

Abstract
Background.
The majority of glioma-associated microglia/macrophages have been identified as M2-type macrophages with immune suppressive and tumor supportive action. Recently, the extracellular adenosine deaminase protein Cat Eye Syndrome Critical Region Protein 1 (CECR1) was shown to regulate macrophage maturation. In this study, we investigate the role of CECR1 in the regulation of the glioma-associated macrophage response.
Methods.
Expression of CECR1 was assessed in human glioma samples. CECR1-mediated macrophage response was studied in vitro, using donor derived CD14+ monocytes and the THP-1 monocytic cell line. The response of the human glioma cell line U87 to conditioned medium of macrophages preconditioned with recombinant human CECR1 or CECR1 silencing was also assessed.
Results.
CECR1 was strongly expressed in high-grade gliomas (P < .001) and correlated positively with the M2 phenotype markers in tumor-associated microglia/macrophages (TAMs) (overall, P < .05). In vitro studies confirmed the presence of a significantly higher level of CECR1 expression in M2-like macrophages exposed to U87 conditioned medium (P < .001). CECR1 knockdown or stimulation of macrophages affected differentiation toward the M2-like phenotype. Stimulation of U87 cells with conditioned medium of CECR1 knockdown or stimulated macrophages affected tumor cell proliferation and migration, coinciding with altered intracellular signaling of mitogen-activated protein kinase (MAPK). In glioma tissue samples, CECR1 expression correlated with Ki67 and MAPK signaling protein.
Conclusions.
CECR1 is a potent regulator of TAM polarization and is consistently highly expressed by M2-type TAMs, particularly in high-grade glioma. Paracrine effects induced by CECR1 in M2-like TAMs activate MAPK signaling and stimulate the proliferation and migration of glioma cells.
Keywords: CECR1, glioma, glioblastoma, tumor associated macrophages (TAMs)
Importance of the study
M2 TAMs are enriched in human high-grade gliomas and express high levels of CECR1.
CECR1 mediates TAM differentiation toward M2.
CECR1 promotes M2 TAM–mediated glial tumor cell growth and migration.
CECR1 is a drug target with high potential for use in immune modulation therapy for treatment of patients with high-grade gliomas.
The heterogeneity of glial tumor cells and the immune privileged environment of the central nervous system hamper the development of effective treatment strategies. The immune system plays a complex and largely undiscovered role in the development of gliomas. Infiltrating myeloid cells, including monocytes and macrophages, are key players in promoting tumor progression and recurrences,1,2 as their robust presence with the accumulation of microglial cells in glioblastoma multiforme (GBM) facilitates an immune suppressive and oncogenic microenvironment. Macrophages in GBM are heterogeneous and display considerable plasticity, complicating their characterization and analysis.2 To ease data interpretation, tumor-associated macrophages (TAMs) are divided into M1- and M2-like cells. M1-like cells express high levels of cluster of differentiation (CD)80, CD86, and major histocompatibility complex (MHC) II molecules and secrete higher levels of tumor necrosis factor α (TNF-α), interleukin (IL)-1 β, and IL-12. M1-like TAMs are reportedly antitumoral and associated with better prognosis.3,4 However, it has been reported that (paracrine) stimulation of glial tumor cells and the hypoxic microenvironment trigger resident microglial cells and infiltrating macrophages to adopt the M2-like phenotype.5 Indeed, M2-like TAMs outnumber their M1 counterparts in GBM. M2-like TAMs are defined by high cell surface levels of CD163, CD204, and CD14; lower levels of CD80 and MHC II; and mediation of the immunosuppressive response by secreting cytokines like IL-10, chemokine C-C ligand (CCL)20, CCL22, and prostaglandin-E2.5 M2-like glioma associated microglia/macrophages are also known to mediate tumor angiogenesis by producing pro-angiogenic factors such as vascular endothelial growth factor A (VEGF-A) and urokinase-type plasminogen activator and are associated with high-grade tumors and poor clinical outcomes.6,7 Thus, microglia/macrophages are relevant to glial tumorigenesis and could become targets of therapeutic intervention.
In humans, the Cat Eye Syndrome Critical Region Protein 1 (CECR1) is highly expressed by macrophages.8 Patients with genetic loss of function of CECR1 show reduced monocyte polarization into the M2 macrophage subtype, which coincides with loss of vessel integrity.9,10 CECR1 is a member of the adenyl-deaminase growth factor family, known for its immune modulatory function.11 As a secreted protein, CECR1 is mainly produced by myeloid cells, with a rise in CECR1 adenosine deaminase activity reported in response to hypoxia, inflammation, and oncogenesis.12 CECR1 has also been shown to promote macrophage proliferation, and CECR1 deficiency was previously associated with neutrophil activation, vascular damage, and inflammation.12,13
However, to date, the role of CECR1 in TAMs regulation in gliomas has not been explored. In this study, we investigated the role of CECR1 specifically with respect to TAMs in glial tumors.
Materials and Methods
Patient Samples
Patient samples as listed in Supplementary Tables 1 and 2 were obtained from the Biobank of the Department of Pathology, Erasmus Medical Center, Rotterdam. The use of these samples was approved by the medical ethical committee of the Erasmus MC.
Small Interfering RNA Transfection
Small interfering (si)RNA targeting CECR1 and scrambled nontargeting siRNAs were obtained from Dharmacon (GE Health Care). THP-1 derived macrophage cultures were transfected after 48 hours of para-Methoxyamphetamine (PMA) treatment, following the manufacturer’s protocol (using Dharmafect transfection reagent). Efficiency of CECR1 knockdown was assessed after 24 and 48 hours at transcriptional level and protein level. For functional assays or to obtain macrophage-derived conditioned medium, siRNA transfected cells were used 24–48 hours post transfection.
