

Abstract
Purpose
To characterize a light damage paradigm and establish structural and immunocytochemical measures of acute and protracted light-induced retinal degeneration in the rhodopsin (RHO) T4R dog model of RHO–autosomal dominant retinitis pigmentosa (ADRP).
Methods
Retinal light damage was induced in mutant dogs with a 1-minute exposure to various light intensities (0.1–1.0 mW/cm2) delivered with a Ganzfeld stimulator, or by fundus photography. Photoreceptor cell death was assessed by TUNEL assay, and alterations in retinal layers were examined by histology and immunohistochemistry 24 hours and 2 weeks after light exposure. Detailed topographic maps were made to document changes in the outer retinal layers of all four retinal quadrants 2 weeks post exposure.
Results
Twenty-four hours post light exposure, the severity of photoreceptor cell death was dose dependent. Immunohistochemical analysis revealed disruption of rod outer segments, focal loss of the RPE integrity, and an increase in expression of endothelin receptor B in Müller cells with the two highest doses of light and fundus photography. Two weeks after light exposure, persistence of photoreceptor death, thinning of the outer nuclear layer, and induction of Müller cell gliosis occurred with the highest doses of light.
Conclusions
We have characterized outcome measures of acute and continuing retinal degeneration in the RHO T4R dog following light exposure. These will be used to assess the molecular mechanisms of light-induced damage and rescue strategies in this large animal model of RHO-ADRP.
Keywords: retinal degeneration, light damage, rhodopsin, ADRP, canine model
Decades of exposure to light is considered to influence retinal damage in AMD,1 and it may play a role in modifying the disease course in human patients and animal models of RP2,3 as well as in a subset of inherited retinal diseases in animal models such as Usher's syndrome4 and Leber congenital amaurosis.5,6 Autosomal dominant RP accounts for 30% to 40% of all RP patients, and of these, 30% carry a mutation in the rhodopsin gene (RHO). These patients have been functionally categorized into class A and class B.7 Class A patients exhibit early panretinal rod dysfunction; but the onset of disease in class B patients occurs in adulthood and has a slow progression that affects the inferonasal and pericentral areas more severely than the superotemporal retinal region (subclass B1). This patient subclass also exhibits a delay in dark adaptation kinetics after exposure to light that bleaches >95% of available rhodopsin,8 and a potential for light exposure to trigger and/or contribute to photoreceptor death.3,9 Experimental evidence for increased susceptibility to light damage has been demonstrated in several animal models carrying class B1 RHO mutations, including T4R,9 T4K,10,11 T17M,12 P23H,13–15 Y102H,16 I307N,16 and S334ter.17 These and other studies have fueled the debate about the detrimental impact of constant light “pollution” in our modern societies,18 and the need to consider light restriction measures particularly in patients affected with some forms of retinal degeneration.3
The naturally occurring RHO T4R mutation in the dog8 presents an extreme model of such light-induced damage, where severe membrane disruption of the rod outer segments (OS) can be observed within minutes of acute clinical light exposure, followed by the onset of photoreceptor cell death 6 hours later.9,19 The mutant retina undergoes rapid degeneration with short-duration light intensities commonly found under normal environmental conditions. For this reason, it is an optimal large animal model in which to study the molecular pathways that link a photoreceptor-specific gene mutation with light-induced photoreceptor cell death, and to test rescue strategies such as corrective gene therapy or neuroprotective/pharmacologic treatments.
In previous studies, we established doses that cause light damage, examined cell death pathways during the first 6 hours following light exposure, and demonstrated that photoreceptor loss continues for several weeks following the initial insult.20–23 In the present study, to establish structural outcome measures that can be used to assess the effect of therapeutic intervention for RHO-ADRP in this large animal model, we examined the topography of light-induced photoreceptor loss in the first 2 weeks after exposure and identified concomitant and secondary alterations in other retinal layers that contribute to the degenerative process.
Materials and Methods
Animals and Rearing Conditions
Retinas from 15 mastiff crossbred RHO T4R (homozygous or heterozygous) mutant and 2 wild-type (WT) control dogs were used (see Table 1). All dogs (age range, 11–75 weeks) were maintained at the Retinal Disease Studies facility (Kennett Square, PA, USA). The studies adhered to the ARVO Statement for the Use of Animals in Ophthalmic and Vision Research and were approved by the Institutional Animal Care and Use Committee of the University of Pennsylvania. The dogs' genotypes were determined either from the known status of their parents, or from genetic testing for the disease-causing mutation.8
Table 1.
