

Abstract
Post-stroke cognitive impairment is a major cause of long-term neurological disability. The prevalence of post-stroke cognitive deficits varies between 20% and 80% depending on brain region, country, and diagnostic criteria. The biochemical mechanisms underlying post-stroke cognitive impairment are not known in detail. Cyclin-dependent kinase 5 is involved in neurodegeneration, and its dysregulation contributes to cognitive disorders and dementia. Here, we administered cyclin-dependent kinase 5-targeting gene therapy to the right hippocampus of ischemic rats after transient right middle cerebral artery occlusion. Cyclin-dependent kinase 5 RNA interference prevented the impairment of reversal learning four months after ischemia as well as neuronal loss, tauopathy, and microglial hyperreactivity. Additionally, cyclin-dependent kinase 5 silencing increased the expression of brain-derived neurotrophic factor in the hippocampus. Furthermore, deficits in hippocampal long-term potentiation produced by excitotoxic stimulation were rescued by pharmacological blockade of cyclin-dependent kinase 5. This recovery was blocked by inhibition of the TRKB receptor. In summary, these findings demonstrate the beneficial impact of cyclin-dependent kinase 5 reduction in preventing long-term post-ischemic neurodegeneration and cognitive impairment as well as the role of brain-derived neurotrophic factor/TRKB in the maintenance of normal synaptic plasticity.
Keywords: CDK5 RNAi, long-term post-ischemia, neuroprotection, cognitive impairment, BDNF, neuronal plasticity
Introduction
Stroke is responsible for more than five million deaths each year worldwide and is the second leading cause of death and a major cause of physical and mental disability.1 Stroke is also a risk factor for cognitive impairment and dementia.2 Post-stroke cognitive impairment is characterized by the decline of various cognitive domains, such as learning, memory, constructional abilities3,4 and executive function, over both the short term and long term.5 However, the pathogenesis of long-term post-ischemic cognitive effects is not yet clearly understood. In rats, CA1 neurons and their dendritic processes have been reported to partially disappear months after cerebral ischemia, and the entire CA1 region appears to shrink. This long-term post-ischemic neurodegeneration is associated with the accumulation of large mineralized calcium deposits and extensive neuroinflammatory and astroglial reactions.6 However, more studies are needed to characterize the long-term neurological damage in detail and to establish a link between it and cognitive decline.
Epidemiological studies have shown that the prevalence of cognitive impairment in ischemic stroke patients is nine-fold higher than in controls at three months7 and 4–12 times higher than in controls four years after an infarct.8 However, that prevalence has not been interpreted as a direct consequence of the primary ischemic damage9 because of the multifactorial consequences of ischemia/reperfusion. Furthermore, outside the thrombolytic therapeutic window, there is no gold standard treatment to prevent long-term physical and mental disabilities after a stroke. Thus, the development of an effective therapy to prevent and treat short- and long-term post-stroke cognitive impairment is urgently needed.
Cyclin-dependent kinase 5 (CDK5) is involved in cerebral ischemia-related cognitive deterioration.10,11 CDK5 is a proline-directed serine/threonine kinase that is found ubiquitously throughout the nervous system and plays important roles in synaptic plasticity and neurotransmission; in addition, CDK5 over-regulation is involved in neurodegeneration.12 Many studies have shown that CDK5 hyperactivation is involved in tau hyperphosphorylation and the subsequent development of cognitive impairment in neurodegenerative diseases,13 and multiple pieces of evidence have implicated CDK5 in the progression of neurodegeneration after cerebral ischemia. We have recently demonstrated that silencing CDK5 in the hippocampus prevented learning, memory, and reversal learning deficits at one month post-ischemia and reduced tauopathy and other hallmarks of neurodegeneration.14 Part of the central aim of the present study was to determine whether CDK5 RNAi-induced neuroprotection is maintained four months after ischemia/reperfusion, an intermediate timepoint for long-term deterioration according to previous reports. We also sought to identify a potential preventive therapy based on the silencing of pathogenic genes, as CDK5, which has been strongly implicated in post-stroke cognitive disorders and dementia.
Materials and methods
All animal procedures were conducted in concordance with ARRIVE guidelines, the Guide for the Care and Use of Laboratory Animals, 8th edition, published by the National Institutes of Health (NIH) at SUNY-Downstate, and Colombian standards (law 84/1989 and resolution 8430/ 1993). The procedures were also approved by the Ethics Committee for Animal Experimentation of the University of Antioquia, Medellín, Colombia.
Animal model
Male Wistar albino rats from our in-house, pathogen-free colony at the vivarium at SIU (Sede de Investigación Universitaria), University of Antioquia, Medellin, Colombia, were kept on a 12:12-h dark:light cycle and were provided food and water ad libitum. Special care was taken to minimize animal suffering and to reduce the number of animals used. Three-month-old rats weighing 250–310 g were used. To evaluate spatial learning and memory, 12–15 rats per experimental group were used. In addition, 5–6 rats per experimental group were used for the histological and biochemical assessments.
