

 The synthesized novel 6β-pyridinyl amidomorphines exhibit long-lasting antinociception.
The synthesized novel 6β-pyridinyl amidomorphines exhibit long-lasting antinociception.
Abstract
It was previously reported that 6β-aminomorphinan derivatives show high affinity for opiate receptors. Novel 6β-heteroarylamidomorphinanes were designed based on the MOR selective antagonist NAP. The 6β-aminomorphinanes were prepared by stereoselective Mitsunobu reaction and subsequently acylated with nicotinic acid and isonicotinic acid chloride hydrochlorides. The receptor binding and efficacy were determined in vitro and the analgesic activity was studied in vivo. The in vitro studies revealed moderate selectivity for the MOR. At least two compounds in this series exhibited a long-lasting analgesic response when administered subcutaneously and intracerebroventricularly. When the substances were given intracerebroventricularly to mice, they showed analgesic potency comparable to morphine.
Introduction
The perception of pain is a consequence of several complex neurochemical processes both in the peripheral and central nervous system. For the treatment of pain, there are several classes of drugs available. For serious or chronic malignant pains, opioids are the first choice.1 Although opioids are very effective analgesics, their use is associated with deleterious side effects such as dependence, constipation, respiratory depression, addiction liability and abuse.2–4
Opioids act on opioid receptors μ, κ, and δ, which belong to the family of G-protein coupled receptors (GPCRs). Activation of the μ-opioid receptor (MOR) is primarily responsible for the desired analgesic effect. Selective MOR agonists can be useful medications for the treatment of pain and can also serve as probes to better understand the underlying biochemical mechanisms of MOR activation. In 6β-aminomorphinanes, the hydroxyl group in the C-6 position is substituted by an amino group while the configuration of stereogenic C-6 carbon is inverted. Caruso et al. synthesized the first 6-aminomorphinane derivative, chloroxymorphamine, an irreversible MOR agonist.5 It was previously reported that derivatives of 6β-aminomorphinans show selectivity toward opiate receptors, for example, a selective KOR agonist (nalfurafine),6 a selective MOR antagonist (clocinnamox),7 and a dual KOR/DOR agonist (MP1104).8 Several studies have been reported about 6β-aminomorphine derivatives developed in order to achieve higher selectivity to the MOR to mitigate the side effects of opiate analgetics.9,10
McDougall et al. prepared a series of 6β-morphine arylamides by the reaction of 6β-aminomorphine with aryl and heteroaryl acid chlorides.11 These derivatives were found to bind to the MOR with significant potency. Functional assays showed that the compounds were full MOR agonists. Li et al. carried out molecular modeling studies on the MOR and a new lipophilic binding domain was identified.12 Based on the modeling, a series of 6β-heteroaryl naltrexamine analogues were designed and synthesized with 6β side chains including nicotinic, isonicotinic, and isoquinoline carboxylic acid amides. One of the most potent and selective to the MOR in the series was the nicotinic acid derivative, NAP (1), which was chosen as the lead molecule for further studies.12 The compound was found to be a potent peripheral MOR selective antagonist based on in vitro and in vivo pharmacological studies.
We have previously reported the synthesis of a series of novel 6β-cinnamoyl-morphinamines carrying various cinnamoyl side chains.13In vitro and in vivo characterization of the synthesized compounds revealed high affinity for the MOR. The analgesic activity of 6β-cinnamoylmorphinamine (2) was found to be comparable to morphine, but with significantly reduced respiratory depression, a major side effect of commonly used opioids.
Here we report the design, synthesis, in vitro and in vivo evaluation of a series of selective MOR agonists, 6β-heteroarylamidomorphinanes, containing nicotinamide or isonicotinamide moieties at the 6β position (Fig. 1).
Fig. 1. Structures of 6β-amidomorphinanes.

