by the platform. The drug effect is measured on both the proliferating and nonproliferating cell subsets. The cell dye, CFDA SE, labels leukocytes selectively in a whole sample environment, allowing differential analysis of leukemic cells and the residual populations. Proliferation is detected by dye dilution.
CLL behavior and outcome also depend on stimuli present in the leukemic microenvironment.19 The critical role of external stimulation is shown by the remarkable therapeutic activity of drugs interfering with the CLL microenvironment, namely, inhibitors of signaling through the clonotypic B cell receptors. These novel drugs constitute a major paradigm shift in CLL treatment,20 and idelalisib, tested here, is one of them. To more closely reproduce and re-create the complexity of the in vivo microenvironment in CLL, we have developed an ex vivo assay that best promotes CLL proliferation and viability with the combination of CpG + IL2 + HS5 stromal cells + 10% human serum, that approximates a lymph node environment.21 Human stromal cell lines, such as HS5, have been reported to prevent spontaneous death and promote proliferation of tumor cells.22 The CLL patient samples are co-cultured with the HS5 stromal cell line at a ratio of 1:100 (HS5:CLL cells) for the incubation period. The effect of the PI3Kδ inhibitor idelalisib is tested in 19 CLL samples on both proliferating and nonproliferating fractions. We note a potent inhibition of proliferation ( Fig. 2C ) with a limited induction of apoptosis ( Fig. 2D ), suggesting a marked, nearly complete antiproliferative effect of idelalisib, at clinically achievable concentrations with a reduced proapoptotic activity of the drug. If this new assay would correlate with patient treatment response, it could become a valuable tool to personalize antiproliferating drug treatments for hematological patients.
NE FCM I-O Assays
I-O has emerged as a powerful therapeutic approach to treat cancer. From the different modalities to reinforce the immune system, the development of bispecific T cell–engaging (BiTE) antibodies represents one of the most attractive ones. They force the formation of an immunologic synapse between polyclonal T cells and tumor cells by binding to a surface antigen on cancer cells with one arm and recruit T cells with the CD3 domain on the other arm. This leads to a potent leukemic cell lysis by activated T cells (CD4 or CD8) independent of T cell receptor specificity, co-stimulation, or peptide antigen presentation. An increasing number of bispecific antibodies are currently in clinical practice or clinical trials with promising early results.23–25 However, no in vitro assays have been developed and validated to potentially predict the in vivo responses. The PharmaFlow platform, preserving both the whole BM or whole PB environment and counting the absolute number of cells, measures the activity of these activated T cells in killing tumor cells. For this purpose, 16 AML samples were tested with the CD3–CD123 BiTE at eight concentrations ranging from 0.2 ng/mL to 3.5 µg/mL at different time points (72, 96, 120 h). Use of the whole sample is integral here, as the sample will contain both the tumor cells and the T cells. Most of the samples present an effective lysis of the tumor cells ( Fig. 3A ) that is simultaneous with T cell activation and proliferation ( Fig. 3B ). Interestingly, results show different in vitro T cell cytotoxicity effects between patients that could allow us to better select candidates for adoptive antitumor immunotherapy with BiTEs. The PharmaFlow platform’s flow cytometric base is highly adapted to efficiently count both the number of tumor cells that are killed and the number of activated T cells.
Figure 3.

Dose-response curves from 3.5 µg/mL to 0.2 ng/mL to assess the CD3–CD123 bispecific antibody activity at different time points (72, 96, 120 h) in acute myeloid leukemia samples (n = 16). For both panels, each colored line represents a patient. An appropriate and specific eight-color flow cytometry staining includes Annexin V for apoptosis identification, a blast marker (CD117, CD64, or CD33) for leukemic cells, and a monoclonal antibody cocktail with CD4, CD5, CD3, CD45, CD8, and CD25 to simultaneously identify the leukemic cells, the activated (CD25+) T cells (CD4+ or CD8+), and the residual normal cells. (A) Leukemic cell depletion curves. The survival index (y-axis) ranges from 100% to 0% displaying the leukemic cell depletion after exposure to a dose response of CD3-CD123 bispecific antibody concentrations (x-axis). (B) Simultaneous T cell activation and proliferation at different incubation times. Absolute counts of T cells (y-axis) after exposure to the CD3–CD123 bispecific antibody.