Summary of Dogs and Light-Exposure Conditions
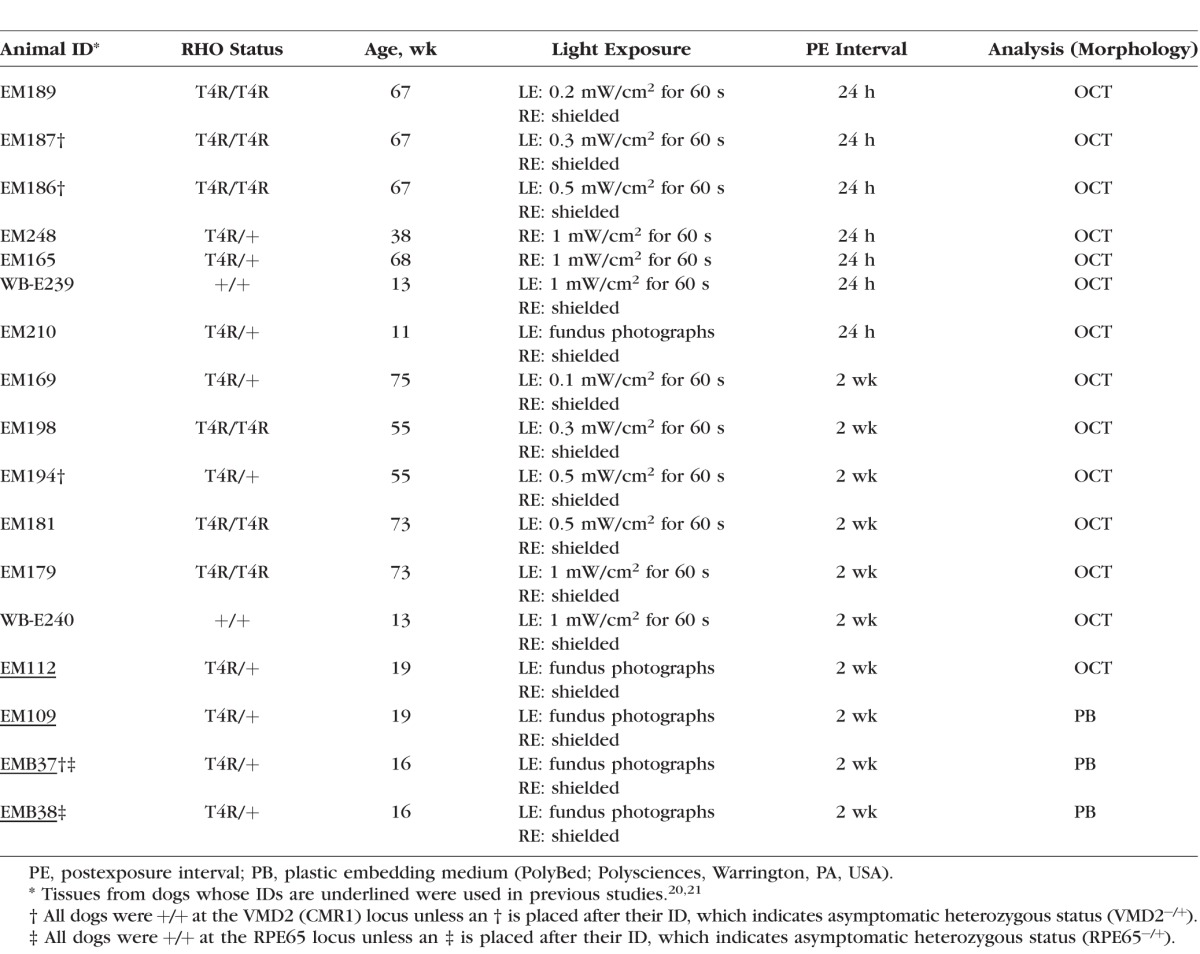
Animals were housed as previously described20 in kennels with dim cyclic illumination, consisting of 12 hours of white light per day (175 to 350 lux at the level of the “standard dog eye”) and 12 hours of darkness (lights on at 6 AM and off at 6 PM). In the procedure room, light intensities range from 350 to 700 lux. These ambient light levels do not cause retinal damage in normal or homozygous mutant prcd dogs that do not carry the RHO T4R mutation.20 To avoid any potential light-induced damage, none of the animals underwent a clinical ophthalmic or ERG examination.
Retinal Illumination
Animals were dark adapted overnight. On the following morning, the dogs were prepared for retinal illumination as previously described.22 With two exceptions, the right eye (RE) was shielded and the left eye (LE) was exposed to light.
Two different approaches were used to illuminate the retinas for light-damage experiments. The first approach used the background white light (6500 K) of an Espion electrophysiology system (Diagnosys LLC, Lowell, MA, USA) delivered through a monocular Ganzfeld stimulator (ColorBurst; Diagnosys LLC). The retinas were exposed for 1 minute with different intensities measured with a luminometer (IL1700; International Light Technologies, Peabody, MA, USA), which resulted in corneal irradiance values of 1, 0.5, 0.3, 0.2, and 0.1 mW/cm2. When measured as illuminance, these light intensities correspond to 1590, 820, 551, 319, and 170 lux, respectively. The second method to light expose the retinas consisted in photographing the retina with a fundus camera. This approach has been previously reported in other studies to induce light damage in the RHO T4R model.9,20,21 In brief, 15 to 17 overlapping retinal photographs of microsecond duration were taken over a period of approximately 5 to 7 minutes with a handheld fundus camera (RC-2; Kowa Ltd., Nagoya, Japan). Light intensities used to view and photograph various retinal regions (tapetal versus nontapetal) were previously described.9 Both methods used to create light damage in the RHO T4R mutant retina do not cause any retinal lesions in normal retinas.20,22,23
Following light exposure, dogs were kept under dim-red illumination until the following day when they were either humanely euthanized 24 hours post exposure or returned to the kennel for 2 weeks. Dogs were humanely euthanized in a dark room under dim-red illumination with an intravenous injection of pentobarbital sodium. After enucleation, a slit incision was made at the level of the ora serrata, and the entire globe was fixed in paraformaldehyde, cryoprotected, and embedded in optimal cutting temperature (OCT) medium as previously described.24
TUNEL Assay for Cell Death
The occurrence of cell death was assessed on retinal cryosections (10-μm thick) extending from the optic nerve head to the ora serrata along the four meridians (superior, inferior, nasal, and temporal) at 24 hours and 2 weeks following light exposure, by imaging DNA fragmentation using a TUNEL assay as previously reported.24 In some retinal regions, the overwhelming number of photoreceptors that were TUNEL-labeled precluded any manual or automated cell count; therefore, a semiquantitative grading system (see Table 2; Fig. 1A) was established for the 24-hour postexposure time point and used to produce topographic maps of the distribution and severity of cell death in the outer nuclear layer (ONL).
Table 2.
Grading System Used to Establish the Incidence of TUNEL-Labeling in 10-μm–Thick Retinal Sections 24 Hours Following Light Exposure

Figure 1.

Spatial distribution of photoreceptor cell death 24 hours following light exposure in RHO T4R mutant dogs. (A) Grading system used to map the incidence of TUNEL-positive cells in the ONL. (B1–B4, C) Schematic color-coded representation of grades of TUNEL-labeling in retinal sections extending from the edge of the optic nerve head to the superior, inferior, nasal, and temporal ora serrata: (B1–B4) four RHO T4R dogs exposed to decreasing doses of light by means of a Ganzfeld stimulator; (C) a RHO T4R dog exposed to a series of overlapping fundus photographs. LE, left eye; RE, right eye.
