

Abstract
Background.
To elucidate molecular features associated with disproportionate survival of glioblastoma (GB) patients, we conducted deep genomic comparative analysis of a cohort of patients receiving standard therapy (surgery plus concurrent radiation and temozolomide); “GB outliers” were identified: long-term survivor of 33 months (LTS; n = 8) versus short-term survivor of 7 months (STS; n = 10).
Methods.
We implemented exome, RNA, whole genome sequencing, and DNA methylation for collection of deep genomic data from STS and LTS GB patients.
Results.
LTS GB showed frequent chromosomal gains in 4q12 (platelet derived growth factor receptor alpha and KIT) and 12q14.1 (cyclin-dependent kinase 4), and deletion in 19q13.33 (BAX, branched chain amino-acid transaminase 2, and cluster of differentiation 33). STS GB showed frequent deletion in 9p11.2 (forkhead box D4-like 2 and aquaporin 7 pseudogene 3) and 22q11.21 (Hypermethylated In Cancer 2). LTS GB showed 2-fold more frequent copy number deletions compared with STS GB. Gene expression differences showed the STS cohort with altered transcriptional regulators: activation of signal transducer and activator of transcription (STAT)5a/b, nuclear factor–kappaB (NF-κB), and interferon-gamma (IFNG), and inhibition of mitogen-activated protein kinase (MAPK1), extracellular signal-regulated kinase (ERK)1/2, and estrogen receptor (ESR)1. Expression-based biological concepts prominent in the STS cohort include metabolic processes, anaphase-promoting complex degradation, and immune processes associated with major histocompatibility complex class I antigen presentation; the LTS cohort features genes related to development, morphogenesis, and the mammalian target of rapamycin signaling pathway. Whole genome methylation analyses showed that a methylation signature of 89 probes distinctly separates LTS from STS GB tumors.
Conclusion.
We posit that genomic instability is associated with longer survival of GB (possibly with vulnerability to standard therapy); conversely, genomic and epigenetic signatures may identify patients where up-front entry into alternative, targeted regimens would be a preferred, more efficacious management.
Keywords: genome sequencing, glioblastoma, outlier, responders, survival difference
Importance of the study
Standard of care treatment for all GB patients is radiation with concomitant temozolomide therapy. However, factors that predict response by individual GB patients to standard of care treatment remain unclear. In this manuscript, we elucidate molecular features associated with disproportionate survival in GB patients ("GB outliers"). Using deep genomic, transcriptomic, and methylomic comparative analysis (exome, whole genome sequencing, RNA and DNA methylation) of a cohort of patients receiving standard therapy, our integrated study unmasks a number of genetic differences between LTS and STS GB patients and provides an important molecular foundation for developing actionable signatures from GB biopsies of patients who show exceptionally good (or poor) outcomes from standard of care therapy.
Current treatment options for glioblastoma (GB) patients are limited and largely palliative. Mechanism(s) driving the development and recurrence of GB are poorly understood, limiting improved management. Standard treatment includes maximal safe surgical resection followed by concurrent radiation and chemotherapy with the DNA alkylating agent temozolomide (TMZ), which extends median survival to approximately 14.7 months.1 Unfortunately, GB manifests resistance to standard therapy regimen and recurrence is virtually assured, due largely to highly invasive cells that aggressively disperse into surrounding normal brain.
However, a small percentage of GB patients respond to standard treatment and benefit with an average survival time greater than 2 years. To date, it is unclear why individuals with the same diagnosis of GB die quickly, while others have extended survival. Thus, studying the genomics and transcriptomics of these “outlier” GB patients could inform prognosis and may suggest ways to better treat GB patients.
Several factors besides tumor size and location determine patients’ survival. These include age at diagnosis (where younger patients often receive more aggressive treatment that is multimodal), functional status or Karnofsky performance score at presentation (which has a significant negative correlation with age), and histologic and genetic markers.2 Among these factors, genetic markers could provide prognostic prediction of survival; balancing prognosis in the arms of clinical trials or prioritizing patients with poor prognosis into more innovative regimens are 2 meaningful outcomes from understanding prognostic markers.
Previous studies, where large-scale genomic characterization was implemented, have associated altered retinoic acid signaling,3 enhanced immune-related gene expression,4 distinct DNA methylation profiles,5 and O6-DNA methylguanine-methyltransferase (MGMT) methylation and isocitrate dehydrogenase (IDH)1/2 mutation status6 with long-term survival in GB. To date, there is no genomic study that comprehensively examines the outliers in the 2 tails of the survival spectrum of primary GB patients, all of whom received standard therapy.
Although The Cancer Genome Atlas (TCGA) GB database provides genomic data from primary GB tumors, samples with “multi-omics” data, including copy number variants (CNVs), exome sequencing, mRNA expression profiles, and global methylation data for the GB outliers, are available for only 6 cases (Supplementary Fig. 1). Here, using the Ohio Brain Tumor Study (OBTS),7 we identified 2 cohorts of glioma patients: long-term survivors (LTS, average 33 mo overall survival [OS]) and short-term survivors (STS, average 7 mo OS). We employed genomic analyses (exome, whole genome sequencing), transcriptomic sequencing, and methylation profiling to assemble an integrated genomic landscape for gaining insight into the underpinning mechanism(s) associated with the survival differences. Our integrated study unmasks a number of genetic differences between LTS and STS GB patients and provides an important molecular foundation for developing actionable signatures from GB biopsies of patients who show exceptionally good (or poor) outcomes from therapy.