Results and discussion
Chemistry
The 6β-aminomorphinans (6a, 6c, 12) were synthesized using the Mitsunobu reaction as described previously.13,14 Morphine (3c) and 14-hydroxy-dihydromorphine (3a) were selectively protected in the C-3 position with acetic anhydride.15 Two acetyl protected derivatives (4a, 4c) and codeine (10) were reacted in the Mitsunobu reaction with phthalimide and diisopropyl azodicarboxylate (DIAD) in the presence of triphenylphosphine to yield 6β-phthalimido morphinans (5a, 5c, 11). The desired 6β-aminomorphinan derivatives (6a, 6c, 12) were obtained upon treatment with hydrazine hydrate. The synthesis of 6β-aminodihydromorphine (6e) was accomplished from 6β-azidodihydromorphine (7). Bognár and Makleit reported the reduction of 6β-azidodihydromorphine (7) by lithium aluminum hydride in diethyl ether.16 We elaborated an efficient new method for this reduction, utilizing hydrazine and RANEY® nickel in ethanol.17 With the new method, higher yields were achieved, work-up was easier and the final product did not require further purification (Fig. 2).
Fig. 2. Synthesis of 6β-amidomorphinans 9a–g. a) Acetic anhydride, NaHCO3 water r.t. 2 h; b) benzene/Ph3P, phthalimide, DIAD, r.t. 1 h; c) ethanol/hydrazine monohydrate, heating 3 h; d) hydrazine monohydrate/RANEY® nickel, ethanol, r.t. 2 h; e) dichloromethane/acyl chloride, triethylamine; f) K2CO3/methanol, heating 4 h.

The 6β-aminomorphinans (6a–e) were reacted with nicotinic acid chloride and isonicotinic acid chloride in dry dichloromethane in the presence of triethylamine to yield the appropriate 6β-amidomorphinans. The acyl chlorides were synthesized as described in the literature.18 In the case of molecules containing a free phenolic group (5a–e), the ester by-products were hydrolyzed with potassium carbonate in methanol.
Binding studies
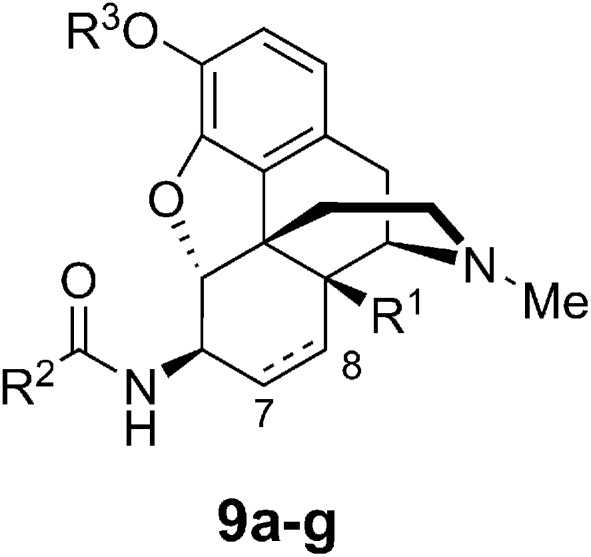
The seven compounds used in in vitro and in vivo studies are summarized in Table 1. Codeine derivative 9g showed a significantly lower binding affinity compared with the morphine derivatives, suggesting that the free phenolic hydroxyl group is necessary for affinity at the receptor (Fig. 3).
Table 1. Structure of the studied compounds.
| Compound | C7–C8 bond | R1 | R2 | R3 |
| 9a | Single | OH | Isonicotinic | H |
| 9b | Single | OH | Nicotinic | H |
| 9c | Double | H | Isonicotinic | H |
| 9d | Double | H | Nicotinic | H |
| 9e | Single | H | Isonicotinic | H |
| 9f | Single | H | Nicotinic | H |
| 9g | Double | H | Isonicotinic | Me |
Fig. 3. General structure of the studied compounds.