Immunohistochemistry
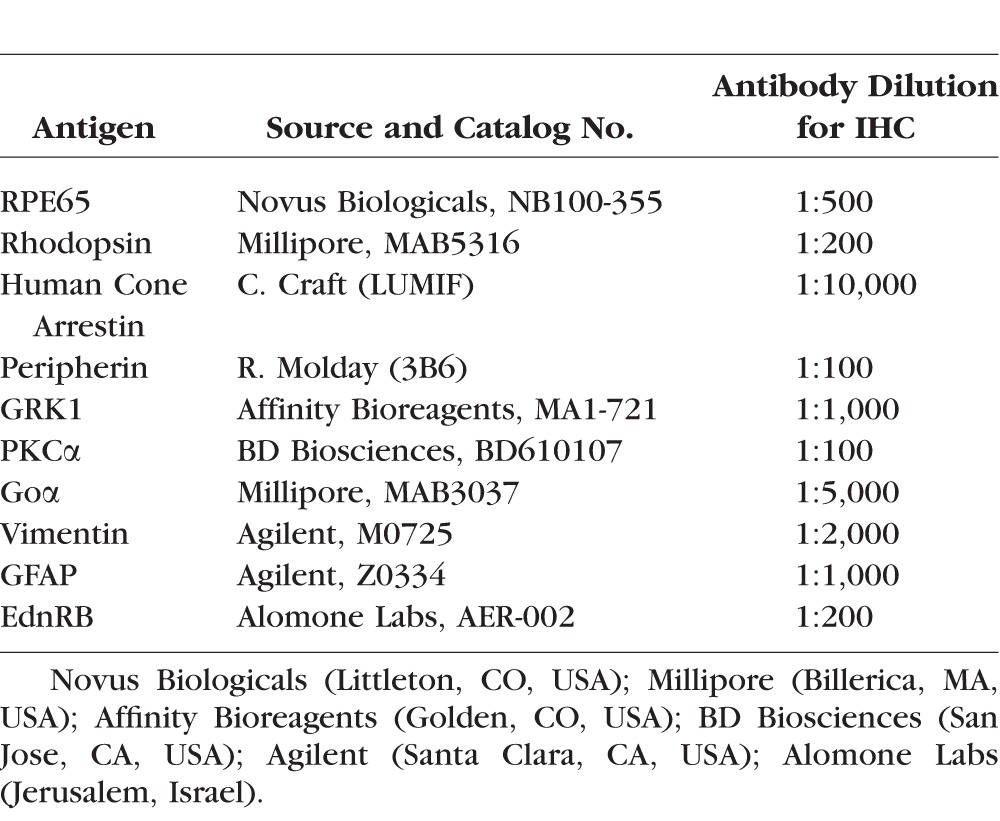
Assessment of the morphology and immunoreactivity of several retinal cell populations was done at 24 hours and 2 weeks following light exposure, by fluorescence immunohistochemistry (IHC) using a battery of cell-specific antibodies (see Table 3) on 10-μm-thick cryosections that extended along the superior (tapetal) and inferior (nontapetal) meridians. Specificity of immunostaining with endothelin receptor B (EdnRB) was demonstrated by incubating 1 μg EdnRB antibody with 1 μg commercially available blocking peptide for 1 hour at room temperature prior to incubation on the sections. DAPI stain was used to label nuclei. Sections were viewed under a widefield epifluorescent microscope (Axioplan; Carl Zeiss Meditec GmbH, Oberkochen, Germany), and images were digitally captured (Spot 4.0 camera; Diagnostic Instruments, Inc., Sterling Heights, MI, USA) and imported into a graphics program (Photoshop; Adobe, Mountain View, CA, USA) for display. A detailed protocol has been described previously.24
Table 3.
List of Antibodies Used for IHC
Retinal Morphology and Morphometry
Retinal cryosections (10-μm thick) extending along the four meridians were stained with hematoxylin and eosin (H&E) and examined under bright field microscopy (Axioplan; Carl Zeiss Meditec) with a 40× objective. Outer nuclear layer thickness was based on the number of ONL nuclei counted every 1000 μm. In addition, the location and morphology of the photoreceptor inner segments (IS) and OS, the RPE, and the tapetum lucidum (a cellular multilayered reflective structure located in the superior region of the choroid) were observed, manually recorded, and reported in a graphic format. Specific criteria examined included density and/or length of IS and OS, pigmented status of the RPE and altered morphology (e.g., hypertrophy), and presence or absence of a tapetum.
Results
Light Exposure Causes Acute Dose-Dependent Photoreceptor Cell Death in the RHO T4R Retina
A dose-dependent cell death response was observed in the ONL of RHO T4R mutant retinas 24 hours after a 1-minute exposure period of varying intensities of white light delivered by means of a Ganzfeld stimulator (Figs. 1B1–B4). With the highest dose of light (1 mW/cm2 for 1 minute), there was massive TUNEL-labeling of photoreceptors (grade 4) in the central region of the retina with a decreasing gradient of TUNEL-positive cells from center to periphery (Fig. 1B1). With a 50% decrease in light dose (0.5 mW/cm2 for 1 minute), the most severe damage (grade 4) was limited to the central region of both the superior and temporal meridians, while there were fewer TUNEL-labeled cells (grades 3 and 2) in the nasal and inferior meridians (Fig. 1B2). A dose of 0.3 mW/cm2 for 1 minute caused moderate cell death in the central retina that did not exceed grade 2 (Fig. 1B3). After exposure with the lowest dose (0.2 mW/cm2 for 1 minute), a limited number of TUNEL-labeled photoreceptors (grade 1) were found, predominantly in the tapetal region (superior and temporal meridians) (Fig. 1B4). No cell death was seen in a WT retina exposed to the highest dose (1 mW/cm2 for 1 minute) of light (Supplementary Figs. S1A–C).
Light damage triggered by a series of overlapping fundus photographs caused a pattern of cell death severity that was similar to that obtained after Ganzfeld illumination at 1 mW/cm2 for 1 minute. Grade 4 was observed in the central retina throughout the four meridians (Fig. 1C). One difference between the two light-damage paradigms appeared to be the more limited distribution of cell death throughout the retinal expanse, which likely resulted from anatomic limitations because it is more difficult to visualize and photograph the more peripheral regions with a handheld fundus camera in the awake dog.