Materials and Methods
Ethics Statement and Sample Collection
Informed consent was obtained for each patient enrolled on the ongoing OBTS (approved by University Hospitals Case Medical Center institutional review board protocol no. CASE 1307-CC296). After assessing the OS distribution for all GBs consented to the OBTS, we defined STS as the lowest quartile and LTS as the upper quartile of the overall OBTS survival distribution. All selected patients were primary GB cases, deceased, and received standard of care therapy. Clinical data elements include gender, age at diagnosis/surgery, pathology (ie, pretreatment/recurrence/secondary tumor), therapy class, vital status, OS and progression-free survival. Tissue specimens and matched blood samples were collected fresh frozen and maintained below −80°C until nucleic acid extraction.
DNA and RNA Isolation
Tumor specimens were collected from 18 treatment-naïve primary GB patients who subsequently received surgery and standard of care treatment (10 STS and 8 LTS). Genomic DNA and total RNA from fresh frozen tissue specimens were isolated using kits described in the Supplementary material.
Next-Generation Sequencing
All next-generation sequencing (NGS) data acquisition and analysis was carried out using previously described methods.8 Methods for whole genome sequencing, exome sequencing, RNA sequencing, and data analyses are described briefly in the Supplementary material.
Data Availability
Binary sequence alignment/map (BAM) files from whole genome, whole exome sequencing, as well as RNA-seq data are available from the EMBL-EBI European Nucleotide Archive database (http://www.ebi.ac.uk/ena/) with accession number PRJEB10881 and are accessible via http://www.ebi.ac.uk/ena/data/view/PRJEB10881. The sample accession numbers are from ERS848749 to ERS848765 for RNA sequencing. For the whole genome and exome sequencing, the sample accession numbers are ERS848748–ERS853219 and ERS872925–ERS872960 for exome and genome, respectively. Methylation data are available with accession number ERS1205964. The file name ending with “T” indicates tumor sample and the file name ending with “N” indicates matched normal.
Alignment and Variant Calling
Whole Genome and Whole Exome
For whole genome and exome sequencing, fastq files were aligned with BWA 0.6.2 to GRCh37.62 and the SAM outputs were converted to a sorted BAM file using SAMtools 0.1.18. BAM files were then processed through insertion/deletion (indel) realignment, mark duplicates, and recalibration steps in this order with GATK 1.5, where dpsnp135 was used for known single nucleotide polymorphism (SNPs), and 1000 Genomes’ ALL.wgs.low_coverage_vqsr.20101123 was used for known indels. Lane level sample BAMs were then merged with Picard 1.65 if they were sequenced across multiple lanes. Comparative variant calling for exome data was conducted with Seurat.9
Previously described copy number and translocation detection were applied to the whole genome long insert sequencing data.10 Briefly, copy number detection was based on a log2 comparison of normalized physical coverage (or clonal coverage) across tumor and normal whole genome long-insert sequencing data, where physical coverage was calculated by considering the entire region a paired-end fragment span on the genome, then the coverage at 100 bp intervals was kept. Normal and tumor physical coverage was then normalized, smoothed, and filtered for highly repetitive regions prior to calculating the log2 comparison. To quantify the copy number aberrations, CNV score was calculated based on the intensity of copy number change (log ratio) as well as the range of such alterations. Genomic Identification of Significant Targets in Cancer (GISTIC) was then used to identify regions of the genome that were significantly amplified or deleted across the LTS and STS groups.11 GISTIC calculated a statistic (G-score) for the frequency of occurrence and the amplitude of the aberration. The statistical significance of each aberration was computed by comparing the observed G-score with the results expected by chance. Regions with false discovery rate q-values less than 0.25 were considered statistically significant.
Translocation detection was based on discordant read evidence in the tumor whole genome sequencing data compared with its corresponding normal data. In order for the structural variant to be called, there needs to be greater than 7 read pairs mapping to both sides of the breakpoint. The unique feature of the long-insert whole genome sequencing was the long overall fragment size (~1 kb), whereby two 100 bp reads flank a region of ~800 bp. The separation of forward and reverse reads increases the overall probability that the read pairs do not cross the breakpoint and confound mapping.
RNA
For RNA sequencing, lane level fastq files were appended together if they were sequenced across multiple lanes. These fastq files were then aligned with STAR 2.3.1 and TopHat 2.0.8 to GRCh37.62 using ensembl.63.genes.gtf as a GTF file. Changes in transcript expression were calculated with Cuffdiff 2.1.1 in FPKM (fragments per kilobase of exon per million fragments mapped) format using upper-quartile normalization. Genes with mean FPKM less than 0.1 were filtered out and surrogate variable analysis (SVA) was applied to remove batch effect.12 Student’s t-test was then used to call differentially expressed genes (DEGs) between LTS and STS groups using a P-value of .05 as cutoff. For novel fusion discovery, reads were aligned with TopHat-Fusion 2.0.8. Clustering was performed using the R Heatmap.2 package with Euclidean Distance and the McQuitty clustering method.
Unsupervised hierarchical clustering was performed using expression of genes known to be related to genome instability and are included in the Chromosomal Instability (CIN)70 gene list.13 Gene set variation analysis (GSVA)14 was used to determine the subtype of GB based on previously published signatures.15,16 Additionally, to identify specific molecular programming that might be driving outcome to standard of care treatment, ontology and pathway enrichment analysis was carried out using genes differentially expressed between LTS and STS groups.