Compounds 9a–f show similar nanomolar affinity for the MOR and much lower affinity for the DOR and KOR. In all cases, the difference in MOR affinities between the compounds possessing nicotinic acid and isonicotinic acid moieties was minimal; however, there are greater differences in affinities for the DOR and KOR. Compounds 9a and 9b having the 14-hydroxy dihydromorphine skeleton showed the lowest affinity to the MOR. The dihydromorphine derivatives 9e and 9f had the highest MOR/DOR selectivity among the studied molecules. The high MOR selectivity reported for the NAP, however, was not seen in this group. 9c and 9d had the highest affinity to the MOR. 9a only differs from NAP (1) in the substituent of the tertiary amine. The cyclopropylmethyl substituent of NAP (1) is responsible for the antagonistic effect; on the other hand, the methyl substituent in our compounds results in an agonistic effect. These results revealed that the saturation of the double bond and the presence of the OH group at 14-position are not beneficial in terms of binding (Table 2).
Table 2. Binding affinities of compounds (9a–f) a .
| Compound | MOR | KOR | DOR | M/K | M/D |
|
K
i (nM) | |||||
| 9a | 7.02 ± 0.77 | >100 | 173.4 ± 43.6 | >14.2 | 24.7 |
| 9b | 6.91 ± 0.88 | >100 | 103.7 ± 17.0 | >14.5 | 15.0 |
| 9c | 2.00 ± 0.17 | 77.9 ± 31 | 65 ± 19.0 | 39.0 | 32.5 |
| 9d | 1.25 ± 0.23 | 18.97 ± 5.3 | 34.4 ± 5.00 | 15.2 | 27.5 |
| 9e | 3.73 ± 0.14 | >100 | 198.4 ± 69 | >26.8 | 53.2 |
| 9f | 3.29 ± 0.36 | 119 ± 50.4 | 130.3 ± 37 | 36.2 | 39.6 |
| 9g | 40.59 ± 3.65 | >100 | 423.9 ± 88 | >2.5 | 10.4 |
| 1 b | 0.37 ± 0.07 | 277.51 ± 7.97 | 60.72 ± 5.58 | 747 | 163 |
| 2 c | 0.10 ± 0.02 | 2.90 ± 0.66 | 10.26 ± 6.76 | 29.0 | 102.6 |
| Morphine | 4.60 ± 1.81 | ||||
a[125I]BNtxA (0.1 nM) competition binding assays were performed in membranes prepared from CHO cells expressing mouse MOR, DOR, or KOR, as previously described. Protein concentrations were between 10–20 μg mL–1 and the incubation time was 90 minutes.19Ki was determined by nonlinear regression analysis (GraphPad Prism). Assays were performed at least 3 times and means ± SEM are reported.
bValues are taken from ref. 12.
cValues are taken from ref. 13.
The [35S]GTPγS-binding assay was used to characterize the functional activity of compounds 9a–fin vitro on opioid transfected cell lines. In the [35S]GTPγS assay, all compounds were full agonists. 9c and 9d sharing the same morphine skeleton had the highest potencies. The EC50 value on the KOR was only determined for 9d, since no other analogs had considerable affinity for this receptor. It is well known in the literature that the saturation of the double bond and the presence of the 14-OH group increase the affinity of morphine derivatives. In our case, saturation of the double bond and the presence of the 14-OH group were not advantageous as these modifications decreased both the affinity and activity (Table 3).20,21
Table 3. In vitro efficacy of the compounds in the [35S]GTPγS assay a .
| Compound | MOR |
KOR |
||
| EC50 (nM) | %Emax | EC50 (nM) | %Emax | |
| 9a | 26.42 ± 3.17 | 115.07 ± 7.3 | nd b | nd b |
| 9b | 25.7 ± 1.68 | 120.89 ± 2.55 | nd b | nd b |
| 9c | 4.62 ± 0.28 | 114.36 ± 7.06 | nd b | nd b |
| 9d | 2.83 ± 0.41 | 113.63 ± 4.54 | 58.83 ± 7.49 | 87.88 ± 4.9 |
| 9e | 8.16 ± 2.64 | 114.85 ± 1.26 | nd b | nd b |
| 9f | 7.98 ± 2.35 | 117.16 ± 1.98 | nd b | nd b |
| 2 | 1.38 ± 0.8 | 95.3 ± 0.8 | 36.5 ± 19.2 | 106.3 ± 8.5 |
| DAMGO | 19 ± 7.0 | nd b | nd b | nd b |
| U50,488H | nd b | nd b | 17 ± 6.1 | nd b |
aFunctional properties were studied using agonist-induced stimulation of the [35S]GTPγS binding assay. Potency is represented as EC50 (nM) and efficacy as percent maximal stimulation (%Emax) relative to standard agonist DAMGO (MOR), or U50,488H (KOR) at 100 nM. The protein concentration was 50 μg mL–1 and the incubation time was 90 minutes. All values are expressed as mean ± SEM of three separate assays performed in triplicate.