In addition, we can also measure other key parameters to aid in predicting the in vitro response after bispecific antibody (or similar formats) exposure, including tumor antigen expression (ie, CD123, CD19), T cell expansion, or expression of immune checkpoints on target and effector cells before and after the cell culture. The integration of these parameters, together with the pharmacological data (EC50, Emax, AUC), allow us to generate a predictive model of in vitro response. This could discriminate between the best patient candidates for adoptive antitumor immunotherapy with bispecific antibodies, those with higher T cell cytotoxicity, and those patients with a remarkable immune resistance. Especially, but not exclusively for those patients with immune resistance, the PharmaFlow platform represents a viable approach to evaluate multiple combinations of BsAbs with immunomodulatory checkpoint inhibitors that could enhance the BsAb cytotoxicity. In our assay, bispecific activity can also be tested in normal cells that do not express the tumor-associated antigen reflecting the tumor specificity of the activated T cells.
Pharmacological Analysis, PKPD Models, and Synergy
In a previous work, we have used the PharmaFlow platform to measure the pharmacological activity of drugs in AML patients.6 Now, we have accumulated dose-response data in more than 3000 patients for more than 80 approved drugs in hematological diseases, creating a reference database and PKPD models to extrapolate differences between patients for personalized medicine purposes or to compare new compounds versus standard of care. We have derived the pharmacological evaluation of single drugs and drug combinations across patients with hematological malignancies as a tool for individualized treatment selection. To this purpose, the efficacy of a treatment for an individual patient has been assessed relative to its activity in the patient population included in our database. As an example, the pharmacological activity for a specific drug, cytarabine, is shown for 463 AML patient samples ( Fig. 4A ). There is a large range of interpatient variability in the response to this drug, one that is a standard treatment for AML.26 A combination of potency (EC50), represented by a dose that kills 50% of cells and efficacy (Emax), represented as the percentage of leukemic cells killed, can be used for determining the pharmacological behavior of a drug for an individual patient by comparing it to the entire sample population. Those AML patients whose curves are shifted more to the left of the average curve will be more suitable for receiving lower doses of the drug for killing the leukemic blast cells, whereas those patients with curves shifted more to the right of the average curve will need higher doses to kill the blast cells. Patients at the extremes will be more suitable for predicting sensitive (left extreme) or resistance (right extreme) to a given treatment. We have achieved a high correlation with clinical outcome in first-line AML in an observational clinical trial13 (Montesinos et al., unpublished manuscript), which suggests that these ex vivo assays can be used as biomarkers or companion diagnostics, substantially improving the outcome of clinical trials of new drugs.
Figure 4.
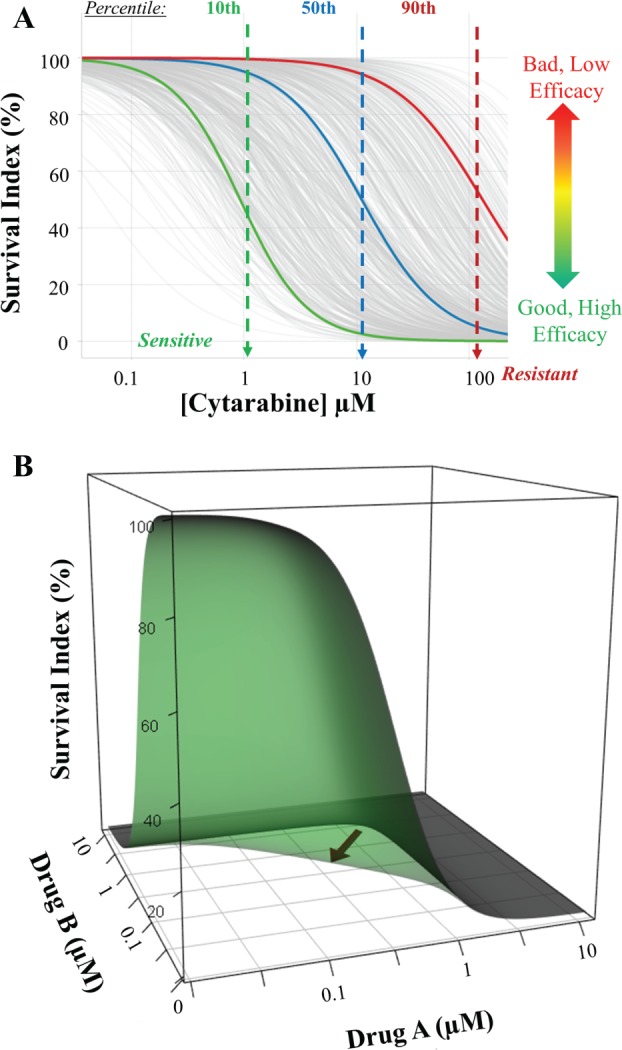
Pharmacological ex vivo activity across individual acute myeloid leukemia (AML) patients to cytarabine. (A) High interpatient heterogeneity between 463 AML patients (gray lines) to cytarabine after a 48 h incubation period. The blue line depicts the average response. The integration of both potency (EC50) and efficacy (Emax) determines for each individual the in vitro effectiveness of the drug referred to in the population models. (B) Three-dimensional (3D) plots showing the interaction surface models for additive (gray) and synergistic (green) interactions. Synergy is graphically observed through the shift of the surface toward the axis origin. Sigmoidal curves on each side of the 3D object represent the dose-response curve of each single drug.