DNA Methylation Analysis
Global DNA methylation was evaluated using the Infinium HumanMethylation450 Beadchip Array (Illumina). Briefly, 1 µg of each DNA sample underwent bisulfite conversion using the EZ DNA methylation kit according to the manufacturer’s recommendation for the Illumina Infinium Assay. Bisulfite-treated DNA was then hybridized to arrays according to the manufacturer’s protocol. GenomeStudio V2011.1 (Illumina) for methylation was used for data assembly and acquisition. Methylation levels for each cytosine–phosphate–guanine (CpG) residue are presented as β values, estimating the ratio of the methylated signal intensity over the sum of the methylated and unmethylated intensities at each locus. The average β value reports a methylation signal ranging from 0 to 1 representing completely unmethylated to completely methylated values, respectively. Methylation data were preprocessed in R using the Illumina Methylation Analyzer.17,18 Data preprocessing included background corrections, probe scaling to balance Infinium I and II probes, quantile normalization, and logit-transformation. Additionally, probes with P-values >.05 in 25% or more of samples, probes on X and Y chromosomes, and probes situated within 10 bp of putative SNPs were removed. Differential methylation on logit-transformed values was performed to compare LTS tumors with STS samples in the Illumina Methylation Analyzer. Wilcox rank test was conducted between LTS and STS samples and P-values were corrected by calculating the false discovery rate by the Benjamini–Hochberg method. Subsequent to differential methylation, logit-transformed values were detransformed to beta values for simpler assessment of the magnitude of methylation change. Probes with adjusted P-values <.05 and delta β values ≥0.2 or ≤−0.2 were considered statistically significant and differentially methylated.
Biological Concept Enrichment Analysis
Biological concept enrichment analysis was performed on the DEG list using the ClueGO v2.1.5 + CluePedia v1.1.5 cytoscape plugin. Enrichment was performed with the following ontologies/pathway gene sets: GO Biological Process, GO Cellular Component, KEGG, Reactome, and WikiPathways. Advanced term/pathway selection options were set at Go Tree Interval Minimum level 3 and Max level 8 and minimum number of genes at 3. The kappa score was set at 0.4. A 2-sided hypergeometric test was used with Bonferroni step down correction. Common genes for each enriched term were projected onto an enriched network figure using CluePedia cytoscape plugin.
Results
Outlier Cohort
Our LTS and STS cohorts consisted of diagnoses of primary GB in patients, taken from the highest and lowest quartiles, respectively, of the overall GB survival distribution for the OBTS. All tumor specimens were treatment naïve and contained an average of 75% tumor cellularity (range, 50%–95%). Long-term survivors are defined as patients with GB with an average OS of 33 months (range, 18–57 mo; Table 1), and short-term survivors are patients with an average OS of 7 months (range, 3–11 mo; Table 1).
Table 1.
Clinical characteristics of the primary GB patients in the study
| Patient # | Path Diagnosis | Age | Gender | Race | Estimate of Resection | Post-Resection Therapy (Rad. + TMZ) | IDH Status | MGMT Methylation Status | G-CIMP Status | Overall Survival (days) | Recurred | Time to Progression (days) | Treatment after Recurrence | Survival Cohort |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | GB | 69 | F | White | GTR | Y | WT | M | -ve | 102 | N | – | – | STS |
| 2 | GB | 51 | M | White | GTR | Y | WT | UM | -ve | 148 | Y | 87 | Surgery | STS |
| 3 | GB | 70 | M | White | GTR | Y | WT | UM | -ve | 151 | N | — | — | STS |
| 4 | GB | 56 | M | White | STR | Y | WT | UM | -ve | 157 | Y | 91 | Surgery | STS |
| 5 | GB | 71 | M | White | GTR | Y | WT | UM | -ve | 184 | N | — | — | STS |
| 6 | GB | 71 | M | White | STR | Y | WT | M | -ve | 229 | Y | 92 | TMZ | STS |
| 7 | GB | 51 | M | White | GTR | Y | WT | UM | -ve | 282 | Y | 74 | Surgery | STS |
| 8 | GB | 50 | M | White | STR | Y | WT | UM | -ve | 304 | Y | 172 | Surgery | STS |
| 9 | GB | 70 | M | White | STR | Y | WT | M | -ve | 307 | Y | 105 | None | STS |
| 10 | GB | 70 | M | White | GTR | Y | WT | M | -ve | 331 | Y | 176 | Avastin and Thalidomide | STS |
| 11 | GB | 83 | F | White | STR | Y | WT | M | -ve | 596 | Y | 441 | None | LTS |
| 12 | GB | 55 | F | Black | GTR | Y | WT | M | -ve | 748 | Y | 110 | Avastin and Radiosurgery | LTS |
| 13 | GB | 68 | F | White | STR | Y | WT | UM | -ve | 749 | Y | 107 | Radiosurgery, GDC- 0449 and Avastin | LTS |
| 14 | GB | 63 | F | White | GTR | Y | WT | M | -ve | 772 | N | — | — | LTS |
| 15 | GB | 71 | M | Other | GTR | Y | WT | UM | -ve | 1042 | Y | 495 | Radiosurgery | LTS |
| 16 | GB | 55 | F | White | GTR | Y | WT | M | -ve | 1208 | Y | 899 | Surgery and TMZ | LTS |
| 17 | GB | 56 | F | White | GTR | Y | WT | M | -ve | 1267 | Y | 648 | Isotretinoin | LTS |
| 18 | GB | 61 | M | White | STR | Y | WT | UM | -ve | 1713 | N | — | — | LTS |
GTR = gross total resection (>95% by volume), STR = subtotal resection (≤95% by volume), Y = Yes, N = No, F = Female, M = Male, M = Methylated, UM = Unmethylated, WT = wild type.