Cell Cultures
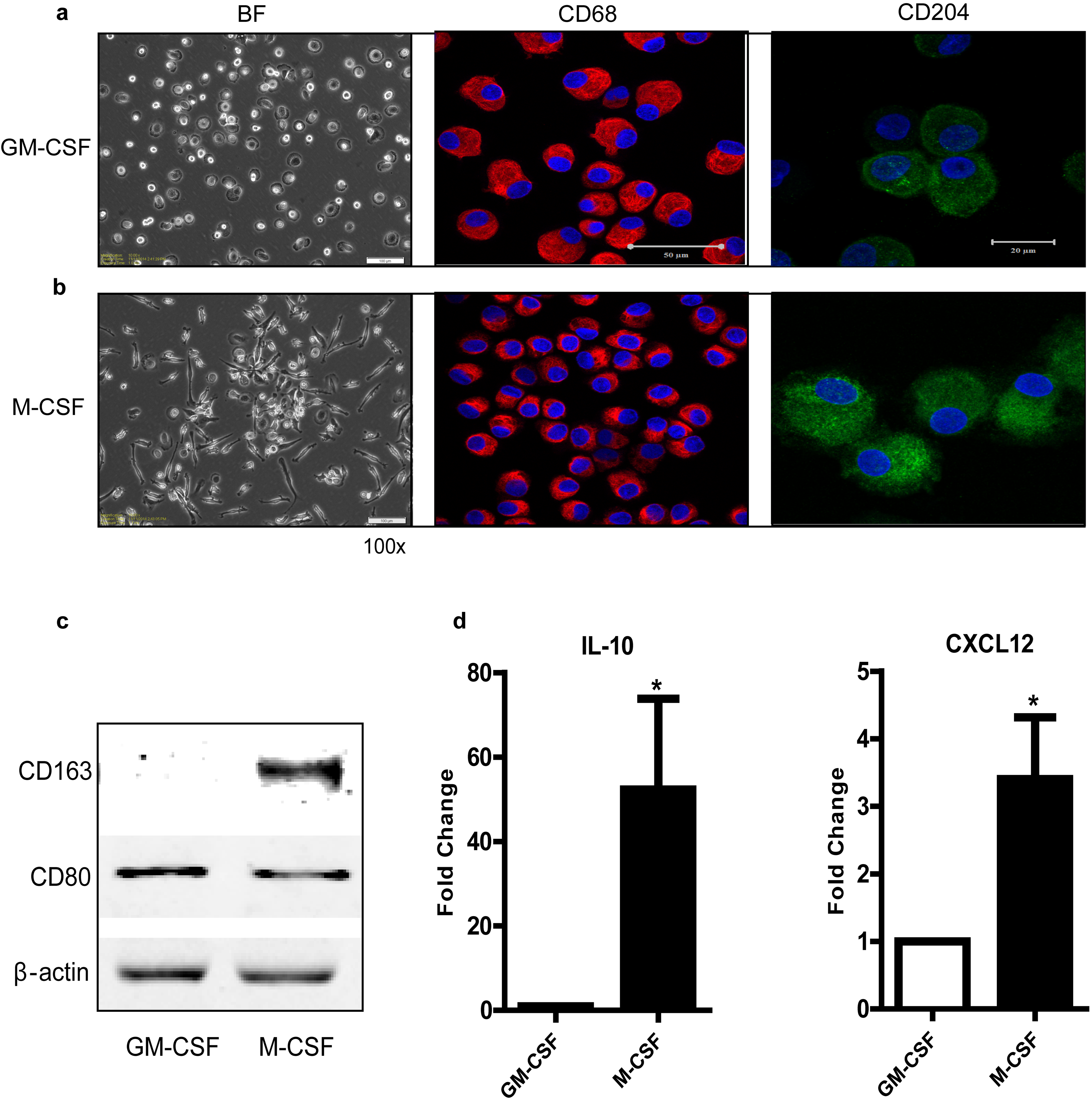
CD14+ monocytes were obtained according to the manufacturer’s protocols. Cells were induced with 10 ng/mL granulocyte-macrophage colony-stimulating factor (GM-CSF) (R&D) or 10 ng/mL macrophage colony-stimulating factor (M-CSF) (Immunotools) as previously reported.14
THP-1 cells were cultured in Roswell Park Memorial Institute (RPMI) 1640 medium supplemented with antibiotics and 10% fetal calf serum and matured by PMA (Sigma). Interferon (IFN)-γ/lipopolysaccharide (LPS), IL-4, and IL-10 were applied to generate M1 and M2a/M2c phenotypes in vitro. Recombinant human (rh)CECR1 was used for macrophage treatment for 96 hours.
Human glioblastoma cell lines U87, U373, and U251 were cultured in Dulbecco’s modified Eagle’s medium supplemented with 10% fetal calf serum and antibiotics. U87-derived conditioned medium (U87-CM) was used to stimulate macrophages for 48 hours.
MTT Assay
U87, U373, and U251 cells were seeded in a 96-well setup followed by treatment with macrophage conditioned medium. The growth of U87 cells was monitored by MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) (Sigma) for 5 days. One plate without macrophage conditioned medium was measured at the same day for reference (see Supplementary material).
Migration Assay
The Oris cell migration assay kit (Platypus Technologies) was used to measure migration of PKH-67 labeled U87, U373, and U251 cells according to manufacturer’s protocols in a 96-well plate setup. Macrophage conditional medium was added and after 24 hours of incubation, pictures were taken at 0 and 24 h and quantified using Image J (see Supplementary material).
RNA Isolation and Quantitative Real-Time PCR
Total RNA was isolated and reversely transcripted to cDNA from cryo-materials and macrophages. For the cryo-samples, we measured transcripts of CECR1, CD68, CD86, CD16, CD204, CD206, nitric oxide synthase (NOS)-2, IL-10, IL-12p35, and β-actin (reference gene). For in vitro experiments, transcripts of CECR1, IL-10, IL-12p35, IL-6, C-X-C motif chemokine ligand (CXCL)12, CCL18, and CCL22 were measured and normalized to β-actin level. Primers are listed in Supplementary Table 3.
Western Blot Analysis
Total protein was loaded onto sodium dodecyl sulfate–polyacrylamide gel electrophoresis gel and blotted to nitrocellulose membranes followed by blocking and immunoblotting using specific antibodies. Incubation of secondary antibodies and detection of signals was followed using the Odyssey imaging system (Licor Bioscience) (see Supplementary material).
Flow Cytometry Analysis
Macrophages were harvested and incubated with primary antibody against CD163 (1:400; Abd Serotec) followed by Alexa Fluor-555 conjugated secondary antibody. Cells were counterstained with Hoechst 33258 and analyzed by fluorescence activated cell sorting with a Becton-Dickinson FACS Aria III (see Supplementary material).
Immunostaining
Adjacent (cryo)sections were used for immunohistochemical and immunofluorescence analysis. Immunostaining and slide scanning were performed according to the protocol described previously.15
Macrophages and U87 cells were fixed using 4% paraformaldehyde/phosphate buffered saline followed by antibody incubation. Signal areas were quantified using Image J (see Supplementary material and Table 4).
Statistics
Data from clinical samples were analyzed by Mann–Whitney U-test and Spearman correlation using SPSS 21.0. All in vitro data were tested using unpaired 2-tailed Student’s t-test (significance levels P < .05). All data are presented as means ± SEM, unless otherwise stated.
Results
Expression of CECR1 Is Skewed to High-Grade Astrocytoma and Associated with the M2-like Macrophage Phenotype
Transcription of CECR1 was assessed in astrocytomas of various malignancy grades, using 2 different online Gene Expression Omnibus databases (GDS 4467, GDS1813). CECR1 was mainly expressed in GBM (Fig. 1A). Quantitative (q)PCR analysis of astrocytoma samples from our biobank (19 astrocytomas grade II, 5 astrocytomas grade III, and 19 GBM) shows that expression of the M2-microglia/macrophage specific markers CD16, CD204, and IL-10 were significantly higher in the grade III astrocytomas and GBM than in the grade II astrocytomas (Supplementary Fig. 1B). The microglia/macrophage markers CD68 (pan macrophage marker), CD86 (M1 marker), CD206 (M2 marker), the inducible isoform of NOS (iNOS; M1 marker), and IL-12p35 (M1 marker) were equally expressed in the low- and high-grade tumors (Supplementary Fig. 1C, D).
Fig. 1.

CECR1 is highly expressed by M2-like macrophage in GBM. (A) Immunohistochemistry for CECR1, CD204, and CD206 in autopsy brain, astrocytoma grade II (AII), and grade IV (GBM) (scale bar: 200 µm). (B) Box plots displaying the mean percentages of CECR1, CD204, CD206-positive areas per image view in autopsy brains, AII, and GBM. *P < .05, ***P < .005. (C) Heat map summarizing the Spearman correlation coefficients between CECR1 and CD68, IL-12p35, iNOS, CD86, CD206, CD16, CD204, IL-10 in autopsy, astrocytoma grades II and III, and GBM. *P < .05, **P < .01. (D) Confocal images showing the colocalization of CECR1 with CD68 and Iba-1 in GBM (scale bar: 20 µm). (E) (F) Confocal images showing the colocalization of CECR1 with CD204 and CD163 in GBM (scale bar: 50 µm for the low magnification field; 10 µm for high magnification inlet).
To further investigate the relation between CECR1 and microglia/macrophages in human glioma, 7 autopsy brains, 6 astrocytomas grade II, and 8 astrocytomas grade IV (GBM) were immunostained for CECR1, CD204, CD206, and CD16. CECR1 overlapped with CD204+, CD206+, and CD16+ perivascular cells in autopsy brain and low-grade glioma (Fig. 1A, Supplementary Fig. 2A). In GBM, the CECR1 signal mainly overlapped with CD204+ and CD16+ cells at both perivascular and tumor parenchymal locations. Areas where CECR1− cells with features of M2-like macrophages were located were also detected (Supplementary Fig. 3A, B). Overlap between CECR1 and CD206+ cells were detected at only the peripheral perivascular locations. Quantitation of the sections revealed significant higher numbers of CECR1, CD204, CD206, and CD16+ cells in GBM versus the autopsy brains and grade II astrocytomas (Fig. 1B, Supplementary Fig. 2B).