As many treatment protocols for hematological malignancies are composed of multiple drugs, an additional method for evaluating an individual patient’s response to a treatment is to measure drug synergism. A representative example of synergism and additivity is shown in Figure 4B using a three-dimensional (3D) plot that represents a fit to an interaction surface model17 in which the synergism between drugs can be seen by the scoop out toward the origin of the axis in the synergy surface (green) in contrast to the additivity surface (gray). These 3D surfaces model the response effect, expressed as a percentage of survival, of each single-drug concentration as well as the effect from mixtures of both drugs at different concentration ratios. This process would also be useful in analyzing drug combinations for potential new protocols or for assessing drug candidates and their interaction with approved drugs.
We have performed a pharmacological profile for all the patients included according to drug potency, percentage of resistant cells, and synergy of the treatments. This information may be useful in selecting the optimal treatment for individual patients, especially relapse/refractory patients in need of therapeutic alternatives. By testing the drugs used in the treatment protocols for hematological malignancies directly on patient samples, a pharmacological based model has been developed to infer drug resistance or sensitivity, patient by patient.
Conclusions
PharmaFlow Platform
We have developed a new, scalable, and automated FCM technology, with robust data analysis, using whole patient samples to advance precision medicine tests and evaluate new candidates for drug discovery ( Fig. 5 ). The use of whole sample, retaining the NE, maintains key factors for drug screening such as RBCs, neutrophils, platelets, cytokines, and other serum components. Ex vivo drug pharmacology using whole PB or BM samples instead of classical long-term cultures with isolated leukocytes may offer results with higher relevance. We have previously reported that the pharmacological drug behavior is significantly different between both methods.6 In addition, we have observed that spontaneous apoptosis after cell culture is reduced by whole-sample incubations in comparison with isolated leukocytes, as also described in the literature.27 We intend that this ex vivo system simulates more closely the conditions in the leukemic microenvironment, overcoming the obstacles for ex vivo testing of drugs that treat cancer. Several groups have developed microenvironment-mimicking assays with varying degrees of success.9,28–32 We have now developed assays for drug screening that better mimic the TME. The use of the PharmaFlow platform with our NE assays allows us to perform different cellular analyses, from simple tumor cell identification to more complex drug MOAs, by monitoring immunophenotyping, depletion, apoptosis, proliferation, differentiation, and immune modulation. Figure 6 shows an overview of our NE assays system from sample reception and validation to final analysis, and Table 2 includes a partial list of drugs that have been successfully tested. The use of the PharmaFlow platform allows us to work with patient’s cells in their NE, achieving clinical relevance with a high level of correlation to clinical results.13
Figure 5.

PharmaFlow automated platform overview. The PharmaFlow platform is automated and capable of measuring up to 2000 points per sample using the whole sample with its native environment. It can evaluate effects selectively in cancer cell subpopulations and analyze data with pharmacokinetic and pharmacodynamic population models that fit dose responses for all samples simultaneously, achieving high clinical correlation.
Figure 6.

Workflow of our native environment assays.