Genomic Landscape
The genomic sequencing coverage was more than 100× for exome and 10× for whole genome for tumor and germline genomes (Supplementary Table 1). Somatic mutations, including single nucleotide variations (SNVs), indels, translocations, intrachromosomal rearrangements (inversion, etc), and copy number alterations, were determined from sequencing of tumor and germline pairs. Overall, tumors from the LTS subgroup demonstrated a higher number of genomic events compared with tumors from the STS subgroup (Fig. 1). Notably, abundant large structural changes across the genomes of LTS tumors showed that the LTS subgroup displayed a 2-fold increase in CNV loss with an average of 155 CNV loss/tumor compared with STS with an average of 74 CNV loss/tumor. However, the LTS and STS cohorts displayed an average of 18 and 12 CNV gains/tumors, respectively. A summary of the total copy number changes in LTS and STS cohorts is presented in Fig. 2A. Both LTS and STS showed similar frequency of some of the most frequently observed copy number alterations in primary GB, such as focal amplification of epidermal growth factor receptor (EGFR) at 7p12.1, focal deletion of cyclin-dependent kinase inhibitor (CDKN)2A/B at 9p21.3, and phosphatase and tensin homolog (PTEN) deletion at Chr. 10 (Fig. 2A). Interestingly, in our collection, focal amplification of platelet derived growth factor receptor alpha (PDGFRA) and KIT (chromosome 4q12) and 12q14.1 (cyclin-dependent kinase 4 [CDK4]) was found only in LTS and was observed in 3 tumor samples (Fig. 2A). In contrast, STS GB showed frequent deletion in 9p11.2 (forkhead box D4-like 2 and aquaporin 7 pseudogene 3) and 22q11.21 (Hypermethylated In Cancer 2). These observations were further confirmed by GISTIC analysis (Fig. 2B). The sum of CNV events is much greater in the LTS samples, with the greatest difference being the high number of deletions. The most frequently observed CNVs were the classic GB events such as EGFR amplification and CDKN2A deletion. LTS and STS groups demonstrated an average of 93 and 73 translocations/tumors, respectively (Supplementary Table 2).
Fig. 1.
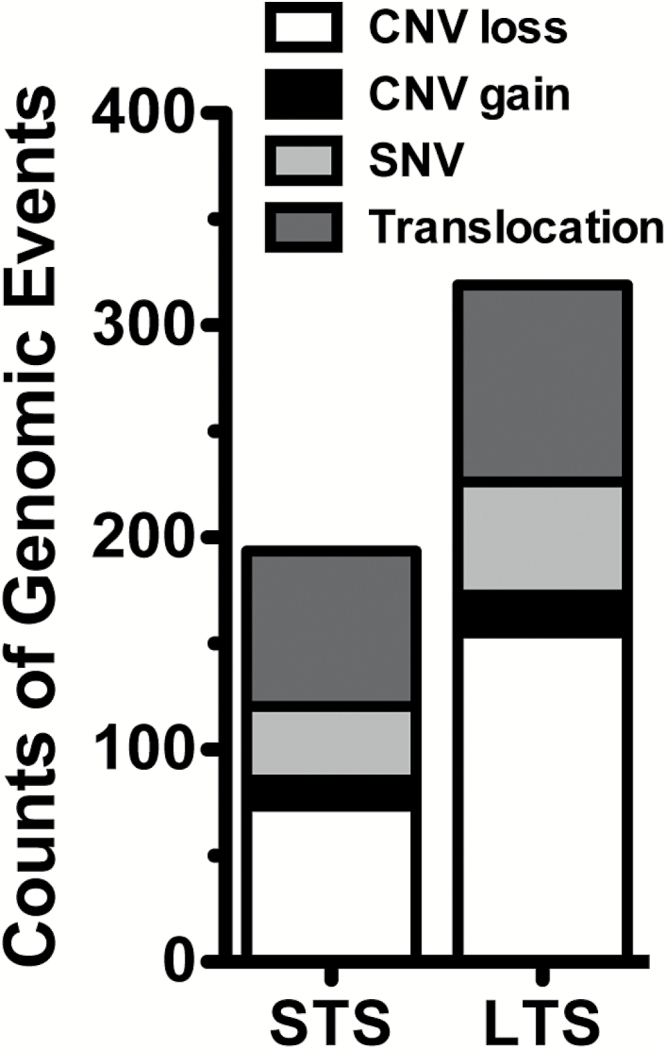
Number of genomic alteration events between STS and LTS groups.
Fig. 2.

(A) CNV compilation plot: LTS are highlighted in green on the y-axis. STS are highlighted in red on the y-axis. X-axis defines the chromosomes. Each sample line represents DNA copy number data as the log2 ratio of normalized coverage in the tumor specimen relative to the matched normal DNA. CNVs can be identified in spikes where the log2 ratio is greater than +0.6 or less than −0.6. Amplifications are marked in red and deletions marked in green. While both cohorts have classic GB CNVs (Chr 7 gain, EGFR amplification, Chr 10 loss) the long-term samples have many more CNV events. (B) GISTIC analysis plot: Each plot shows the frequency of CNVs for each cohort (left: STS right: LTS). In each plot the bottom axis defines the frequency of CNV amplifications in red. The top axis of each plot defines the frequency of CNV deletions. The x-axis of each graph represents the genome with chromosomes marked with alternating white and gray regions. The vertical green bars mark a threshold of significance. Annotations of a variety of significant regions are highlighted in boldface. Q-values for amplified (red) and deleted (blue) regions are displayed along the x-axis on the bottom and top of figures, respectively. (C) Validation of copy number alterations in TCGA dataset. (a&c) Boxplot showing the CNV score of STS (n = 8) and LTS (n = 10) in outlier dataset. (b&d) Boxplot illustrating the difference of CNV score in STS (n = 28) and LTS (n = 44) patients who met the same criteria. P-value was calculated using one-tailed t-test.