Three-month old male C57BL/6 mice from a pathogen-free colony at the vivarium of SUNY Downstate Medical Center, New York, USA were used for the electrophysiological studies. All animal-related procedures were conducted in accordance with the guidelines of the National Institutes of Health (Guide for the Care and Use of Laboratory Animals) and were approved by the SUNY IACUC committee.
Middle cerebral artery occlusion
The rats were anesthetized with an intraperitoneally administered mixture of ketamine (60 mg/kg; Holliday Scott S.A. Int. Neyer, Buenos Aires, Argentina) and xylazine (5 mg/kg; Synthesis LTDA & CIA S.C.A., Bogotá, Colombia) and subcutaneously administered atropine (100 mg/kg; ERMA S.A., Bogotá, Colombia). The rats received a mixture of 2–4% isoflurane (Baxter, Deerfield, IL, USA) and 96% oxygen via an inhalation anesthesia machine. Body temperature was monitored throughout the surgery with a rectal thermometer, and the rats were maintained in a condition of mild hypothermia (34–35℃). The right common carotid artery was exposed and dissected. The right external carotid artery and the right internal carotid artery were exposed, and the first arterial branches were cauterized by electrocoagulation (AARON bipolar cautery, Albany, NY, USA). A 4-0 monofilament nylon suture (Corpaul, Bogotá, Colombia) was inserted into the internal carotid artery from the external carotid artery to occlude the right middle cerebral artery (MCA) at its origin. The nylon filament tips had been previously rounded by passing them through a flame and were coated with a poly-l-lysine solution (0.1% w/v in deionized water; Sigma, St Louis, MO, USA). The suture was removed to enable reperfusion after 60 min, and the wound was then closed. Sham-operated animals underwent identical procedures, excluding insertion of the suture.
Short hairpin RNAmiR delivery
The RNAi (short hairpin RNAmiR, shRNAmiR) sequences for CDK5 silencing (CDK5miR), the scrambled control RNA sequence (SCRmiR), the viral particle production and validation of the in vitro silencing model were based on Piedrahita et al.15 The AAV particles were obtained from the Davidson Laboratory (University of Iowa Viral Vector Core). The right hippocampus (bregma coordinates: −2.56 antero-posterior, 0.8 lateral, and 4.1 depth) of the animals was injected with 2.5 µL of AAV2.5.shSCRmiR.GFP or AAV2.5.shCDK5miR.GFP. The injections were performed during the transient middle cerebral artery occlusion (tMCAO), 30 min after the filament was inserted (therapeutic intervention performed during the ischemic phase). The intrahippocampal injections were performed with a 10 µL Hamilton syringe at a rate of 0.2 µL/min, and the syringe was held in place for 5 min after the infusion before it was withdrawn.
Neurological evaluation
The animals were evaluated from 6 h to 15 days and from 100 days to 102 days after MCAO (n = 12–15 per experimental group) in each experimental group. Neurological function was scored on an 18-point scale.11 The composite neuroscore comprises six different neurologic tests: (1) spontaneous activity, (2) symmetry in limb movement, (3) forepaw outstretching, (4) climbing, (5) body proprioception, and (6) response to vibrissae touch. Each test was scored with a maximum of three points based on a set of predetermined criteria described elsewhere. The scores for each test were summed for a highest possible score of 18, indicating no neurologic deficits, and lowest score of 3, for animals with the most severe impairment. Neurological scoring was performed each day and in the same order for all rats.
Rotarod activity
Locomotor activity was measured in rats (n = 12–15 per experimental group) from 100 days to 103 days after MCAO post-ischemia by rotarod test apparatus (IITC Life Science, Woodland Hills, CA, USA). The rod is a drum, 7 cm in diameter, elevated 25 cm above the bottom of the apparatus, driven by a motor to control speed. Briefly, rats were trained for two days. On the first training day, rats were placed on the rotarod at a constant speed of 4 r/min for 1 min. On the second training day, animals were trained with accelerated speed from 4 to 10 r/min for 3 min. Thereafter, rats were subjected to two trials per day. Animals were scored for their latency to fall (in seconds, máximum 180 s) distance (m) and velocity (r/min) with accelerated speed from 4 to 40 r/min for 3 min.
Water maze test
Four months after the induction of ischemia, the rats (n = 12–15 per experimental group) were trained and evaluated on the Morris water maze.16 The apparatus consisted of a black plastic tank (1.8 m in diameter and 0.6 m in height) that was filled with water (22 ± 2℃) to a depth of 35 cm. The platform (12 cm diameter) was located 3 cm below the water’s surface during the spatial learning session and 2 cm above the water’s surface during the visible session. The extra-maze visual cues located around the room remained in a fixed position throughout the experiment. The first phase involved two daily learning sessions. Each session consisted of four successive trials, and each trial began with the pseudorandom placement of the rat into one of four randomly located starting positions. Before the initial trial, the animals were trained to stay on the platform for 30 s. If the rat did not find the platform after a maximum of 90 s, then it was gently guided to the platform. Eight learning sessions were conducted to evaluate the animals’ spatial learning performance. Two days after the end of the learning phase, the animals were tested for learning retention with a 60-s probe trial without the platform. During the probe trial, the latency to reach the platform’s previous location and the number of crossings over the previous site of the platform were determined. One day after the probe trial, the platform was moved to the opposite quadrant, and the animals were subjected to a transfer test to assess their cognitive flexibility and ability to solve a new spatial problem, and also the speed was measured. To control for any differences in visuomotor abilities or motivation between the experimental groups, the latency to reach the platform was evaluated using a visible platform (four trials) at the end of the retention test. The animals’ behavior was recorded by an automated system (Viewpoint, Lyon, France). Finally, the animals were killed for histological and biochemical evaluation.