bNot determined.
In vivo studies
All animal studies have been reviewed and approved by the IACUC. The animal care systems of the MSKCC are fully accredited by AAALAC and USDA are in compliance with the “Guide For the Care and Use of Laboratory Animals”. We are also in compliance with the Animal Welfare Act and agree to adhere to the Public Health Service “Principles for the Use of Animals” (NIH Manual Chapter 4206). The antinociceptive activity of the four compounds (9c–f) with the highest affinity and agonism for the MOR was determined supraspinally in mice (Fig. 4) in a tail flick antinociception assay. While 9d was as potent as morphine (ED50 = 0.53 μg (0.27, 1.0)) given icv, the other three compounds were 3–4× less potent antinociceptives. Upon subcutaneous administration, the compounds did exhibit an antinociceptive response at 30 mg kg–1 (solubility limitations didn't permit testing at higher doses). At this high dose, 9c and 9d exhibited about 30% MPE, while 9e and 9f exhibited ∼80% MPE (Fig. 5A). The antinociceptive ED50 of morphine was 2.5 mg kg–1 (1.8, 3.4) in the same assay. We evaluated 9e and 9f further. Both compounds showed exceptionally long-lasting antinociception. In contrast to morphine's 3 h long antinociceptive response, the effect of 9e and 9f lasted more than 24 h subcutaneously (Fig. 5B). Similarly, upon icv dosing, unusually long antinociception was observed, lasting more than 24 h. This shows good correlation with the sc time course. Antinociception of both 9e and 9f was antagonized by β-FNA (Fig. 5D), a mu selective antagonist, confirming that the antinociception is mediated by mu opioid receptors.
Fig. 4. Two independent determinations of the cumulative dose–response curves were performed on groups of mice (n = 5) for antinociception in the tail flick assay with the compounds given intracerebroventricularly. The animals were tested 15 min later at the peak effect to generate the antinociceptive dose–response curve. Each point represents mean ± SEM for 10 mice. ED50 values (and 95% CI) were: 9c: 2.0 μg (1.1, 3.6); 9d: 0.51 μg (0.3, 0.9); 9e: 0.7 μg (0.4, 1.2); 9f: 1.8 μg (1.0, 3.2); morphine: 0.53 μg (0.27, 1.0).

Fig. 5. A) Antinociceptive effect of compounds 9c–f. CD1 mice (n = 10) were given the compounds sc at 30 mg kg–1 and the antinociceptive effects were assessed 30 min post-injection using the radiant heat tail-flick method. Results (mean ± SEM): 9c: 38% ± 13%; 9d: 35% ± 10%; 9e: 78% ± 11%; 9f: 83% ± 11%. B) Time course of the antinociceptive effect of morphine (4.5 mg kg–1), compound 9e (30 mg kg–1), and 9f (30 mg kg–1) following sc administration to CD1 mice. Two independent determinations were performed on groups of mice (n = 10) for antinociception in the tail flick assay. C) Time course of the antinociceptive effect of morphine (1 μg), compound 9e (7.5 μg), and 9f (7.5 μg) following icv administration to CD1 mice. Two independent determinations were performed on groups of mice (n = 10) for antinociception in the tail flick assay. D) Reversal of antinociception by selective antagonists. Groups of CD1 mice (n = 10) received 9e (30 mg kg–1, sc), or 9f (30 mg kg–1) with or without β-funaltrexamine (β-FNA; 40 mg kg–1, sc). β-FNA was administered 24 hours before agonist testing. All antinociception testing was performed 30 min after the administration of 9e and 9f. Similar results were observed in two independent replications. Antinociception was antagonized by β-FNA (two-way ANOVA followed by the Bonferroni post hoc comparison test, p < 0.05). The means in each determination were determined as the percentage maximal possible effect (%MPE) [(observed latency – baseline latency)/(maximal latency – baseline latency)] × 100. The baseline latencies were between 2–3 s and the maximal latency was 10 s to avoid tissue damage.