To validate our observation of increased genomic alteration in the LTS cohort, we examined the copy number alterations in GB samples in the TCGA database. To ensure the sample cohorts are similar, we selected the cohort from TCGA with similar criteria as our outlier cohort, which includes: (i) patient must be diseased, (ii) patient must have received standard of care (surgery followed by radiation and TMZ therapy), and (iii) patient survival (in days) must be within one standard deviation of outlier cohort survival (in days) (Supplementary Table 3). Based on these criteria, we identified 44 LTS and 28 STS in the dataset of TCGA. Examination of the CNV alteration showed that the LTS cohort displayed increased genomic alterations, with more CNV loss compared with STS (Fig. 2C.d; P = .025), thus corroborating our GB outlier dataset (Fig. 2C.c; P = .035). The CNV gain comparison is not significant in TCGA (Fig. 2C.b), which indicates that higher CNV in the LTS group is largely due to deletions.
The list of somatic SNVs and small indels detected is provided in Supplementary Table 4. Overall, the LTS group exhibited a total of 446 somatic coding mutations, with an average of 56 somatic coding mutations/tumors (range, 36–82), whereas the STS group showed a total of 359 somatic coding mutations, with an average of 36 somatic coding mutations/tumors (range, 2–56). Additionally, when we compared mutational landscapes in the outlier cohort and TCGA outlier cohort with the most frequent genomic alterations known to be present in primary GB,19 we detected similar alteration frequency in glioblastoma multiforme (GBM) signature events such as EGFR gain and CDKN2A loss, but we observed a higher frequency of mutations in LTS compared with STS (Fig. 3 and Supplementary Fig. 2). Nonsynonymous SNVs identified in more than 2 tumors for LTS include PDGFRA, tumor protein (TP)53, ankyrin repeat domain 36, and neurofibromatosis type 1(NF1), and unique missense mutations were detected in at least one LTS tumor, including BRAF, alpha thalassemia/mental retardation syndrome X-linked, calcitonin receptor, CD3e molecule associated protein, collagen type I alpha 2 chain, glutathione peroxidase 5, HEAT repeat-containing protein 7B2, and transient receptor potential vanilloid type 5. In contrast, in STS tumors, detection of missense mutations was observed in genes including leucine zipper like transcription regulator 1 and trichohyalin for at least one tumor.
Fig. 3.

Genomic alterations identified in outlier cohort . The spectrum of alterations identified within the 2 patient groups was mapped against the subset of frequently altered genes previously identified in primary GB. Red indicates copy number gain, green is copy number loss, an asterisk (*) indicates nonsynonymous mutation, where missense mutations are colored blue, nonsense mutations colored gray, and frameshift mutations colored yellow. The order of patients is ranked by their survival days in a descending order. We added a few known drivers in primary GB and no IDH1 mutation was found in our cohort.
Global Methylation Patterns
We assessed global DNA methylation patterns in the outlier GB LTS and STS cohorts using the 450K-methylation platform. A logit transformation was performed on each sample, where logit transformation converts otherwise heteroscedastic beta values (bounded by 0 and 1) to M values following a Gaussian distribution. The analysis revealed 89 differentially methylated CpG loci (DML) encompassing 69 unique genes (Supplementary Table 5). Normalized z-scores were used for generating box plots to represent overall methylation levels across DML for LTS and STS. Overall methylation was significantly lower in STS (β = 0.374) than in LTS (β = 0.472) (P = .0429) (Fig. 4A), indicating hypomethylation in STS. Examination of the overall methylation levels of the GB outliers in TCGA (selected based on the criteria described in the previous section) also showed an overall significant hypomethylation status in STS, corroborating our data (P < .0001) (Fig. 4B).
Fig. 4.

Differential methylation analysis in GB outlier cohort. (A) Boxplot showing the higher beta value of differentially methylated loci in LTS compared with STS in the GB outlier dataset. (B) Boxplot illustrating the similar observations in TCGA dataset. (C, D) Functional distribution analysis of differentially methylated loci in the GB outlier. (E) Heatmap of differentially methylated probes found with an absolute delta beta value greater than 0.2 in LTS and STS samples. Y-axis lists the differentially methylated probes and the x-axis shows the samples with LTS samples highlighted in green and STS samples highlighted in red.
The regional and functional CpG distributions of DML in the outlier GB cohorts were queried. Functional distribution relates CpG position to transcription start sites (TSS −200 to −1500 bp), the 5′ untranslated region, exon 1 for coding genes, or gene bodies. The distribution of probes differed between hypomethylated and hypermethylated probes albeit the majority of DMLs were situated in gene bodies (Fig. 4C).