A qPCR dataset showed in astrocytomas grades III and IV (GBM) positive correlations of CECR1 expression with CD68, CD86, CD16, CD204, and IL-10. In astrocytomas grade III, CECR1 expression showed a negative correlation with IL-12p35. In contrast, no significant correlations between the markers iNOS, IL-12p35, CD206, and CECR1 were detected in high-grade GBM samples (Fig. 1C).
In GBM, CECR1 colocalized with the pan macrophage markers CD68 and Iba-1 (Fig. 1D). CECR1 colocalized with CD68 in both perivascular areas and sites remote from the vasculature in autopsy brains (Supplementary Fig. 4A–D), but only at perivascular locations in grade II astrocytoma (Supplementary Fig. 4E, F). No CECR1 signal was detected in CD68+ macrophages remote from blood vessels (data not shown).
In addition, CECR1 colocalized with markers of M2-like TAMs (CD204, CD163, CD16) in GBM (Fig. 1E and F; Supplementary Fig. 2C and D). CECR1+/CD163+ cells were detected in only the perivascular areas in autopsy brains and in grade II astrocytomas (Supplementary Fig. 5A–D).
CECR1 Is Preferentially Expressed in M2-like U87 Stimulated Macrophages In vitro
M-CSF induced (M2-like) macrophages showed a higher protein level of CECR1 than did GM-CSF induced (M1-like) macrophages (Fig. 2A), associated with higher protein levels of M2 phenotype markers (CD204, CD163) and higher mRNA levels of M2 cytokines (IL-10, CXCL12) (Supplementary Fig. 6A–D).
Fig. 2.

CECR1 is highly expressed in M2-like, U87 stimulated macrophage. (A) Western blot of CECR1 protein in GM-/M-CSF induced macrophages (MΦ) with/without U87-CM from 3 experiments. Numbers above bands indicate quantified levels normalized to β-actin. (B) Western blot of CECR1 protein in THP-1 derived macrophages (MΦ) in response to RPMI-1640, LPS-INFγ (M1), IL-4 (M2a), IL-10 (M2b), and U87-CM from 3 experiments. Numbers above bands indicate quantified levels normalized to β-actin. (C) Representative staining of CECR1 and CD163 in GM-CSF and M-CSF induced macrophages with/without U87 stimulation for 96 hours (scale bar: 20 µm). (D) Quantification of fluorescent signal of CECR1 per cell in different groups. Data are presented as mean ± SEM and were obtained from 3 independent experiments. *P < .05, **P < .01, ***P < .005. (E) Quantitative PCR of CECR1 in macrophages at 24 h posttransfection with CECR1 knockdown and control. Data were obtained from 3 experiments and presented as mean ± SEM; *P < .05. (F) Western blot of CECR1 protein in THP-1 macrophages at 48 h posttransfection, showing siCECR1, siSham, and nontransfected control. (G) CD163 immunostaining in siCECR1 and siSham transfected THP-1 macrophages (scale bar: 20 µm). (H) Quantified CD163 signal per cell in siCECR1 and siSham control groups are presented as mean ± SEM and were obtained from 3 independent experiments. **P < .01. (I) IL-10, CD163, and CD86 expression in siCECR1, siSham, and nontransfected THP-1 macrophages as measured by real-time qPCR. Data are presented as mean ± SEM and obtained from 3 experiments. *P < .05.
CECR1 protein levels in both types of macrophages were further upregulated in response to stimulation with U87 conditioned medium (U87-CM) (1.4- and 2.4-fold increase in GM-/M-CSF induced macrophage, respectively; Fig. 2A). Similarly, expression of CECR1 was detected in THP-1 derived macrophages (Fig. 2B). Elevated levels of CECR1 protein were detected in M2a and M2c macrophages (1.2- and 1.6-fold increase in IL-4 and IL-10 stimulated groups, respectively) and U87-CM stimulated macrophages (1.8-fold increase) compared with control group (RPMI). In contrast, in M1 macrophages (LPS+INFγ group), a lower level of CECR1 (0.8-fold change) was detected (Fig. 2B).
CECR1 protein levels were further assessed in GM-/M-CSF induced macrophages (Fig. 2C and D). CECR1 protein detected by immunostaining was significantly higher in M-CSF induced (M2-like) macrophages than in GM-CSF induced (M1-like) counterparts. Furthermore, stimulation of U87-CM increased CECR1 protein levels in both M-CSF and GM-CSF induced macrophages and enhanced the protein signal of the M2 marker CD163 (Fig. 2C and D).
CECR1 Expression Induces the M2-like Macrophage Phenotype In vitro
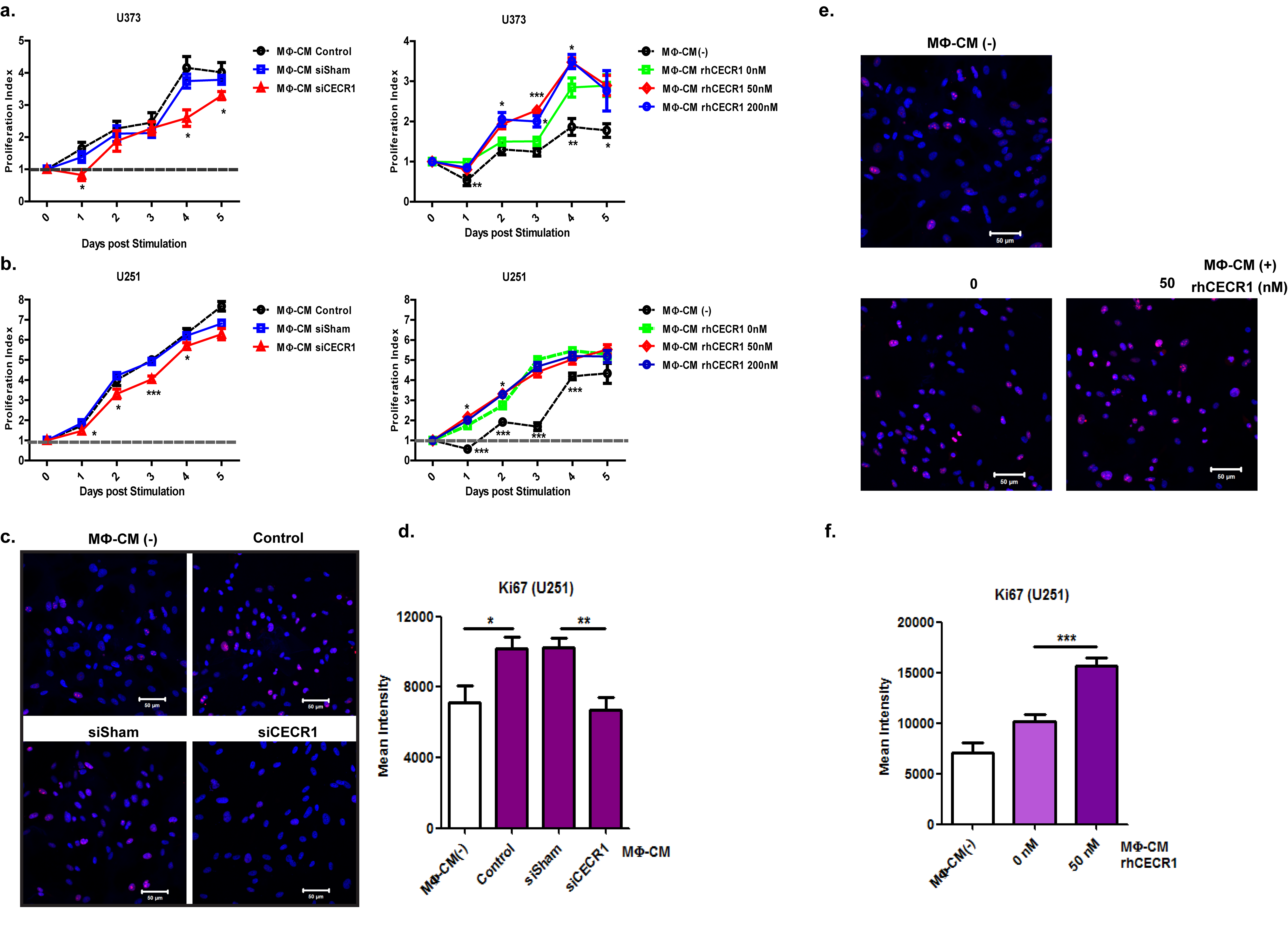
Effective siRNA mediated silencing of CECR1 was validated by qPCR, western blotting, and immunofluorescent staining (Fig. 2E and F and Supplementary Fig. 7A and B) compared with nontargeting siRNA treated controls (siSham). Quantitative PCR, western blot analysis, and immunostaining revealed lower RNA and protein levels of the M2 marker CD163 in THP-1 macrophages silenced for CECR1 compared with the siSham controls (Supplementary Fig. 7C and Fig. 2G, H, I). In addition, mRNA levels of the M2 marker IL-10 decreased and those of the M1 marker CD86 increased, upon silencing for CECR1 (Fig. 2I). Stimulation with rhCECR1 raised the levels of CD163 transcript and protein in THP-1 macrophages in a dose responsive manner (Fig. 3A and B). Similar observations were made at the protein level using the M-CSF induced (M2-like) macrophages (Fig. 3C–E). FACS analysis revealed increased expression of CD163 in M-CSF induced macrophages following treatment with rhCECR1 during M2 macrophage differentiation (Supplementary Fig. 7D). Increased numbers of CD163+ and CD204+ cells were observed in the GBM samples with high CECR1 signals (>5% CECR1+ area per image view) compared with those with low CECR1 signals (<5% CECR1+ area per image view) (Supplementary Fig. 7E and F; Fig. 8A and B.