Thus, these drugs do exhibit systemic antinociception, albeit at a higher dose. Their long-lasting antinociception makes these compounds exciting for further evaluation beyond thermal pain assays such as inflammatory and neuropathic pain models. The next set of analogs in this series will aim to optimize the potency of the compounds while maintaining long-lasting antinociception.
Several studies have been reported about NAP (1) as a selective peripheral MOR antagonist based on its in vitro/in vivo pharmacological assays and pharmacokinetic studies.12,22,23 To our knowledge, antinociceptive C-6 arylamido morphine with 3′-pyridyl or 4′-pyridyl as the aryl moiety has not been reported before. The pharmacology on C-6 amido morphinans has indeed been extensively studied but the SAR of novel compounds cannot be predicted based on that of known compounds. Subtle changes in the structure can have a huge impact on the in vivo pharmacology. It is well established that compounds with an N-methyl substituent such as morphine and oxymorphone are agonists/antinociceptives. N-Allyl and N-cyclopropylmethyl (N-CPM) lead to the antagonists naloxone and naltrexone, respectively. Some C-6 arylamido morphinans do follow this trend where the N-methyl leads to agonists/antinociceptives.11,13 Compounds with an N-CPM like NAP (ref. 12) from the Zhang group are antagonists and N-CPM analogs24 are partial agonists. IBNtxA (ref. 25) and MP 1104 (ref. 8) are agonists/antinociceptives even with an N-CPM moiety. The exceptionally long-lasting antinociception seen with 9e and 9f described in this paper has never been reported and is unexpected based on the published work on C-6 amido morphinans, suggesting the possibility of developing novel therapeutics with this template after further optimization of potency.
Conclusions
In summary, a series of 6-desoxymorphine-6β-acylamides containing nicotinic or isonicotinic acid moieties were synthesized. The acyl side chains were chosen based on selective MOR antagonists reported in the literature. We found that the synthesized compounds are MOR full agonists with low affinity for the DOR and KOR. The compounds exhibited potent antinociception supraspinally and showed exceptionally long-lasting mu opioid receptor-mediated antinociception when given subcutaneously.
Supplementary Material
Acknowledgments
This work was supported by a research grant from the National Institute on Drug Abuse (DA034106) to SM. This research was funded, in part, through the NIH/NCI Cancer Center Support Grant P30 CA008748.
Footnotes
†The authors declare no competing interests.