The regional distribution of DML was assessed based on proximity to the closest CpG island. In addition to island cores, shores are 0–2 kb from CpG islands, shelves are 2–4 kb away, and open sea regions are isolated loci without a designation. When comparing the STS with the LTS cohorts, the majority of hypomethylated DML in STS were in islands (62.73%) and shores (24.54%) (Fig. 4D); the majority of hypermethylated loci (59.15%) were located in the open seas (Fig. 4D). These data show that STS have methylation trends that differ from LTS, which are consistent with greater overall hypomethylation and focal gene body hypermethylation in CpG islands. Unsupervised clustering analysis of DML demonstrated a distinct separation of LTS and STS samples, consisting of 89 probes (Fig. 4E). Using the outlier cohort of TCGA to validate the 89 probe sets, only 2 targets (DOCK2 and miRNA-886) were found to be consistently hypomethylated in STS cases, which are also known to be predictors of poor survival in other cancer types.13,20–22 Such a disagreement between methylation signatures of 2 cohorts also indicates complexity of GB and the dynamic nature of epigenetic regulation in cancer cells. However, when we used outlier 89 probe set signatures for identifying positively and negatively correlated GB samples in TCGA with methylation and survival data (n = 79) using GSVA,14 “LTS-like” group (n = 22) showed significantly higher survival compared with “STS-like” group (n = 20) (Supplementary Fig. 3 and Supplementary Table 6). Thus, our methylation signature could be useful in identifying a portion of GB STS patients.
Transcriptomic Profiling
Gene expression analysis was performed using Cufflinks/Cuffdiff to identify DEGs in outlier cohorts. The comparison identified 615 DEGs (Supplementary Table 7); a heatmap is presented in Fig. 5A. To assess the genes that are potentially epigenetically regulated, we integrated differential gene expression and methylation data; SLC10A4 and FAM24B were consistently hypomethylated and overexpressed in the STS cohort compared with the LTS cohort (Supplementary Fig. 4). We also performed gene expression validation by DEG hierarchical clustering using TCGA outlier samples. Although the clustering did not show clear separation of the LTS and STS groups, we did see a subset of samples with enrichment of short-term survivors (Supplementary Fig. 5, highlighted with orange box). It thus suggests that our signature could be useful in characterizing a portion of GB patients with short-term survival.
Fig. 5.

(A) Analysis of differentially expressed genes in outlier cohort. Hierarchical clustering of 615 genes distinguishing the LTS from the STS group. (B) Biological concept enrichment analysis of outlier cohort. Biological concept analysis of differentially expressed genes between LTS and STS. The nodes in the diagram represent an enriched ontology category, pathway, or gene. Blue nodes are enriched for LTS samples and red are enriched for STS samples. Nodes with enrichment for both are represented as pie charts. Node size corresponds to how many genes in that ontology/pathway concept. Edges represent the statistical association between the nodes based on gene membership and location with gene ontology.
Ingenuity pathway analysis (IPA; Ingenuity Systems) of DEGs between LTS and STS cohorts revealed functional pathways, biological functions, and/or diseases distinct for each outlier cohort (Supplementary Table 8). The results were ranked based on activation or inhibition z-scores to identify the most relevant distinguishing categories with respect to upregulated and downregulated genes; 22 functional pathways were altered between LTS and STS subgroups. Four of the 22 functional annotations mapped to “inositol biosynthesis,” including 3-phosphoinositide, which is associated with high proliferation.3,23 Of IPA enriched biological functions, 59 out of 65 categories are mapped to “Cancer” and “Neurological Disease,” consistent with the biology and anatomic origin of the samples (Supplementary Table 8).
To determine whether common transcriptional regulators may account for the DEGs, we examined results of the upstream regulator analysis (Supplementary Table 8). Six upstream regulators were identified as activated and 3 were identified as inhibited using a z-score of 1 as the filtering threshold. Among the activated regulators in STS were STAT5a/b, NF-κB, and IFNG; 3 inhibited regulators were observed in STS including MAPK1, ERK1/2, and ESR1. Two interesting highly scored transcription factors, NF-κB and IFNG, share 2 regulated genes (nitric oxide synthase 2, proteasome subunit beta type-9), which are illustrated in a combined network (Supplementary Fig. 6).
In order to determine network-based representative biology associated with the DEG between LTS and STS samples, we performed biological concept enrichment analysis using ClueGO software. DEGs for LTS and STS were analyzed as 2 separate gene lists using GO BiologicalProcess, GO CellularComponent, KEGG, Reactome, and WikiPathways. Differentially enriched pathways were detected between the 2 gene lists and visualized in Fig. 5B. The representative genes enriched for LTS and STS are found in Supplementary Table 9.
The network-based modeling takes knowledge from prebuilt canonical pathways as well as potential network rules from each sample. Those analyses revealed a number of biological concepts associated with LTS and STS gene expression changes. The LTS enriched biological concepts include those associated with development and morphogenesis, as well as the mammalian target of rapamycin signaling pathway. The STS concepts are centered on metabolic processes, anaphase-promoting complex degradation, and immune processes associated with major histocompatibility complex class I antigen presentation (Fig. 5B).
Discussion
GB is a highly aggressive brain cancer with median survival of just 14 months with standard of care treatment; although rarely, some GB patients survive far beyond this. Previous studies have found more prevalence of some molecular aberrations, in particular MGMT promoter methylation and IDH1/2 mutation, and distinct gene expression and methylation profiles in long-term survivors than in unselected patients.3–6,24,25 Expanded molecular subclassification is beginning to reveal survival differences across these GB subtypes, leaving open to study whether patient response to therapy may also vary predictably across these subtypes. From a prognostic standpoint, it would be beneficial to understand molecular differences between these 2 survival outlier groups in GB. Therefore, in our study, we conducted a comprehensive genomics analysis using NGS technology to measure alterations at the level of DNA copy number, DNA methylation, DNA somatic mutation, and mRNA expression in a set of GB STS versus LTS.