Table 2.
Overview of Drugs with Several Different Mechanisms of Action That Have Been Tested with the PharmaFlow Platform.
| Mechanism of Action | Compound | Assay |
|---|---|---|
| Alkylating activity | Chlorambucil/melphalan | NE cell depletion |
| Nucleic acid synthesis inhibitor | Fludarabine/cytarabine | NE cell depletion |
| Topoisomerase inhibitor | Idarubicin/mitoxantrone | NE cell depletion |
| Vinca alkaloid | Vincristine/vinblastine | NE cell depletion |
| Corticosteroid hormone receptor agonist | Prednisolone/dexamethasone | NE cell depletion |
| Anti-CDx monoclonal antibody | Ofatumumab/rituximab | NE cell depletion |
| Proteasome inhibitor | Bortezomib/carfilzomib | NE cell depletion |
| HDAC inhibitor | Panobinostat/vorinostat | NE cell depletion |
| Hsp90 inhibitor | Tanespimycin | NE cell depletion |
| Farnesyltransferase inhibitor | Tipifarnib | NE cell depletion |
| p53-MDM2 inhibitor | Idasanutlin | NE cell depletion |
| BCL-2 inhibitor | Venetoclax | NE cell depletion |
| Multikinase inhibitor | PKC412 (midostaurin)/sorafenib | NE cell depletion |
| Tyrosine kinase inhibitor | Imatinib/crizotinib | NE cell depletion |
| AKT inhibitor | Perifosine | NE cell depletion |
| mTOR inhibitor | Rapamycin/everolimus | NE cell depletion |
| JAK1/2 inhibitor | Ruxolitinib | NE cell depletion |
| HIV inhibitor | Ritonavir | NE Cell depletion |
| FLT-3 inhibitor | Quizartinib/crenolanib | NE Cell depletion |
| Syk inhibitor | Entospletinib | NE Cell depletion |
| MEK inhibitor | Trametinib | NE Cell proliferation |
| PLK1 inhibitor | Volasertib | NE Cell proliferation |
| BTK inhibitor | Ibrutinib | NE Cell proliferation |
| PI3K inhibitor | Idelalisib | NE Cell proliferation |
| Hypomethylating activity | Decitabine/5-azacytidine | NE Cell depletion and proliferation |
| Immunomodulator | Thalidomide/lenalidomide | NE cell depletion and proliferation |
| Immune checkpoint inhibitor | Nivolumab | NE I-O |
| Bispecific antibodies | Blinatumomab/CD3-CD123 | NE I-O |
The PharmaFlow platform introduces important key innovations for overcoming issues and supplying sound pharmacology. It is the first automated FCM platform to incorporate well-mixing cycles with plate temperature control and also uses acoustic sample handling for dispensing the drugs and drug combinations for analysis. Moving forward, this system could be used in an iterative process, testing up to 1000 drugs, including cytotoxic, antiproliferative, or new MOA drugs, in patient samples. The PharmaFlow platform could sequentially characterize the best compounds in multiple cycles, until the 10 best are identified. These will then be tested in 50 patient samples ( Fig. 7 ). The synergism drug ratios between them could be established, analyzing the complementarity in monotherapy and performing kinetic and PKPD population models as a useful tool for personalize treatment protocols and drug discovery. The PharmaFlow platform is a reliable tool for overcoming the limitations of ex vivo testing in drug discovery and development in hematological malignancies that can bridge the translational gap. We are pioneers in the simultaneous incorporation of NE assays and automated FCM together with robust and novel data analysis.
Figure 7.

Representation of the iterative drug discovery and development processes.
Acknowledgments
We especially thank the patients, hematologists, and hospitals for providing the samples for the realization of these studies.
Footnotes
Declaration of Conflicting Interests: All authors are employees of Vivia Biotech.
Funding: The authors received no financial support for the research, authorship, and/or publication of this article.