Fig. 3.

CECR1 promotes macrophages towards a M2-like phenotype. (A) Western blot of CD163 in THP-1 derived macrophages treated with rhCECR1 for 96 hours. Numbers above bands indicate quantified protein levels normalized to β-actin. (B) Quantitative PCR analysis of CD163 expression in THP-1 macrophages treated with rhCECR1. Data are from 3 experiments, normalized to β-actin, presented as mean ± SEM; *P < .05, ***P < .005. (C) Representative CD163 immunostaining of M-CSF induced macrophages with or without stimulation with CECR1 (100 nM) for 96 hours. Experiments were repeated 3 times independently (scale bar: 100 µm). (D) Representative western blot of CD163 in M-CSF induced macrophages with or without CECR1 stimulation for 96 hours from 3 experiments. Numbers above bands indicate quantified protein normalized to β-actin. (E) Quantified CD163 intensity in M-CSF induced macrophages with or without CECR1 (100 nM) stimulation is shown in mean ± SEM; ***P < .005. (F) Quantitative PCR analysis of M2-macrophage associated genes; IL-10, CXCL12, CCL18, CCL22, IL-6, and inflammatory gene IL-12p35 in THP-1 derived macrophages treated with CECR1 for 96 hours. Data normalized to β-actin are from 3 independent experiments and shown as mean ± SEM; *P < .05, **P < .01. (G) Quantitative PCR analysis of IL-10, CXCL12, CCL18, IL-6, IL-12p35, and CCL22 in M-CSF induced macrophages treated by CECR1(100 nM) for 96 hours. Data are from 3 experiments and shown as mean ± SEM in fold change compared with the CECR1 free condition. *P < .05, **P < .01.
Cytokines specifically expressed by M2-like or M1-like TAMs were selected for qPCR analysis. In response to rhCECR1, THP-1 macrophages showed a dose responsive upregulation of CD163, IL-10, CXCL12, CCL18, and CCL22. Increased IL-6 was only observed in response to the highest concentration of rhCECR1 (Fig. 3F). RhCECR1 stimulation of M-CSF induced macrophages stimulated the expression of CD163, IL-10, CXCL12, CCL18, and IL-6 (Fig. 3G), but suppressed the expression of CCL22 (Fig. 3G). The discrepancy with CCL22 may be the result of intrinsic differences between THP-1 derived macrophages and M-CSF induced macrophages.16 The M1 cytokine IL-12p35 was downregulated by CECR1 in both THP-1 and peripheral M2 macrophages (Fig. 3F, G).
CECR1 Regulates Proliferation and Migration of Glioma Cells via M2-like Macrophages
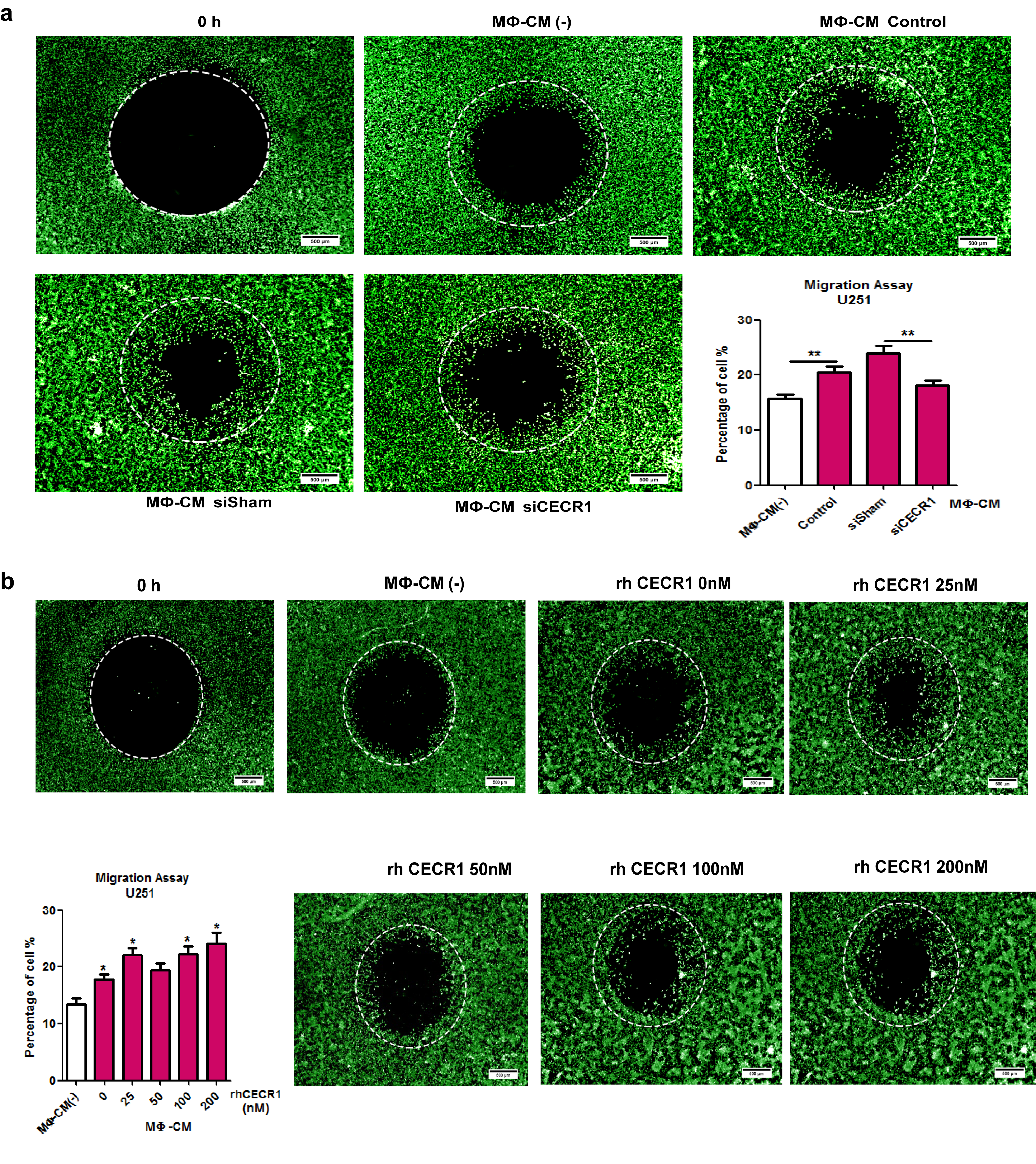
We used the GBM cell lines U87, U373, and U251 (implemented in studies with macrophages previously17–19), and in order to study the effects of the macrophages on tumor progression, we first measured the effects of conditioned medium derived from THP-1 macrophages on the proliferation rates. GBM cells that received conditioned medium from rhCECR1 treated THP-1 macrophages showed higher proliferation rates (Supplementary Figs. 9A, B and 10A, B). GBM cells treated by the macrophage (MФ)-CM from THP-1 macrophage with CECR1 silencing (siCECR1 CM) showed a significantly lower proliferation rate than cells stimulated with MФ-CM from THP-1 macrophage with siSham treatment (siSham CM) (Fig. 4B; Supplementary Fig. 10A, B). A significant reduction in Ki67 was observed in U87 and U251 exposed to siCECR1 CM in comparison to those exposed to siSham CM (Fig. 4D and F; Supplementary Fig. 10C and D). U87 and U251 cells treated with medium from THP-1 macrophages exposed to rhCECR1 resulted in rising levels of Ki67 (Fig. 4D and F; Supplementary Fig. 10C and F). Direct stimulation with rhCECR1 without macrophages did not affect the proliferation rate of U87 cells (Supplementary Fig. 13A). Next, all 3 GBM cell lines were exposed to siCECR1 CM to assess the effects on cell migration. SiCECR1 CM induced a weaker migration response than exposure to siSham CM (Fig. 4C). Medium derived from rhCECR1 treated THP-1 macrophages enhanced the migration of GBM cells (Supplementary Fig. 9C and D; Supplementary Figs. 11 and 12).