Copy number analysis demonstrated that both STS and LTS have similar frequency of common gains and losses of GB such as EGFR (chromosome 7), CDKN2A (chromosome 9), and PTEN (chromosome 10). Beyond these generic regions of GB aberrancy, LTS showed significantly “noisier” genomes; specifically samples from the LTS cohort demonstrated significantly higher CNV loss compared with samples from the STS cohort. These observations of higher frequency of genomic alterations at various levels were validated in TCGA cohorts of similar survival features. It also corroborates recent findings by Andor et al, which demonstrated that copy number alterations affecting a high fraction (>75%) of the tumor genome predicts reduced risk of patient death across different cancer types.26 Whole exome sequencing detected frequently mutated genes similar to previous studies (EGFR, NF1, TP53, PTEN, etc) for both groups but displayed a trend of higher number of somatic mutation in LTS. Methylation analysis also presented distinct epigenetic patterns between STS and LTS, which may affect key regulatory functions. Network analysis of DEGs reveals enriched biological processes associated with development in LTS and metabolic processes in STS. The findings on methylation and expression differences observed in the outlier cohort did not validate in the matched TCGA cohort. Overall, our findings indicate that tumors in patients who survived >18 months have high numbers of CNVs without any association to distinct or specific gene expression or methylation signatures.
Increased genomic instability in the LTS group may be associated with heightened vulnerability to standard of care treatment. Tumor cells with greater numbers of genetic abnormalities may be more vulnerable to DNA damaging interventions. Such a trade-off between growth and drug vulnerability is inevitable owing to limited resources in organisms. In the face of changing environments (treatment in our case), tumors usually have greater competency than other cells to adapt in the short term, while accruing mutations later. Our findings suggest that excess mutational burden compromises tumor cell survival in the face of therapy, contributing to a survival benefit to the patient. Contradictory to our finding, a recent study by Reifenberger and coworkers6 showed no difference between CNVs in long-term survivors compared with other groups in GB. However, they used array comparative genomic hybridization to identify CNVs compared with our findings, which derive from significantly high resolution NGS methods. To identify mechanism(s) behind heightened genetic abnormalities in the LTS group compared with the STS group, we looked at a previously reported gene signature predictive of chromosomal instability (CIN70)13 and found a very modest trend suggesting CIN70 gene signature was overexpressed in the STS group compared with the LTS group (Supplementary Fig. 7). Importantly, we were not able to find any other specific link on why genetic abnormalities are overrepresented in the LTS group compared with the STS group.
Additionally, in our outlier cohort, we noticed a slight gender inequality. The majority of female patients were long-term survivors and, on the contrary, most male patients were short-term survivors. In order to investigate the role of gender in predicting survival, we performed statistical testing to see if such gender effect also holds in a matched TCGA cohort (Supplementary Fig. 8) and found no significant difference (P = .332) between survival of males and females in the cohort from TCGA. Moreover, even within the STS and LTS, the dataset of TCGA did not show a significant divergence between male and female patients (STS P = .8581; LTS P = .9628).
Our long-term goal is to improve the treatment and prognosis for patients with GBM. Those patients who are categorized to be potential LTS would benefit from standard therapy, whereas for patients with STS signature, their treatment selection may benefit from molecular profiling of targetable mutations and gene pathways that vary among patients. In light of this, molecular/genomic signatures in patient tumors may direct optimal or effective therapy selection, thereby enabling personalized treatment planning. The net result of this approach will be to have more effective therapy directed to identify features in profiled patient cancer specimens as opposed to the current paradigm of indiscriminately exposing patients to chemotherapeutic toxins and hoping for a response. Our studies have highlighted a number of genetic and epigenetic alterations occurring in STS and LTS, which indicate targetable mutations and hold promise for better clinical outcomes.
Supplementary Material
Supplementary material is available at Neuro-Oncology online.
Funding
This work was supported by The Ben & Catherine Ivy Foundation.
Supplementary Material
Acknowledgments
The original idea and project objectives for the outlier genomic investigation was developed by M.E.B., N.L.T., and J.S.B-S. Laboratory studies were carried out by B.A., J.R., B.S., A.E.S., and Q.T.O. Informatics analysis strategy was developed and executed by S.P., J.K., C.L., and B.S. The manuscript was drafted by S.P., H.D., B.S., N.L.T., and M.E.B. All authors read and approved the final manuscript. The authors would like to thank Andrew Sloan, MD, FACS, for his assistance in collection of tissues from the Ohio Brain Tumor Study patients used in this study.
Conflict of interest statement. The authors declare that they have no competing interests.