References
- 1. Rogowski W. H., Grosse S. D., Khoury M. J. Challenges of Translating Genetic Tests into Clinical and Public Health Practice. Nat. Rev. Genet. 2009, 10, 489–495. [DOI] [PubMed] [Google Scholar]
- 2. Burke W. Genetic Tests: Clinical Validity and Clinical Utility. Curr. Protoc. Hum. Genet. 2014, 81, 9.15.1–9.15.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Williams P. M., Lively T. G., Jessup J. M., et al. Bridging the Gap: Moving Predictive and Prognostic Assays from Research to Clinical Use. Clin. Cancer Res. 2012, 18, 1531–1539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Gockeritz E., Kerwien S., Baumann M., et al. Efficacy of Phosphatidylinositol-3 Kinase Inhibitors with Diverse Isoform Selectivity Profiles for Inhibiting the Survival of Chronic Lymphocytic Leukemia Cells. Int. J. Cancer 2015, 137, 2234–2242. [DOI] [PubMed] [Google Scholar]
- 5. Tina E., Prenkert M., Hoglund M., et al. Topoisomerase IIalpha Expression in Acute Myeloid Leukaemia Cells That Survive after Exposure to Daunorubicin or Ara-C. Oncol. Rep. 2009, 22, 1527–1531. [DOI] [PubMed] [Google Scholar]
- 6. Bennett T. A., Montesinos P., Moscardo F., et al. Pharmacological Profiles of Acute Myeloid Leukemia Treatments in Patient Samples by Automated Flow Cytometry: A Bridge to Individualized Medicine. Clin. Lymphoma Myeloma Leuk. 2014, 14, 305–318. [DOI] [PubMed] [Google Scholar]
- 7. McMillin D. W., Negri J. M., Mitsiades C. S. The Role of Tumour-Stromal Interactions in Modifying Drug Response: Challenges and Opportunities. Nat. Rev. Drug Discov. 2013, 12, 217–228. [DOI] [PubMed] [Google Scholar]
- 8. Pottier C., Wheatherspoon A., Roncarati P., et al. The Importance of the Tumor Microenvironment in the Therapeutic Management of Cancer. Exp. Rev. Anticancer Ther. 2015, 15, 943–954. [DOI] [PubMed] [Google Scholar]
- 9. Buggins A. G., Pepper C., Patten P. E., et al. Interaction with Vascular Endothelium Enhances Survival in Primary Chronic Lymphocytic Leukemia Cells via NF-kappaB Activation and De Novo Gene Transcription. Cancer Res. 2010, 70, 7523–7533. [DOI] [PubMed] [Google Scholar]
- 10. Sounni N. E., Noel A. Targeting the Tumor Microenvironment for Cancer Therapy. Clin. Chem. 2013, 59, 85–93. [DOI] [PubMed] [Google Scholar]
- 11. Bosanquet A. G., Richards S. M., Wade R., et al. Drug Cross-Resistance and Therapy-Induced Resistance in Chronic Lymphocytic Leukaemia by an Enhanced Method of Individualised Tumour Response Testing. Br. J. Haematol. 2009, 146, 384–395. [DOI] [PubMed] [Google Scholar]
- 12. Piatkowska M., Styczynski J., Kolodziej B., et al. Individualized Tumor Response Testing Profile Has a Prognostic Value in Childhood Acute Leukemias: Multicenter Non-Interventional Long-Term Follow-Up Study. Leuk. Lymphoma 2013, 54, 1256–1262. [DOI] [PubMed] [Google Scholar]
- 13. Montesinos P., Ballesteros J., Martinez-Cuadron D., et al. An Ex Vivo Native Environment Precision Medicine Test Shows High Clinical Correlation with Responses to First Line Acute Myeloid Leukemia Treatment. Haematologica 2016, 101(suppl. 1), 216. [Google Scholar]
- 14. Kalina T., Flores-Montero J., van der Velden V. H., et al. EuroFlow Standardization of Flow Cytometer Instrument Settings and Immunophenotyping Protocols. Leukemia 2012, 26, 1986–2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. van Dongen J. J., Lhermitte L., Bottcher S., et al. EuroFlow Antibody Panels for Standardized n-Dimensional Flow Cytometric Immunophenotyping of Normal, Reactive and Malignant Leukocytes. Leukemia 2012, 26, 1908–1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Chou T. C. Drug Combination Studies and Their Synergy Quantification Using the Chou-Talalay Method. Cancer Res. 2010, 70, 440–446. [DOI] [PubMed] [Google Scholar]