‡Electronic supplementary information (ESI) available. See DOI: 10.1039/c6md00450d
References
- Trescot A. M., Datta S., Lee M., Hansen H. Pain Physician. 2008;11:S133. [PubMed] [Google Scholar]
- Hayes A. G., Tyers M. Br. J. Pharmacol. 1983;79:731. doi: 10.1111/j.1476-5381.1983.tb10011.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiara G. D., North R. A. Trends Pharmacol. Sci. 1992;13:185. doi: 10.1016/0165-6147(92)90062-b. [DOI] [PubMed] [Google Scholar]
- Loh H. H., Liu H.-C., Cavalli A., Yang W., Chen Y.-F., Wei L.-N. Mol. Brain Res. 1998;54:321. doi: 10.1016/s0169-328x(97)00353-7. [DOI] [PubMed] [Google Scholar]
- Caruso T., Takemori A., Larson D., Portoghese P. Science. 1979;204:316. doi: 10.1126/science.86208. [DOI] [PubMed] [Google Scholar]
- Kumagai H., Ebata T., Takamori K., Muramatsu T., Nakamoto H., Suzuki H. Nephrol., Dial., Transplant. 2010;25:1251. doi: 10.1093/ndt/gfp588. [DOI] [PubMed] [Google Scholar]
- Comer S. D., Burke T. F., Lewis J. W., Woods J. H. J. Pharmacol. Exp. Ther. 1992;262:1051. [PubMed] [Google Scholar]
- Váradi A., Marrone G. F., Eans S. O., Ganno M. L., Subrath J. J., Le Rouzic V., Hunkele A., Pasternak G. W., McLaughlin J. P., Majumdar S. ACS Chem. Neurosci. 2015;6:1813. doi: 10.1021/acschemneuro.5b00153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caruso T. P., Larson D. L., Portoghese P. S., Takemori A. E. J. Pharmacol. Exp. Ther. 1980;213:539. [PubMed] [Google Scholar]
- Ward S. J., LoPresti D., James D. W. J. Pharmacol. Exp. Ther. 1986;238:625. [PubMed] [Google Scholar]
- MacDougall J. M., Zhang X.-D., Polgar W. E., Khroyan T. V., Toll L., Cashman J. R. Bioorg. Med. Chem. 2004;12:5983. doi: 10.1016/j.bmc.2004.08.019. [DOI] [PubMed] [Google Scholar]
- Li G., Aschenbach L. C., Chen J., Cassidy M. P., Stevens D. L., Gabra B. H., Selley D. E., Dewey W. L., Westkaemper R. B., Zhang Y. J. Med. Chem. 2009;52:1416. doi: 10.1021/jm801272c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Váradi A., Hosztafi S., Le Rouzic V., Tóth G., Urai Á., Noszál B., Pasternak G. W., Grinnell S. G., Majumdar S. Eur. J. Med. Chem. 2013;69:786. doi: 10.1016/j.ejmech.2013.09.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon C., Hosztafi S., Makleit S. Synth. Commun. 1992;22:913. [Google Scholar]
- Welsh L. H. J. Org. Chem. 1954;19:1409. [Google Scholar]
- Bognár R., Makleit S. Acta Chim. Acad. Sci. Hung. 1968;58:203. [PubMed] [Google Scholar]
- Malik A. A., Preston S. B., Archibald T. G., Cohen M. P., Baum K. Synthesis. 1989;1989:450. [Google Scholar]
- Christensen J. Molecules. 2001;6:47. [Google Scholar]
- Váradi A., Palmer T. C., Haselton N., Afonin D., Subrath J. J., Le Rouzic V., Hunkele A., Pasternak G. W., Marrone G. F., Borics A., Majumdar S. ACS Chem. Neurosci. 2015;6:1570. doi: 10.1021/acschemneuro.5b00137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbert A.-K., Hosztafi S., Mahurter L., Pasternak G. W. Eur. J. Pharmacol. 2004;492:123. doi: 10.1016/j.ejphar.2004.03.050. [DOI] [PubMed] [Google Scholar]
- Casy A. F. and Parfitt R. T., Opioid Analgesics: Chemistry and Receptors, Springer, US, 2013. [Google Scholar]
- Yuan Y., Li G., He H., Stevens D. L., Kozak P., Scoggins K. L., Mitra P., Gerk P. M., Selley D. E., Dewey W. L., Zhang Y. ACS Chem. Neurosci. 2011;2:346. doi: 10.1021/cn2000348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitra P., Venitz J., Yuan Y., Zhang Y., Gerk P. M. Drug Metab. Dispos. 2011;39:1589. doi: 10.1124/dmd.111.038588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghirmai S., Azar M. R., Polgar W. E., Berzetei-Gurske I., Cashman J. R. J. Med. Chem. 2008;51:1913. doi: 10.1021/jm701060e. [DOI] [PubMed] [Google Scholar]
- Majumdar S., Grinnell S., Le Rouzic V., Burgman M., Polikar L., Ansonoff M., Pintar J., Pan Y.-X., Pasternak G. W. Proc. Natl. Acad. Sci. U. S. A. 2011;108:19778. doi: 10.1073/pnas.1115231108. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.