References
- 1. Wang L, Wei Q, Wang LE, et al. Survival prediction in patients with glioblastoma multiforme by human telomerase genetic variation. J Clin Oncol. 2006;24(10):1627–1632. [DOI] [PubMed] [Google Scholar]
- 2. Walid MS. Prognostic factors for long-term survival after glioblastoma. Perm J. 2008;12(4):45–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Barbus S, Tews B, Karra D, et al. Differential retinoic acid signaling in tumors of long- and short-term glioblastoma survivors. J Natl Cancer Inst. 2011;103(7):598–606. [DOI] [PubMed] [Google Scholar]
- 4. Donson AM, Birks DK, Schittone SA, et al. Increased immune gene expression and immune cell infiltration in high-grade astrocytoma distinguish long-term from short-term survivors. J Immunol. 2012;189(4):1920–1927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Shinawi T, Hill VK, Krex D, et al. DNA methylation profiles of long- and short-term glioblastoma survivors. Epigenetics. 2013;8(2):149–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Reifenberger G, Weber RG, Riehmer V, et al. ; German Glioma Network. Molecular characterization of long-term survivors of glioblastoma using genome- and transcriptome-wide profiling. Int J Cancer. 2014; 135(8):1822–1831. [DOI] [PubMed] [Google Scholar]
- 7. Ostrom QT, McCulloh C, Chen Y, et al. Family history of cancer in benign brain tumor subtypes versus gliomas. Front Oncol. 2012;2:19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Borad MJ, Champion MD, Egan JB, et al. Integrated genomic characterization reveals novel, therapeutically relevant drug targets in FGFR and EGFR pathways in sporadic intrahepatic cholangiocarcinoma. PLoS Genet. 2014;10(2):e1004135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Christoforides A, Carpten JD, Weiss GJ, et al. Identification of somatic mutations in cancer through Bayesian-based analysis of sequenced genome pairs. BMC Genomics. 2013;14:302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Craig DW, O’Shaughnessy JA, Kiefer JA, et al. Genome and transcriptome sequencing in prospective metastatic triple-negative breast cancer uncovers therapeutic vulnerabilities. Mol Cancer Ther. 2013;12(1):104–116. [DOI] [PubMed] [Google Scholar]
- 11. Beroukhim R, Getz G, Nghiemphu L, et al. Assessing the significance of chromosomal aberrations in cancer: methodology and application to glioma. Proc Natl Acad Sci U S A. 2007;104(50):20007–20012. doi: 10.1073/pnas.0710052104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Li S, Łabaj PP, Zumbo P, et al. Detecting and correcting systematic variation in large-scale RNA sequencing data. Nat Biotechnol. 2014;32(9):888–895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Carter SL, Eklund AC, Kohane IS, et al. A signature of chromosomal instability inferred from gene expression profiles predicts clinical outcome in multiple human cancers. Nat Genet. 2006;38(9):1043–1048. [DOI] [PubMed] [Google Scholar]
- 14. Hänzelmann S, Castelo R, Guinney J. GSVA: gene set variation analysis for microarray and RNA-seq data. BMC Bioinformatics. 2013;14:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Verhaak RG, Hoadley KA, Purdom E, et al. ; Cancer Genome Atlas Research Network. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell. 2010; 17(1):98–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Carro MS, Lim WK, Alvarez MJ, et al. The transcriptional network for mesenchymal transformation of brain tumours. Nature. 2010;463(7279):318–325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Wang D, Yan L, Hu Q, et al. IMA: an R package for high-throughput analysis of Illumina’s 450K Infinium methylation data. Bioinformatics. 2012;28(5):729–730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Legendre CR, Demeure MJ, Whitsett TG, et al. Pathway implications of aberrant global methylation in adrenocortical cancer. PLoS One. 2016;11(3):e0150629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Brennan CW, Verhaak RG, McKenna A, et al. ; TCGA Research Network. The somatic genomic landscape of glioblastoma. Cell. 2013;155(2):462–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Wang L, Nishihara H, Kimura T, et al. DOCK2 regulates cell proliferation through Rac and ERK activation in B cell lymphoma. Biochem Biophys Res Commun. 2010;395(1):111–115. [DOI] [PubMed] [Google Scholar]
- 21. Cao J, Song Y, Bi N, et al. DNA methylation-mediated repression of miR-886-3p predicts poor outcome of human small cell lung cancer. Cancer Res. 2013;73(11):3326–3335. [DOI] [PubMed] [Google Scholar]
- 22. Bi N, Cao J, Song Y, et al. A microRNA signature predicts survival in early stage small-cell lung cancer treated with surgery and adjuvant chemotherapy. PLoS One. 2014;9(3):e91388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Fang B, Zhu J, Wang Y, et al. MiR-454 inhibited cell proliferation of human glioblastoma cells by suppressing PDK1 expression. Biomed Pharmacother. 2015;75:148–152. [DOI] [PubMed] [Google Scholar]
- 24. Krex D, Klink B, Hartmann C, et al. ; German Glioma Network. Long-term survival with glioblastoma multiforme. Brain. 2007;130(Pt 10):2596–2606. [DOI] [PubMed] [Google Scholar]
- 25. Hartmann C, Hentschel B, Simon M, et al. ; German Glioma Network. Long-term survival in primary glioblastoma with versus without isocitrate dehydrogenase mutations. Clin Cancer Res. 2013;19(18):5146–5157. [DOI] [PubMed] [Google Scholar]
- 26. Andor N, Graham TA, Jansen M, et al. Pan-cancer analysis of the extent and consequences of intratumor heterogeneity. Nat Med. 2016;22(1):105–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Binary sequence alignment/map (BAM) files from whole genome, whole exome sequencing, as well as RNA-seq data are available from the EMBL-EBI European Nucleotide Archive database (http://www.ebi.ac.uk/ena/) with accession number PRJEB10881 and are accessible via http://www.ebi.ac.uk/ena/data/view/PRJEB10881. The sample accession numbers are from ERS848749 to ERS848765 for RNA sequencing. For the whole genome and exome sequencing, the sample accession numbers are ERS848748–ERS853219 and ERS872925–ERS872960 for exome and genome, respectively. Methylation data are available with accession number ERS1205964. The file name ending with “T” indicates tumor sample and the file name ending with “N” indicates matched normal.