Fig. 4.

CECR1 in macrophages regulates proliferation and migration of U87 cells. (A) Representative images of U87 cells with/without treatment of macrophage conditioned medium (MΦ-CM) at days 1 and 3 post stimulation from 3 experiments (scale bar: 200 µm). Data are obtained from 3 experiments (n = 6 each) and presented as mean ± SEM in fold change compared with MΦ-CM free condition. *P < .05 ***P < .005. (B) Representative figures of U87 cells with treatment of MΦ-CM from nontargeting siSham or siCECR1 transfected macrophages. Pictures were taken at days 1, 3, 5 post stimulation (scale bar: 200 µm). Data are represented from 3 experiments (n = 6 for each), and presented as mean ± SEM in fold change compared with U87 cells exposed to MΦ-CM of siSham treated macrophages. *P < .05 ***P < .005. (C) Left panel: representative pictures of U87 migration into the detection zone (indicated by the white circle) after stimulation with MΦ-CM from macrophage with siSham and siCECR1 (scale bar: 200 µm). Right panel: Quantification of fluorescent cells in the migration zones in siSham and siCECR1 MΦ-CM conditions. Data are represented from 3 experiments and presented as mean ± SEM; *P < .05. (D) Representative images of Ki67 staining in U87 cells, which were treated with MΦ-CM from siSham and siCECR1 transfected macrophages for 48 hours (scale bar: 100 µm). (E) Representative pictures of Ki67 staining in U87 cells, which were treated with MΦ-CM for 48 hours from macrophages treated with rhCECR1 for 96 hours (scale bar: 100 µm). (F) Quantification of Ki67 staining in U87 cells treated with different MΦ-CM for 48 hours. Left graph: Ki67 signal distribution of U87 cells treated with MΦ-CMs of siSham and siCECR1 macrophages. Right graph: Ki67 signal distribution of U87 cells treated with MΦ-CMs of macrophages that were stimulated by CECR1. Data are from 3 experiments. Chi-square test was used for analysis. ***P < .005.
CECR1 Expression in Macrophages Mediates Paracrine Activation of the MAPK Signaling in U87 and U373 Glioma Cells
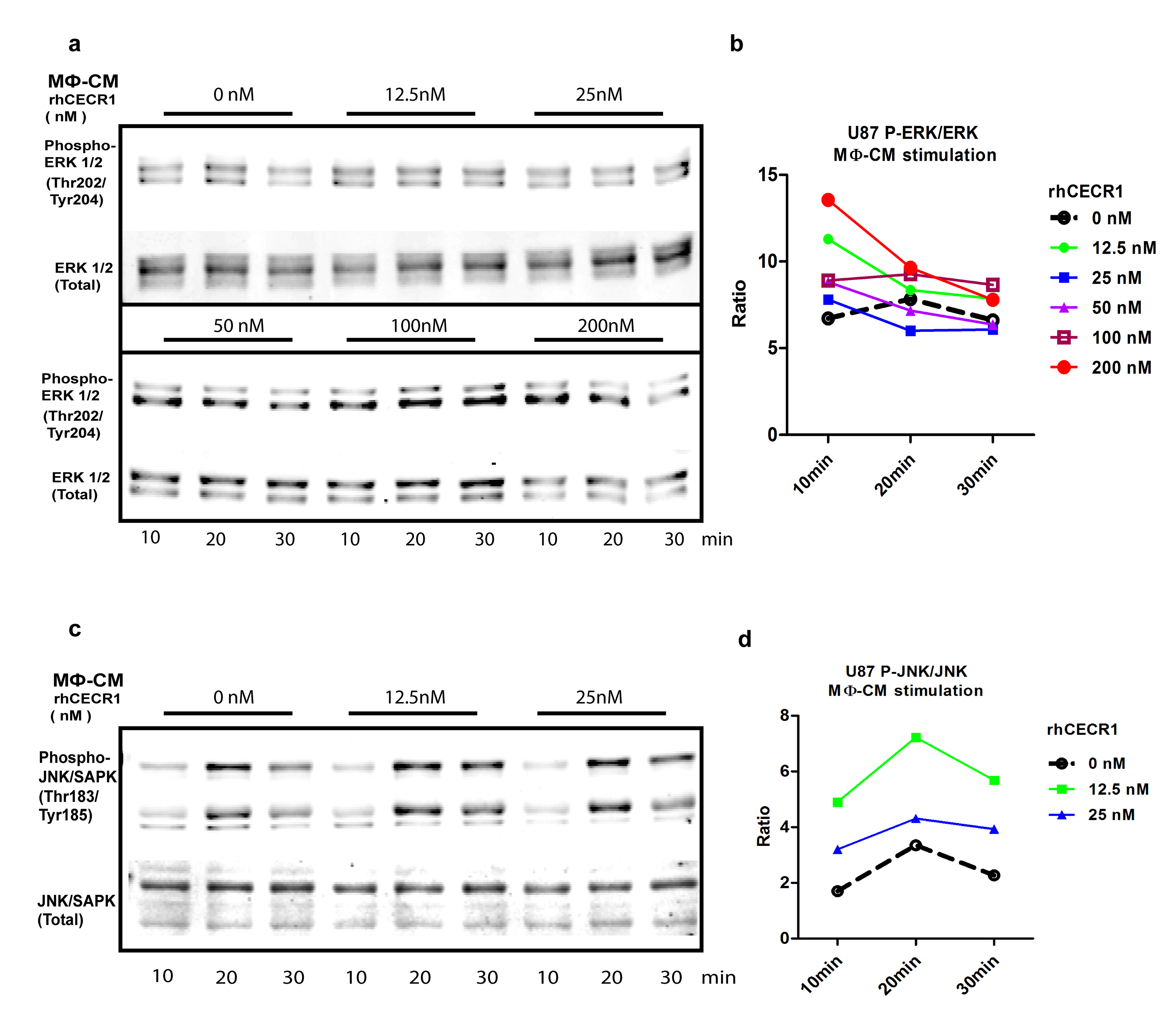
We used the mitogen activated protein kinase (MAPK) pathway consisting of extracellular signal-regulated kinase (ERK)1/2, c-Jun N-terminal kinase (JNK)/stress-activated protein kinase (SAPK), and P38 signaling because it plays crucial roles in tumor cell survival and motility.20,21 Serum-free medium starved glioma cells (U87 and U373) were stimulated for 10 and 20 minutes by siCECR1 CM or siSham CM. Phospho-ERK1/2, total ERK1/2, and phospho-JNK/SAPK/total JNK/SAPK ratios decreased in U87 and U373 cells exposed to THP-1 siCECR1 CM at both time points (Fig. 5A–D and Supplementary Fig. 14A–D). The transcription factor c-Jun is a potential downstream target of JNK/SAPK. The phospho–c-Jun (ser73)/total c-Jun ratio was significantly reduced in the U87 and U373 cells exposed to THP-1 siCECR1 CM at 20 minutes post stimulation only (Fig. 5E–F and Supplementary Fig. 14E, F). Immunostaining for phospho–c-Jun showed a reduced signal in U87 with THP-1 siCECR1 CM treatment (Fig. 5G and H). In contrast, medium derived from THP-1 macrophages treated with rhCECR1 increased phospho-protein/total protein ratios of both ERK1/2 and JNK/SAPK in U87 cells (Supplementary Fig. 15A and D). Direct rhCECR1 treatment of U87 cells without macrophage mediation did not increase phospho-ERK1/2 and phospho-JNK/SAPK ratios (Supplementary Fig. 13B). These data indicate that the MAPK signaling pathways in U87 and U373 cells are activated by a CECR1-mediated paracrine effect from macrophages.