Some morphologic variation in the pattern of TUNEL-labeling could be seen when comparing areas with grade 4 to areas less severely affected. In central regions with grade 4, many TUNEL-labeled figures in the ONL appeared swollen, with an irregular contour, and decreased fluorescence in contrast to the more typical TUNEL-positive nuclei that were seen more peripherally (Figs. 2A1, 2A2). In addition, TUNEL-positive RPE cells were seen with grade 4 in the central retina after fundus photography (Fig. 2A3).
Figure 2.

TUNEL-labeling patterns and ONL swelling. (A1) Irregular TUNEL-labeled nuclear morphology of photoreceptors (white arrows) in the central versus midperipheral retina 24 hours after 1-minute Ganzfeld light exposure. (A2) Regular nuclear morphology of TUNEL-labeled photoreceptors (red arrows) in the midperipheral retina 24 hours after 1-minute Ganzfeld light exposure. (A3) Irregular TUNEL-labeled nuclear morphology of photoreceptors (white arrows) and TUNEL-labeled RPE cells (blue arrows) in central retina 24 hours after fundus photography. (B1, B2) ONL swelling in central versus midperipheral retina 24 hours after 1-minute Ganzfeld light exposure. (B3) ONL swelling in central retina 24 hours after fundus photography. In both cases, the irregular swelling caused undulations of the ONL. T, tapetum lucidum.
Light Exposure Causes Acute Outer Retinal Alterations and Müller Cell Response
We first examined the effect of light at the 24-hour time point following exposure and assessed the effects on the retina using cryosections stained with H&E or immunolabeled. Following both fundus photography and Ganzfeld exposure with the two highest doses of light (0.5 and 1 mW/cm2 for 1 minute), an increase in photoreceptor internuclear spacing was seen centrally, causing an apparent increase in ONL thickness (Figs. 2B1–B3). The RPE layer also showed damage and apparent cell loss centrally as reflected by focal loss of RPE65 immunolabeling (Figs. 3A2, 3A4). Staining with rod-OS specific markers (rhodopsin, peripherin, and GRK1) showed significant disruption of the OS centrally (Figs. 3B2, 3B4, 3C2, 3C4, 3D2, 3D4) but not in a WT dog (Supplementary Fig. S1C). Under the same exposure protocols, the ONL, OS, and RPE were well preserved in the periphery (Figs. 3B3, 3C3, 3D3), and the pattern of immunolabeling was like that seen in the central region of a shielded RHO T4R retina (Figs. 3A1, 3B1, 3C1, 3D1) or in the exposed retinas of WT controls (Supplementary Fig. S1D2, data not shown). At this time point, no obvious changes in the structure of bipolar cells were seen, although some distortion of the lamination of the inner nuclear layer (INL) and shrinkage of the outer plexiform layer (OPL) was observed in the central/exposed RHO T4R retina (Figs. 3E1–E4). These alterations were likely the consequence of the swelling of the overlying ONL. Müller cells, whose radial extension through the retinal thickness was clearly immunolabeled with vimentin, showed no increase in glial fibrillary acid protein (GFAP) staining, a marker of reactive gliosis; however, there was an increase in expression of EdnRB in the central exposed retinas (Figs. 3F1–F4, 3G1–G4). Immunohistochemical alterations were also found in RHO T4R retinas exposed to lower doses of light (0.5 mW/cm2 and 0.3 mW/cm2) for 1 minute, but no obvious changes were seen with exposure to 0.2 mW/cm2 (Table 4).
Figure 3.

Immunohistochemical characterization of light-induced damage 24 hours post exposure in RHO T4R mutant dogs. (A1–A4) RPE-specific protein 65 kDa (RPE65). (B1–B4) RHO and human Cone Arr. (C1–C4) Peripherin. (D1–D4) GRK1. (E1–E4) PKCα and (Goα). (F1–F4) Vim. and GFAP. (G1–G4) Vim. and EdnRB. IPL, inner plexiform layer; NFL, nerve fiber layer; Cone Arr., cone arrestin; PKCα, protein kinase C alpha; GFAP, glial fibrillary acidic protein; Goα, Go-alpha.
Table 4.
Immunohistochemical Alterations in RHO T4R Retina 24 Hours After a 1-Minute Exposure to Light

Light Exposure Causes Rapid Retinal Degeneration in the RHO T4R Dog
To examine the effect of light at 2 weeks post exposure, retinas from dogs exposed to similar light-damage protocols were processed for histologic/IHC evaluation. Biaxial spider graphs that extended along the horizontal (nasal-temporal) and vertical (inferior-superior) axes were constructed. Careful attention to the structure of the IS, OS, and RPE was documented in sections extending from the optic disc to the ora serrata in both tapetal and nontapetal areas. By 2 weeks post exposure, the retina that had received the highest dose of light (1 mW/cm2 for 1 minute) had an ONL thickness reduced to 1 to 2 rows of nuclei over a distance extending more than 10 mm from the optic nerve head and that included the central and midperipheral regions of all four quadrants (Fig. 4A1). This was associated with a loss of photoreceptor IS and OS. Loss of RPE integrity was found particularly in the superior tapetal area, whereas RPE hypertrophy was commonly seen in nontapetal areas (Fig. 5). Exposure to half of the highest dose (0.5 mW/cm2 for 1 minute) caused ONL thinning, particularly in sections extending through tapetal areas (superior and temporal). Loss or shortening of IS and OS was also seen, but the RPE was preserved (Figs. 4A2, 4A3). Exposure to 0.3 mW/cm2 of light for 1 minute caused photoreceptor damage and some ONL thinning that was limited to the central to midperipheral tapetal areas (Fig. 4A4). No evidence of photoreceptor loss was seen in a retina exposed to 0.1 mW/cm2 for 1 minute, corresponding to one-tenth of the highest dose of light, although loss of OS was found in one focal area (Fig. 4A5). Spider graphs of retinas that were exposed to a series of overlapping fundus photographs (Figs. 4B1–B4) showed a loss of ONL, IS, OS, and RPE that was similar to that observed following Ganzfeld exposure to 1 mW/cm2 of light for 1 minute and was uniformly present in the three animals with this exposure paradigm. However, damage was limited to the central retina (approximately 5 mm from the optic nerve head) as more peripheral regions were difficult to light expose using fundus photography as the exposure paradigm.