- 17. Greco W. R., Bravo G., Parsons J. C. The Search for Synergy: A Critical Review from a Response Surface Perspective. Pharmacol. Rev. 1995, 47, 331–385. [PubMed] [Google Scholar]
- 18. Stemberger J., Witt V., Printz D., et al. Novel Single-Platform Multiparameter FCM Analysis of Apoptosis: Significant Differences between Wash and No-Wash Procedure. Cytometry A 2010, 77, 1075–1081. [DOI] [PubMed] [Google Scholar]
- 19. Mittal A. K., Chaturvedi N. K., Rai K. J., et al. Chronic Lymphocytic Leukemia Cells in a Lymph Node Microenvironment Depict Molecular Signature Associated with an Aggressive Disease. Mol. Med. 2014, 20, 290–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Chung C., Lee R. Ibrutinib, Obinutuzumab, Idelalisib, and Beyond: Review of Novel and Evolving Therapies for Chronic Lymphocytic Leukemia. Pharmacotherapy 2014, 34, 1298–1316. [DOI] [PubMed] [Google Scholar]
- 21. Ballesteros J., Scarfo L., Mattsson M., et al. Ex Vivo Lymph Node Native Microenvironment Assay Shows Novel Antiproliferative Activity for Idelalisib and Ibrutinib on CLL Cells. Haematologica 2016, 101(suppl. 1), 426. [Google Scholar]
- 22. Zhang W., Trachootham D., Liu J., et al. Stromal Control of Cystine Metabolism Promotes Cancer Cell Survival in Chronic Lymphocytic Leukaemia. Nat. Cell Biol. 2012, 14, 276–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Viardot A., Goebeler M. E., Hess G., et al. Phase 2 Study of the Bispecific T-Cell Engager (BiTE) Antibody Blinatumomab in Relapsed/Refractory Diffuse Large B-Cell Lymphoma. Blood 2016, 127, 1410–1416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ribera J. M., Ferrer A., Ribera J., et al. Profile of Blinatumomab and Its Potential in the Treatment of Relapsed/Refractory Acute Lymphoblastic Leukemia. Onco. Targets Ther. 2015, 8, 1567–1574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Topp M. S., Gokbuget N., Stein A. S., et al. Safety and Activity of Blinatumomab for Adult Patients with Relapsed or Refractory B-Precursor Acute Lymphoblastic Leukaemia: A Multicentre, Single-Arm, Phase 2 Study. Lancet Oncol. 2015, 16, 57–66. [DOI] [PubMed] [Google Scholar]
- 26. Dombret H., Gardin C. An Update of Current Treatments for Adult Acute Myeloid Leukemia. Blood 2016, 127, 53–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Hodge G., Hodge S., Han P. Increased Levels of Apoptosis of Leukocyte Subsets in Cultured PBMCs Compared to Whole Blood as Shown by Annexin V Binding: Relevance to Cytokine Production. Cytokine 2000, 12, 1763–1768. [DOI] [PubMed] [Google Scholar]
- 28. Ding W., Nowakowski G. S., Knox T. R., et al. Bi-Directional Activation between Mesenchymal Stem Cells and CLL B-Cells: Implication for CLL Disease Progression. Br. J. Haematol. 2009, 147, 471–483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kurtova A. V., Balakrishnan K., Chen R., et al. Diverse Marrow Stromal Cells Protect CLL Cells from Spontaneous and Drug-Induced Apoptosis: Development of a Reliable and Reproducible System to Assess Stromal Cell Adhesion-Mediated Drug Resistance. Blood 2009, 114, 4441–4450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Gill B. J., West J. L. Modeling the Tumor Extracellular Matrix: Tissue Engineering Tools Repurposed towards New Frontiers in Cancer Biology. J. Biomech. 2014, 47, 1969–1978. [DOI] [PubMed] [Google Scholar]
- 31. Mongini P. K., Gupta R., Boyle E., et al. TLR-9 and IL-15 Synergy Promotes the In Vitro Clonal Expansion of Chronic Lymphocytic Leukemia B Cells. J Immunol. 2015, 195, 901–923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Pascutti M. F., Jak M., Tromp J. M., et al. IL-21 and CD40L Signals from Autologous T Cells Can Induce Antigen-Independent Proliferation of CLL Cells. Blood 2013, 122, 3010–3019. [DOI] [PubMed] [Google Scholar]