Fig. 5.

CECR1 function in macrophages activates the MAPK pathway in U87 cells. (A) Western blot of phospho-ERK1/2 and total ERK1/2 in U87 cells at 10 and 20 minutes after stimulation with MΦ-CM from siSham and siCECR1 transfected macrophages. (B) Phospho-ERK/total ERK ratios are presented as mean ± SEM in fold change compared with MΦ-CM derived from macrophages with nontargeted siRNA (siSham) and siRNA targeting CECR1 (siCECR1) from 5 experiments. *P < .05. (C) Western blot of phospho-JNK/SAPK and total JNK/SAPK protein in U87 cells at 10 and 20 minutes after stimulation with MΦ-CM derived from siSham and siCECR1 transfected macrophages. (D) Phospho-JNK/total JNK ratios are presented as mean ± SEM in fold change compared with MΦ-CM derived of macrophages with non-targeted siRNA (siSham) and siRNA targeting CECR1 (siCECR1) from 3 experiments. *P < .05. (E) Western blot of phospho-/total c-Jun in U87 cells at 10 and 20 minutes after stimulation with MΦ-CM derived from siSham and siCECR1 siRNA transfected macrophages. (F) Phospho–c-Jun/total c-Jun ratios at 20 minutes after stimulation are presented as mean ± SEM in fold change compared with MΦ-CM derived from macrophages with nontargeted siRNA (siSham) and siRNA targeting CECR1 (siCECR1) from 3 experiments. *P < .05. (G) Representative immunostainings of phospho–c-Jun in U87 cells of 2 experiments. U87 were stimulated with MΦ-CM derived from siSham and siCECR1 transfected macrophages for 15 and 30 minutes (scale bar: 100 µm). (H) Quantified phospho–c-Jun in U87 cells were obtained from 200 randomly selected cells and presented as mean ± SEM in each condition. **P < .01, ***P < .005.
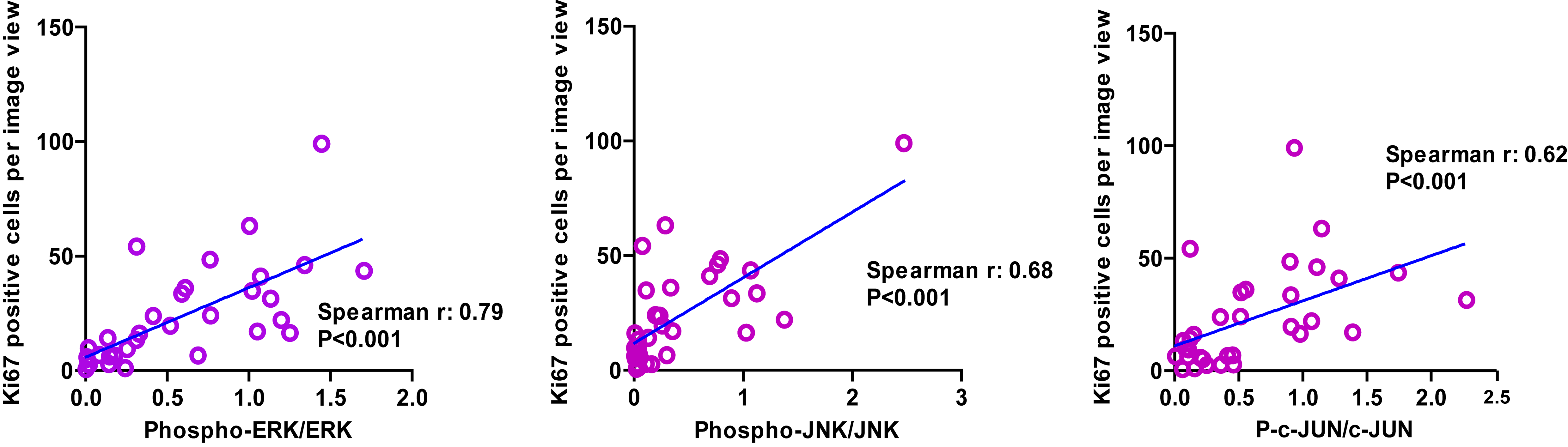
Expression of CECR1 Correlates with Increased Levels of Ki67 Positive Cells and Phosphorylation of ERK1/2 and JNK/SAPK in Patient GBM Samples
Thirty-three GBM surgical samples were selected for immunohistochemical evaluation and divided into 2 groups based on the median of the percentage of CECR1+ areas per image view (Fig. 6A–C). The number of Ki67+ cells and the percentage of positive areas for phospho-ERK1/2, phospho-JNK/SAPK, and phospho–c-Jun (ser73) proteins were analyzed and compared between the 2 groups (Fig. 6A–C). A significantly higher number of Ki67+ cells were counted in the samples with high CECR1 signal (Fig. 6C). The number of Ki67+ cells is positively correlated with the percentage of CECR1+ area (Fig. 6D). In addition, phospho-ERK1/2, phospho-JNK/SAPK, and phospho–c-Jun signals were significantly higher in the CECR1 high group. The ratios of the phospho-ERK1/2–total ERK1/2, phospho-JNK/SAPK–total JNK/SAPK, and phospho–c-Jun/total c-Jun signals correlated positively with the percentage of CECR1+ areas in GBM samples (Fig. 6D). In addition, phospho-ERK1/2–total ERK1/2, phospho-JNK/SAPK–total JNK/SAPK, and phospho–c-Jun/total c-Jun signals were positively correlated with the number of Ki67+ cells in GBM specimens (Supplementary Fig. 16).
Fig. 6.