Figure 4.

Morphometric analysis of retinal changes 2 weeks after light exposure. Spider graphs of ONL thickness and schematic representation of IS, OS, and RPE structure 2 weeks post light exposure of the LE of RHO T4R mutant dogs. The RE was shielded. Data derived from retinal histologic sections extending from the edge of the optic nerve head to the superior, inferior, nasal, and temporal ora serrata. T, tapetum.
Figure 5.

Retinal sections (H&E stained) showing light-induced alterations in the RPE of the RHO T4R mutant dog. (A) Preserved structure and morphology of the RPE in the shielded eye (bracket shows RPE thickness). Microphotograph was taken in the nontapetal region, where the RPE is pigmented. (B) Light-induced RPE hypertrophy (bracket shows RPE thickness) in the nontapetal region of the contralateral eye exposed 2 weeks earlier. (C) Light-induced disruption and loss of RPE monolayer in the tapetal region of the same eye as shown in (B). Dotted line indicates area devoid of RPE nuclei and arrows point to single RPE nuclei. The RPE is nonpigmented in the tapetal region.
With the exception of the 0.1 mW/cm2 exposure, TUNEL-labeled nuclei were present in the central ONL of all retinas 2 weeks after light exposure (Figs. 6A1–A6) but were not observed in the peripheral retina at any light dose. Significant autofluorescent material was observed in the RPE layer (Figs. 6A2–A4, white asterisk) in retinas exposed to damaging doses of light (0.3–1 mW/cm2). RPE65 staining was preserved in all retinas except in that exposed to the highest dose of light, or fundus photography where there was a loss of RPE cells (Figs. 6B1–B6). Complete loss of rhodopsin (Figs. 6C2, C3) was observed at the two highest doses of light, and there was a marked shortening of rod OS in the retina exposed to 0.3 mW/cm2 (Fig. 6C4). At the lowest dose (0.1 mW/cm2), mild shortening of rod OS and rhodopsin mislocalization to the ONL were seen (Fig. 6C5). Cone-arrestin labeling revealed prominent alterations of cone structure (loss of IS and OS) following exposure to the two highest doses of light as well as after fundus photography, with only cone-arrestin labeled remnants remaining (Figs. 6C2, 6C3, 6C6). Although the INL appeared to be well preserved, significant reduction in OPL thickness because of retraction of bipolar cell dendrites was seen in the light-damaged retinas (Figs. 6D2–D4, 6D6). Increase in vimentin and GFAP staining, indicative of reactive gliosis (Figs. 6E2, 6E3), as well as sustained expression of EdnRB in ONL, INL, and retinal ganglion cell layer (GCL) (Figs. 6F2–F6) was observed in light-damaged retinas.
Figure 6.

TUNEL and immunohistochemical characterization of light-induced damage 2 weeks post exposure in RHO T4R mutant dogs. (A1–A6) TUNEL-labeling. (B1–B6) RPE-specific 65 kDa protein (RPE65). (C1–C6) RHO and human Cone Arr. (D1–D6) PKCα and Goα. (E1–E6) Vim. and GFAP. (F1–F6) Vim. and EdnRB. All images were acquired in central retina. Asterisks (A2–A4) indicate RPE autofluorescence.
Discussion
The occurrence of nonsynchronized and protracted cell loss that takes place over years or decades presents a significant challenge in studying the molecular mechanisms of photoreceptor degeneration in RP patients and most animal models. Indeed, at any given time, the low percentage of dying photoreceptors frequently limits the use of tissue-based assays that may not be sensitive enough to detect rare cell death events in a whole retina. Thus, even in models of RP that undergo a transient increased rate of photoreceptor death, single-cell assays or methods that provide cellular resolution have been proposed.25 Light-induced retinal damage, however, can trigger a synchronized and massive burst of photoreceptor cell death that facilitates the characterization of activated cell signaling pathways.26 Among the subset of RHO-ADRP models with increased light sensitivity (for review, see Ref. 22, Supplementary Table S1), the RHO T4R dog retina undergoes rapid and synchronized cell death following a 1-minute duration exposure to white light at levels encountered under normal environmental or clinical conditions.
We have previously shown that clinical fundus photography of RHO T4R dogs triggers an acute retinal degeneration9 that is not activator protein 1 (AP-1) dependent.20,21 In subsequent publications, we have used a 1-minute exposure to 1 mW/cm2 of white light delivered by a Ganzfeld dome to initiate rapid photoreceptor cell death22,23 and ruled out the involvement of an endoplasmic-reticulum (ER) stress response.23 Here, we show by TUNEL assay that the cell-death pattern in the ONL using a Ganzfeld stimulator at 1 mW/cm2 was very similar to that initiated after fundus photography. However, we were able to achieve more widespread retinal damage with the Ganzfeld stimulator compared with the fundus camera and in a much shorter period of time (1 minute by Ganzfeld illumination); in contrast, comparable damage was obtained by fundus photography where ∼10-μsec duration flashes were delivered over a 5- to 7-minute time period for a total cumulative exposure duration of ∼150 to 170 μsec. As previously shown,22 light exposure caused more ONL cell death in the tapetal region versus the nontapetal area. This topographic difference is expected as the tapetum lucidum, a biologic reflector system found in various vertebrates that are active under dim light environment, increases photoreceptor photon catch by reflecting light back onto the OS.27 At 24 hours post light exposure, the central TUNEL-stained nuclei presented an irregular contour and variable labeling intensities, in contrast to the peripheral TUNEL-positive nuclei that were circular with uniform staining. The spatial variability in TUNEL-labeling may indicate differences in the modes of rod cell death in the central versus peripheral retina, or alternatively, the irregularly labeled nuclei may represent those undergoing fragmentation. Since TUNEL staining is not sufficient to discriminate between apoptosis, necrosis, or autolytic cell death,28 additional assays may be needed to identify the possible concomitant occurrence of multiple cell death processes.