CECR1 correlates with Ki67, MAPK signals in GBM samples. (A) (B) Representative figures of GBM samples immunostained for CECR1, Ki67, phospho-/total ERK1/2, phospho-/ total JNK, phospho-/ total c-Jun are shown (scale bar: 100 µm). (C) Left graph: Quantificated CECR1 staining level (%CECR1+ area per image view) in CECR1 high and low groups are presented as mean ± SEM; ***P < .005. Middle graph: Quantificated Ki67 in CECR1 high and low group are presented as mean ± SEM ***P < .005. Right panel: Ratios of phospho-ERK/total ERK, phospho-JNK/total JNK and phospho–c-Jun/total c-Jun. Data are presented as mean ± SEM; ***P < .005. (D) Correlation between %CECR1+ area with ratios of phospho-ERK/total ERK, phospho-JNK/total JNK, phospho–c-Jun/total c-Jun and numbers of Ki67+ cells. Spearman correlation coefficient and P-value are shown.
Discussion
The main function of CECR1 is conversion of extracellular adenosine to inosine by replacing an amino with a hydroxyl group.22 CECR1 itself can also directly bind to, and possibly activate, the adenosine receptors A2AR and A2BR.23 In general, adenosine limits the production of pro-inflammatory factors such as tumor necrosis factor α and IL-12, and increases the production of anti-inflammatory cytokines, including IL-10.24,25 During M1-type inflammations, LPS stimulation of macrophages upregulates the adenosine receptor A2AR, which increases in turn the sensitivity of M1 macrophages to adenosine. This provides a feedback inhibitory mechanism to the M1 response by mediating a switch toward a milder M2 response.26 In GBM, mainly M2 polarized cells are found with high levels of CECR1 production. This should reduce local adenosine levels and promote a more pro-inflammatory M1-based feedback response. However, M1 TAMs are in the minority compared with M2 TAMs in GBM. As indicated by previous studies, CECR1 itself can also directly activate adenosine receptor associated pathways independently of CECR1 catalytic activity and thus is capable of promoting the same anti-inflammatory reaction as observed with adenosine stimulation.23 Previous studies, in addition to our own findings, have highlighted the crucial role of CECR1 in promoting M2 macrophage polarization.9,10 More research on the exact (possibly independent of catalytic activity) working mechanism is required to fully elucidate this process. CECR1 also appears to be critical in the modulation of the immune system in general, as heterozygous CECR1 mutation is associated with increased antibody deficiency and overall immune dysregulation in the affected patients.27 A significant rise in activated CECR1 levels in the circulation of systemic lupus erythematosus relapse patients also highlights an important role for CECR1 in the modulation of the (auto)immune response.28 Other reports have implicated CECR1 as possibly a crucial regulator of neutrophils, as reduced adenosine deaminase activity in biallelic CECR1 mutation patients is an important causative factor for increased neutrophil activity that coincided with systemic vasculitis.13 Others have confirmed that CECR1 mutations with deleterious protein function are indeed strongly associated with systemic vasculopathy in addition to lacunar strokes.9,10 This phenotype was mimicked in zebrafish models in which knockdown of the zebrafish homologue of CECR1 caused intracranial hemorrhages and neutropenia. The clinical pathologies observed in patients with CECR1 deficiency could be partially reduced by anti–tumor necrosis factor therapy,29 further strengthening the association between CECR1 deficiency with a systemic hyperactivated inflammation response.
The link between CECR1 deficiencies and vasculopathy is further strengthened by a report on 2 patients with biallelic CECR1 mutations and histories of vasculitis and intracranial hemorrhages.13 In our current study, CECR1-expressing immune cells were observed at the perivascular areas of the gliomas in particular—possibly reflecting the modulation of immune cells by CECR1 leading to progressive malfunction of the endothelial barrier function of glioma vasculature. Based on these reports and our own observations, we hypothesize that the influence of CECR1 on immune cells, and on macrophage polarization especially, seems to predominantly impact the systemic and cerebral vasculature, and could affect the tumor vasculature in GBM.12
The fraction of resident microglia and TAMs reportedly constitute up to 30% of the total cell population of GBM.30 Glioma cells are capable of actively reprogramming the TAMs in order to support tumor progression.5 Based on expression profiling of mouse gliomas, subtyping beyond the classic M1 and M2 poles has been introduced and M2a (Th2, anti-pathogens response), M2b (Th2, immune regulatory), and M2c (immune modulatory and tissue remodeling)31 TAMs are now being recognized.32 These 3 subtypes can be differentially activated in malignant tumors via IL-4/IL-13 (for M2a),31 CXC receptor 3(+) B cells,33 IL-21/IFN-γ pathways (for M2b),34 and IL-10 or M-CSF stimulation (for M2c).31,35 The M2 subtypes have been shown to contribute to carcinogenesis in various ways, including contact-dependent dispersion of carcinoma cell aggregates (M2a),36 supporting tumor progression (M2b),34 and active suppression of immune surveillance (M2c). Expression analysis of typical M1 and M2a, b, and c subtype markers validated the presence of these TAM subtypes in human gliomas. Of note, over 50% of genes in TAMs in murine glioma do not match with any genes that typically characterize these 4 macrophage subtypes. In addition, a recent study has presented evidence that the TAM phenotype in GBM could also resemble the unpolarized Ω macrophage phenotype.37 It remains unclear whether the glioma TAMs constitute a unique phenotype that is not found in macrophage populations in nonneoplastic tissues. Further investigations of the influence of CECR1 on the expression of the M1 and M2a, b, and c markers may reveal specific CECR1-mediated effects on TAM polarization.
The modest effects of CECR1 knockdown in vitro contrasted with the strong correlation between CECR1 protein expression levels and phospho-JNK, phospho-ERK1/2, and phospho–c-Jun levels that were found in the tumor samples (which were more significant). This discrepancy could be explained by the isolated conditions in our experimental setup in vitro. We designed the study to investigate the direct paracrine effects of CECR1 modulation in macrophages on GBM cell response and chose to use conditioned medium derived from the siCECR1 treated macrophages to stimulate GBM cells. In vivo, direct interaction with CECR1 high-expressing macrophages with the surrounding tumor environment—including tumor cells and other (inflammatory) cells such as subsets of CD3+ T cells,38 microglia,2 and neutrophils39—could further enhance JNK and ERK signaling.
In conclusion, the current study has demonstrated a CECR1-mediated cross-talk mechanism between macrophages and glioma cells. Continuous CECR1 autocrine stimulation of macrophages enables M2-like TAMs to stimulate MAPK and c-Jun signaling in glioma cells via paracrine activation, leading to higher proliferation and motility rates in tumor glioma cells. Based on our findings, CECR1 could become a suitable drug target for selective modulation of the M2-like TAMs in glioma.
Supplementary Material
Supplementary material is available at Neuro-Oncology online.
Funding
C.Z. was supported by the Chinese Scholar Council (201206230102). C.C. was supported by the Netherlands Foundation for Cardiovascular Excellence and by the Netherlands Cardiovascular Research Initiative: an initiative with support of the Dutch Heart Foundation, (CVON2014-11 RECONNECT), VIDI grant 91714302, the Erasmus MC fellowship grant, and the RM fellowship grant of the UMC Utrecht.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgments
The authors would like to thank CardioGenx B.V. for providing the recombinant CECR1. We would also gratefully acknowledge Protein Atlas and Dr A. T. Rowshani for sharing CECR1 antibody and CD163 and CD80 antibodies.
Conflict of interest statement. The authors state that there are no conflicts of interest.
References
- 1. Qian BZ, Pollard JW. Macrophage diversity enhances tumor progression and metastasis. Cell. 2010;141(1):39–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Hambardzumyan D, Gutmann DH, Kettenmann H. The role of microglia and macrophages in glioma maintenance and progression. Nat Neurosci. 2016;19(1):20–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Alvaro T, Lejeune M, Camacho FI, et al. The presence of STAT1-positive tumor-associated macrophages and their relation to outcome in patients with follicular lymphoma. Haematologica. 2006;91(12):1605–1612. [PubMed] [Google Scholar]
- 4. Kinouchi M, Miura K, Mizoi T, et al. Infiltration of CD40-positive tumor-associated macrophages indicates a favorable prognosis in colorectal cancer patients. Hepatogastroenterology. 2013;60(121):83–88. [DOI] [PubMed] [Google Scholar]
- 5. Li W, Graeber MB. The molecular profile of microglia under the influence of glioma. Neuro Oncol. 2012;14(8):958–978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Prosniak M, Harshyne LA, Andrews DW, et al. Glioma grade is associated with the accumulation and activity of cells bearing M2 monocyte markers. Clin Cancer Res. 2013;19(14):3776–3786. [DOI] [PubMed] [Google Scholar]
- 7. Komohara Y, Ohnishi K, Kuratsu J, et al. Possible involvement of the M2 anti-inflammatory macrophage phenotype in growth of human gliomas. J Pathol. 2008;216(1):15–24. [DOI] [PubMed] [Google Scholar]
- 8. Conlon BA, Law WR. Macrophages are a source of extracellular adenosine deaminase-2 during inflammatory responses. Clin Exp Immunol. 2004;138(1):14–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Navon Elkan P, Pierce SB, Segel R, et al. Mutant adenosine deaminase 2 in a polyarteritis nodosa vasculopathy. N Engl J Med. 2014;370(10):921–931. [DOI] [PubMed] [Google Scholar]
- 10. Zhou Q, Yang D, Ombrello AK, et al. Early-onset stroke and vasculopathy associated with mutations in ADA2. N Engl J Med. 2014;370(10):911–920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Footz TK, Brinkman-Mills P, Banting GS, et al. Analysis of the cat eye syndrome critical region in humans and the region of conserved synteny in mice: a search for candidate genes at or near the human chromosome 22 pericentromere. Genome Res. 2001;11(6):1053–1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Caorsi R, Penco F, Schena F, et al. Monogenic polyarteritis: the lesson of ADA2 deficiency. Pediatr Rheumatol Online J. 2016;14(1):51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Belot A, Wassmer E, Twilt M, et al. Mutations in CECR1 associated with a neutrophil signature in peripheral blood. Pediatr Rheumatol Online J. 2014;12:44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Lacey DC, Achuthan A, Fleetwood AJ, et al. Defining GM-CSF- and macrophage-CSF-dependent macrophage responses by in vitro models. J Immunol. 2012;188(11):5752–5765. [DOI] [PubMed] [Google Scholar]
- 15. Zheng PP, van der Weiden M, Kros JM. Fast tracking of co-localization of multiple markers by using the nanozoomer slide scanner and NDPViewer. J Cell Physiol. 2014;229(8):967–973. [DOI] [PubMed] [Google Scholar]
- 16. Schildberger A, Rossmanith E, Eichhorn T, et al. Monocytes, peripheral blood mononuclear cells, and THP-1 cells exhibit different cytokine expression patterns following stimulation with lipopolysaccharide. Mediators Inflamm. 2013;2013:697972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Clark MJ, Homer N, O’Connor BD, et al. U87MG decoded: the genomic sequence of a cytogenetically aberrant human cancer cell line. PLoS Genet. 2010;6(1):e1000832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Siegelin MD, Reuss DE, Habel A, et al. Quercetin promotes degradation of survivin and thereby enhances death-receptor-mediated apoptosis in glioma cells. Neuro Oncol. 2009;11(2):122–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Fukumura D, Xavier R, Sugiura T, et al. Tumor induction of VEGF promoter activity in stromal cells. Cell. 1998;94(6):715–725. [DOI] [PubMed] [Google Scholar]
- 20. Johnson GL, Lapadat R. Mitogen-activated protein kinase pathways mediated by ERK, JNK, and p38 protein kinases. Science. 2002;298(5600):1911–1912. [DOI] [PubMed] [Google Scholar]
- 21. Dhillon AS, Hagan S, Rath O, et al. MAP kinase signalling pathways in cancer. Oncogene. 2007;26(22):3279–3290. [DOI] [PubMed] [Google Scholar]
- 22. Zavialov AV, Engström A. Human ADA2 belongs to a new family of growth factors with adenosine deaminase activity. Biochem J. 2005;391(Pt 1):51–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Zavialov AV, Gracia E, Glaichenhaus N, et al. Human adenosine deaminase 2 induces differentiation of monocytes into macrophages and stimulates proliferation of T helper cells and macrophages. J Leukoc Biol. 2010;88(2):279–290. [DOI] [PubMed] [Google Scholar]
- 24. Haskó G, Cronstein B. Regulation of inflammation by adenosine. Front Immunol. 2013;4:85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Linden J. New insights into the regulation of inflammation by adenosine. J Clin Invest. 2006;116(7):1835–1837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ferrante CJ, Pinhal-Enfield G, Elson G, et al. The adenosine-dependent angiogenic switch of macrophages to an M2-like phenotype is independent of interleukin-4 receptor alpha (IL-4Rα) signaling. Inflammation. 2013;36(4):921–931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Schepp J, Bulashevska A, Mannhardt-Laakmann W, et al. Deficiency of adenosine deaminase 2 causes antibody deficiency. J Clin Immunol. 2016;36(3):179–186. [DOI] [PubMed] [Google Scholar]
- 28. Saghiri R, Ghashghai N, Movaseghi S, et al. Serum adenosine deaminase activity in patients with systemic lupus erythematosus: a study based on ADA1 and ADA2 isoenzymes pattern. Rheumatol Int. 2012;32(6):1633–1638. [DOI] [PubMed] [Google Scholar]
- 29. Nanthapisal S, Murphy C, Omoyinmi E, et al. Deficiency of adenosine deaminase type 2: a description of phenotype and genotype in fifteen cases. Arthritis Rheumatol. 2016;68(9):2314–2322. [DOI] [PubMed] [Google Scholar]
- 30. Kerber M, Reiss Y, Wickersheim A, et al. Flt-1 signaling in macrophages promotes glioma growth in vivo. Cancer Res. 2008;68(18):7342–7351. [DOI] [PubMed] [Google Scholar]
- 31. Martinez FO, Gordon S. The M1 and M2 paradigm of macrophage activation: time for reassessment. F1000Prime Rep. 2014;6:13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Szulzewsky F, Pelz A, Feng X, et al. Glioma-associated microglia/macrophages display an expression profile different from M1 and M2 polarization and highly express Gpnmb and Spp1. PLoS One. 2015;10(2):e0116644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Liu RX, Wei Y, Zeng QH, et al. Chemokine (C-X-C motif) receptor 3-positive B cells link interleukin-17 inflammation to protumorigenic macrophage polarization in human hepatocellular carcinoma. Hepatology. 2015;62(6):1779–1790. [DOI] [PubMed] [Google Scholar]
- 34. Chen MM, Xiao X, Lao XM, et al. Polarization of tissue-resident TFH-like cells in human hepatoma bridges innate monocyte inflammation and M2b macrophage polarization. Cancer Discov. 2016;6(10):1182–1195. [DOI] [PubMed] [Google Scholar]
- 35. Sousa S, Brion R, Lintunen M, et al. Human breast cancer cells educate macrophages toward the M2 activation status. Breast Cancer Res. 2015;17:101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Bai J, Adriani G, Dang T-M, et al. Contact-dependent carcinoma aggregate dispersion by M2a macrophages via ICAM-1 and β2 integrin interactions. Oncotarget. 2015;6(28):25295–25307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Gabrusiewicz K, Rodriguez B, Wei J, et al. Glioblastoma-infiltrated innate immune cells resemble M0 macrophage phenotype. JCI Insight. 2016;1(2):. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Sims JS, Grinshpun B, Feng Y, et al. Diversity and divergence of the glioma-infiltrating T-cell receptor repertoire. Proc Natl Acad Sci U S A. 2016;113(25):E3529–E3537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Liang J, Piao Y, Holmes L, et al. Neutrophils promote the malignant glioma phenotype through S100A4. Clin Cancer Res. 2014;20(1):187–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.