We have recently shown continued ONL loss occurring for weeks after acute light exposure.22 In the current study, we now have confirmed that the light-induced death of rods is not just an acute event but continues for at least 2 weeks post exposure. This finding strongly supports the occurrence of a distinct process of cell death happening long after the acute light insult. Possible causes for this prolonged cell death may include both passive and active processes such as loss of structural integrity of the tissue, “bystander” effect,29,30 and hyperoxia31,32 as well as immune responses to primary cell death.33,34
The two lowest light intensities (170 lux/0.1 mW/cm2 and 319 lux/0.2 mW/cm2) used in this study for acute exposure overlap with the range (175–350 lux) of ambient illuminations in the kennels where these dogs are housed. In our previous publication,22 we suggest that the mild ONL thinning observed at 36 weeks (but not at 2 weeks) post exposure to 1 minute of the lowest light intensity (170 lux/0.1 mW/cm2) may have been triggered by the acute light insult (continuous exposure for 1 minute in a dilated eye). However, we did not exclude the possibility that this might also have been caused by environmental exposure to the standard levels of white light used in the kennels. In this current study, we confirmed the absence of photoreceptor loss 2 weeks after a 1-minute exposure to 170 lux/0.1 mW/cm2 but did observe some TUNEL-labeling in the ONL 24 hours after a 1-minute exposure to 319 lux/0.2 mW/cm2. We are confident that these short-term (24 hours and 2 weeks) post–light-exposure changes are secondary to the experimental acute exposure and not to environmental light, as these were seen exclusively in the exposed but not the shielded eyes. However, these combined results do suggest that prolonged housing under ambient illuminations ranging between 175 and 350 lux may trigger a sustained and slow rate of retinal degeneration. Ongoing studies in which RHO T4R mutant dogs are being housed exclusively under dim-red illumination are being conducted to experimentally confirm the likely contribution of low levels of environmental white light to the slow “natural” course of disease.
Several studies have reported that the disruption of rod OS is one of the very first events of retinal degeneration following light exposure.35,36 In the RHO T4R model, disruption of rod OS (but not cones) occurs within 15 minutes of light exposure at the highest dose (1 mW/cm2 for 1 minute).23 In the current study, there was disruption of rod OS 24 hours after light exposure with the two highest doses of light, as evidenced by the altered labeling of two rod OS integral membrane proteins, rhodopsin and peripherin, and the loss of immunostaining of G protein-coupled receptor kinase 1 (GRK1), a peripheral membrane-associated protein that binds to discs by isoprenylation37 and/or through interaction with phosphodiesterase 6D (PDE6D).38 Two weeks following exposure to the lowest dose of light (0.1 mW/cm2 for 1 minute), there was shortening of rod OS length and rhodopsin mislocalization without any evidence of photoreceptor cell loss. These structural alterations may limit the amounts of RHO protein packaged in discs and subsequently lead to an overflow in other cellular compartments. Transient alterations in IS/OS structure has been previously demonstrated by OCT imaging in RHO T4R dogs exposed to similar low doses of light, suggesting that a repair mechanism of OS may take place over time after a sublethal insult.9
This initial disruption was specific to rods, while cones appeared to be initially preserved. There was also focal loss of the integrity of the RPE layer, particularly in the central retina, and an increase in internuclear spacing in the ONL, both processes indicative of a collapse of structural integrity of the outer retinal layers. Breakdown of the outer blood retinal barrier, which is formed by the tight junctions between RPE cells, has been associated with edema in retinas exposed to blue light.39 As the levels of white light used in this current study did not cause similar alterations in the RPE of WT dogs, it remains to be determined whether RPE susceptibility to light in the RHO T4R dogs is mediated via a direct phototoxic effect or whether it is secondary to the acute damage of mutant rods.
While the initial alterations were seen in the outer retina, an increase in EdnRB expression in Müller cells was also observed at 24 hours post light exposure. At two weeks post exposure, EdnRB immunoreactivity was present in the INL, in particular in cell bodies of Müller cells, as well as in the ONL (Supplementary Fig. S2). To the best of our knowledge, this is the first time that expression of EdnRB in photoreceptors has been reported, and it may suggest an interspecies difference as also found for the receptor of ciliary neurotrophic factor.40 Although increased GFAP immunoreactivity was also seen in Müller cells, the level of reactive gliosis appeared to be milder in the T4R light-induced model of retinal degeneration than in P23H rats or following light damage in BALB/C mice.41,42 Regardless, our observations are consistent with other reports of increased EdnRB expression after acute light damage42,43 and induction of Müller cell gliosis by a wide variety of retinal injuries including photoreceptor degeneration.44,45 While we have not examined the expression of endothelin itself, it has been shown to be increased in photoreceptors after light damage as well as in inherited retinal degeneration.42 Endothelin-2 secretion by photoreceptors and its binding to EdnRB on the surface of Müller cells can induce reactive gliosis.46
In summary, we have further characterized a light-damage paradigm using a Ganzfeld stimulator that extends retinal damage to all four quadrants and induces an acute onset of widespread rod cell death in a large animal model of RHO-ADRP. Death of rods begins by 6 hours,23 is very pronounced at 24 hours, and persists at least up to 2 weeks post light exposure. We also identified various immunohistochemical markers to assess acute as well as progressive photoreceptor loss following a single light-exposure event. This sets the stage to further our efforts20,21,23 at identifying pathogenic mechanisms of light-induced damage as well as to evaluate outcomes of therapeutic interventions in the RHO T4R model of ADRP.
Supplementary Material
Acknowledgments
The authors thank Lydia Melnyk for research coordination, Svetlana Savina for histology support, the Retinal Disease Study Facility staff for animal husbandry, Mary Leonard for graphic support, Cheryl Craft (University of Southern California, Los Angeles, CA, USA) for the human cone arrestin antibody, and Robert Molday (University of British Columbia, Vancouver, Canada) for the peripherin antibody.
Supported by National Institutes of Health Grants R24EY022012, R24EY023937, EY06855, P30EY-001583; the Foundation Fighting Blindness; the National Institutes of Health–Merck/Merial Summer Research Fellowship (NIH T35RR07065); Hope for Vision; and the Van Sloun Fund for Canine Genetic Research.
Disclosure: R. Sudharsan, None; K.M. Simone, None; N.P. Anderson, None; G.D. Aguirre, None; W.A. Beltran, None
References
- 1. Sui GY,, Liu GC,, Liu GY,, et al. Is sunlight exposure a risk factor for age-related macular degeneration? A systematic review and meta-analysis. Br J Ophthalmol. 2013; 97: 389–394. [DOI] [PubMed] [Google Scholar]
- 2. Chang B,, Hawes NL,, Pardue MT,, et al. Two mouse retinal degenerations caused by missense mutations in the beta-subunit of rod cGMP phosphodiesterase gene. Vision Res. 2007; 47: 624–633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Paskowitz DM,, LaVail MM,, Duncan JL. Light and inherited retinal degeneration. Br J Ophthalmol. 2006; 90: 1060–1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Peng YW,, Zallocchi M,, Wang WM,, Delimont D,, Cosgrove D. Moderate light-induced degeneration of rod photoreceptors with delayed transducin translocation in shaker1 mice. Invest Ophthalmol Vis Sci. 2011; 52: 6421–6427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Maeda A,, Maeda T,, Imanishi Y,, et al. Retinol dehydrogenase (RDH12) protects photoreceptors from light-induced degeneration in mice. J Biol Chem. 2006; 281: 37697–37704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. van de Pavert SA,, Kantardzhieva A,, Malysheva A,, et al. Crumbs homologue 1 is required for maintenance of photoreceptor cell polarization and adhesion during light exposure. J Cell Sci. 2004; 117: 4169–4177. [DOI] [PubMed] [Google Scholar]
- 7. Cideciyan AV,, Hood DC,, Huang Y,, et al. Disease sequence from mutant rhodopsin allele to rod and cone photoreceptor degeneration in man. Proc Natl Acad Sci U S A. 1998; 95: 7103–7108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kijas JW,, Cideciyan AV,, Aleman TS,, et al. Naturally occurring rhodopsin mutation in the dog causes retinal dysfunction and degeneration mimicking human dominant retinitis pigmentosa. Proc Natl Acad Sci U S A. 2002; 99: 6328–6333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Cideciyan AV,, Jacobson SG,, Aleman TS,, et al. In vivo dynamics of retinal injury and repair in the rhodopsin mutant dog model of human retinitis pigmentosa. Proc Natl Acad Sci U S A. 2005; 102: 5233–5238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Tam BM,, Noorwez SM,, Kaushal S,, Kono M,, Moritz OL. Photoactivation-induced instability of rhodopsin mutants T4K and T17M in rod outer segments underlies retinal degeneration in X. laevis transgenic models of retinitis pigmentosa. J Neurosci. 2014; 34: 13336–13348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Tam BM,, Moritz OL. The role of rhodopsin glycosylation in protein folding, trafficking, and light-sensitive retinal degeneration. J Neurosci. 2009; 29: 15145–15154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. White DA,, Fritz JJ,, Hauswirth WW,, Kaushal S,, Lewin AS. Increased sensitivity to light-induced damage in a mouse model of autosomal dominant retinal disease. Invest Ophthalmol Vis Sci. 2007; 48: 1942–1951. [DOI] [PubMed] [Google Scholar]
- 13. Organisciak DT,, Darrow RM,, Barsalou L,, Kutty RK,, Wiggert B. Susceptibility to retinal light damage in transgenic rats with rhodopsin mutations. Invest Ophthalmol Vis Sci. 2003; 44: 486–492. [DOI] [PubMed] [Google Scholar]
- 14. Zhang R,, Oglesby E,, Marsh-Armstrong N. Xenopus laevis P23H rhodopsin transgene causes rod photoreceptor degeneration that is more severe in the ventral retina and is modulated by light. Exp Eye Res. 2008; 86: 612–621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Naash ML,, Peachey NS,, Li ZY,, et al. Light-induced acceleration of photoreceptor degeneration in transgenic mice expressing mutant rhodopsin. Invest Ophthalmol Vis Sci. 1996; 37: 775–782. [PubMed] [Google Scholar]
- 16. Budzynski E,, Gross AK,, McAlear SD,, et al. Mutations of the opsin gene (Y102H and I307N) lead to light-induced degeneration of photoreceptors and constitutive activation of phototransduction in mice. J Biol Chem. 2010; 285: 14521–14533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Vaughan DK,, Coulibaly SF,, Darrow RM,, Organisciak DT. A morphometric study of light-induced damage in transgenic rat models of retinitis pigmentosa. Invest Ophthalmol Vis Sci. 2003; 44: 848–855. [DOI] [PubMed] [Google Scholar]
- 18. Contin MA,, Benedetto MM,, Quinteros-Quintana ML,, Guido ME. Light pollution: the possible consequences of excessive illumination on retina. Eye (Lond). 2016; 30: 255–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Zhu L,, Jang GF,, Jastrzebska B,, et al. A naturally occurring mutation of the opsin gene (T4R) in dogs affects glycosylation and stability of the G protein-coupled receptor. J Biol Chem. 2004; 279: 53828–53839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Gu D,, Beltran WA,, Li Z,, Acland GM,, Aguirre GD. Clinical light exposure, photoreceptor degeneration, and AP-1 activation: a cell death or cell survival signal in the rhodopsin mutant retina? Invest Ophthalmol Vis Sci. 2007; 48: 4907–4918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Gu D,, Beltran WA,, Pearce-Kelling S,, Li Z,, Acland GM,, Aguirre GD. Steroids do not prevent photoreceptor degeneration in the light-exposed T4R rhodopsin mutant dog retina irrespective of AP-1 inhibition. Invest Ophthalmol Vis Sci. 2009; 50: 3482–3494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Iwabe S,, Ying GS,, Aguirre GD,, Beltran WA. Assessment of visual function and retinal structure following acute light exposure in the light sensitive T4R rhodopsin mutant dog. Exp Eye Res. 2016; 146: 341–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Marsili S,, Genini S,, Sudharsan R,, Gingrich J,, Aguirre GD,, Beltran WA. Exclusion of the unfolded protein response in light-induced retinal degeneration in the canine T4R RHO model of autosomal dominant retinitis pigmentosa. PLoS One. 2015; 10: e0115723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Beltran WA,, Hammond P,, Acland GM,, Aguirre GD. A frameshift mutation in RPGR exon ORF15 causes photoreceptor degeneration and inner retina remodeling in a model of X-linked retinitis pigmentosa. Invest Ophthalmol Vis Sci. 2006; 47: 1669–1681. [DOI] [PubMed] [Google Scholar]
- 25. Arango-Gonzalez B,, Trifunovic D,, Sahaboglu A,, et al. Identification of a common non-apoptotic cell death mechanism in hereditary retinal degeneration. PLoS One. 2014; 9: e112142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Hao W,, Wenzel A,, Obin MS,, et al. Evidence for two apoptotic pathways in light-induced retinal degeneration. Nat Genet. 2002; 32: 254–260. [DOI] [PubMed] [Google Scholar]
- 27. Ollivier FJ,, Samuelson DA,, Brooks DE,, Lewis PA,, Kallberg ME,, Komaromy AM. Comparative morphology of the tapetum lucidum (among selected species). Vet Ophthalmol. 2004; 7: 11–22. [DOI] [PubMed] [Google Scholar]
- 28. Grasl-Kraupp B,, Ruttkay-Nedecky B,, Koudelka H,, Bukowska K,, Bursch W,, Schulte-Hermann R. In situ detection of fragmented DNA (TUNEL assay) fails to discriminate among apoptosis, necrosis, and autolytic cell death: a cautionary note. Hepatology. 1995; 21: 1465–1468. [DOI] [PubMed] [Google Scholar]
- 29. Cusato K,, Bosco A,, Rozental R,, et al. Gap junctions mediate bystander cell death in developing retina. J Neurosci. 2003; 23: 6413–6422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ripps H. Cell death in retinitis pigmentosa: gap junctions and the ‘bystander' effect. Exp Eye Res. 2002; 74: 327–336. [DOI] [PubMed] [Google Scholar]
- 31. Yu DY,, Cringle S,, Valter K,, Walsh N,, Lee D,, Stone J. Photoreceptor death, trophic factor expression, retinal oxygen status, and photoreceptor function in the P23H rat. Invest Ophthalmol Vis Sci. 2004; 45: 2013–2019. [DOI] [PubMed] [Google Scholar]
- 32. Yu DY,, Cringle SJ. Retinal degeneration and local oxygen metabolism. Exp Eye Res. 2005; 80: 745–751. [DOI] [PubMed] [Google Scholar]
- 33. Zhang C,, Shen JK,, Lam TT,, et al. Activation of microglia and chemokines in light-induced retinal degeneration. Mol Vis. 2005; 11: 887–895. [PubMed] [Google Scholar]
- 34. Langmann T. Microglia activation in retinal degeneration. J Leukoc Biol. 2007; 81: 1345–1351. [DOI] [PubMed] [Google Scholar]
- 35. Vaughan DK,, Nemke JL,, Fliesler SJ,, Darrow RM,, Organisciak DT. Evidence for a circadian rhythm of susceptibility to retinal light damage. Photochem Photobiol. 2002; 75: 547–553. [DOI] [PubMed] [Google Scholar]
- 36. Organisciak DT,, Vaughan DK. Retinal light damage: mechanisms and protection. Prog Retin Eye Res. 2010; 29: 113–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Inglese J,, Glickman JF,, Lorenz W,, Caron MG,, Lefkowitz RJ. Isoprenylation of a protein kinase: requirement of farnesylation/alpha-carboxyl methylation for full enzymatic activity of rhodopsin kinase. J Biol Chem. 1992; 267: 1422–1425. [PubMed] [Google Scholar]
- 38. Zhang H,, Li S,, Doan T,, et al. Deletion of PrBP/delta impedes transport of GRK1 and PDE6 catalytic subunits to photoreceptor outer segments. Proc Natl Acad Sci U S A. 2007; 104: 8857–8862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Putting BJ,, Zweypfenning RC,, Vrensen GF,, Oosterhuis JA,, van Best JA. Blood-retinal barrier dysfunction at the pigment epithelium induced by blue light. Invest Ophthalmol Vis Sci. 1992; 33: 3385–3393. [PubMed] [Google Scholar]
- 40. Beltran WA,, Rohrer H,, Aguirre GD. Immunolocalization of ciliary neurotrophic factor receptor alpha (CNTFRalpha) in mammalian photoreceptor cells. Mol Vis. 2005; 11: 232–244. [PubMed] [Google Scholar]
- 41. Fernandez-Sanchez L,, Lax P,, Campello L,, Pinilla I,, Cuenca N. Astrocytes and Müller cell alterations during retinal degeneration in a transgenic rat model of retinitis pigmentosa. Front Cell Neurosci. 2015; 9: 484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Rattner A,, Nathans J. The genomic response to retinal disease and injury: evidence for endothelin signaling from photoreceptors to glia. J Neurosci. 2005; 25: 4540–4549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Torbidoni V,, Iribarne M,, Ogawa L,, Prasanna G,, Suburo AM. Endothelin-1 and endothelin receptors in light-induced retinal degeneration. Exp Eye Res. 2005; 81: 265–275. [DOI] [PubMed] [Google Scholar]
- 44. Garcia M,, Vecino E. Role of Müller glia in neuroprotection and regeneration in the retina. Histol Histopathol. 2003; 18: 1205–1218. [DOI] [PubMed] [Google Scholar]
- 45. Bringmann A,, Wiedemann P. Müller glial cells in retinal disease. Ophthalmologica. 2012; 227: 1–19. [DOI] [PubMed] [Google Scholar]
- 46. Sarthy VP,, Sawkar H,, Dudley VJ. Endothelin2 induces expression of genes associated with reactive gliosis in retinal Müller cells. Curr Eye Res. 2015; 40: 1181–1184. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.