Histology
Anesthetized animals (n = 5–6 per experimental group) were perfused with 4% paraformaldehyde in 0.1 mol/L phosphate buffer, pH 7.4. The brains were removed and postfixed with 4% paraformaldehyde at 4℃ for 48 h, rinsed with saline buffer and sectioned at 50 µm with a vibrating-blade microtome (VT1000S, Leica Microsystems, Nussloch, Germany). The hippocampus was analyzed in coronal sections located 2.56, 3.60, and 5.20 mm posterior to bregma.
Immunohistochemistry
Briefly, the sections were incubated while free floating with moderate shaking. The sections were quenched endogenous peroxidase activity and permeabilized. The sections were then incubated overnight at 4℃ with anti-NeuN (monoclonal mouse, 1:1000; Millipore, Bedford, MA, USA), anti-PHF-Tau (AT-8, 1:500, Pierce, Rockford, IL, USA), anti-MAP2 (monoclonal mouse, 1:1000, Chemicon, Temecula, CA, USA), anti-GFAP (monoclonal mouse, 1:1000; Chemicon, Temecula, CA, USA), and anti-OX42 (polyclonal rabbit, 1:1000; Millipore) antibodies, which were visualized using diaminobenzidine. NeuN, AT8, MAP2, GFAP, and OX42 immunostaining intensity was evaluated in a total area of 1.2288 mm2 (40×). The images were converted to a binary system, and integrated densities (relative units) were obtained for each image. The background was automatically subtracted from each image to quantify the relative intensity of immunostaining. For more details to review, see Gutierrez-Vargas et al.11
Immunofluorescence
Shortly, the sections at the level of bregma were rinsed in 0.1 M PBS and incubated for 10 min with 50 mM ammonium chloride to minimize autofluorescence. Then, the sections were permeabilized and incubated overnight at 4℃ with the following primary antibodies: anti-CDK5 (C-8) (1:1000; Santa Cruz Biotechnology), anti-NeuN (monoclonal mouse, 1:1000; Millipore), anti-GFAP (monoclonal mouse, 1:1000; Chemicon, Temecula, CA, USA), and anti-brain-derived neurotrophic factor (BDNF) (polyclonal rabbit, 1:500; Chemicon® Temecula, CA, USA). After several washing, the sections were incubated for 90 min at room temperature with a rabbit Alexa Fluor 594-conjugated secondary antibody (1:2,000; Molecular Probes) or a mouse Alexa Fluor 350-conjugated secondary antibody (1:1000; Molecular Probes). The sections were photographed at 40 × magnification, and the images were used to evaluate the fluorescence intensity (FI) of CDK5 immunostaining at 40 × magnification using Cell M (Olympus software, Miami, FL, USA). For more details, see Gutierrez-Vargas et al.11
Western blotting
In brief, the animals (n = 5–6 per experimental group) were sacrificed by decapitation, and their brains were quickly removed. The CA1 ipsilateral to the site of MCAO was dissected and frozen at −80℃ until analysis. The samples were homogenized in lysis buffer. Protein (30 µg) was loaded into each lane with loading buffer After electrophoresis, the proteins were transferred to nitrocellulose membranes (Amersham, Buckinghamshire, UK) at 250 mA for 2 h using an electrophoretic transfer system (mini Trans-blot Electrophoretic Transfer Cell). The membranes were first incubated overnight at 4℃ with the following primary antibodies: rabbit anti-CDK5 (C-8) (1:1000; Santa Cruz, CA, USA), rabbit anti-p35/p25 (1:500; Santa Cruz, CA, USA), rabbit AKT (1:1000; Millipore), mouse pAKT (1:1000; Millipore), rabbit p38 (1:500; Cell Signaling Technology), rabbit cAMP response element-binding protein (CREB; 1:1000; Cell Signaling Technology), mouse pCREB (1:1000; Cell Signaling Technology), rabbit TRKB (1:1000; Millipore); mouse ERK1/2 (1:1000; Cell Signaling Technology), mouse pERK1/2 (1:1000; Millipore), mouse pCAMKII (1:1000; Cell Signaling Technology), and mouse NR2B (1:1000; Millipore). The membranes were then incubated with a goat anti-rabbit IRDye 800WE secondary antibody (Li-COR, Lincoln, NE, USA), and the staining was detected using the ODYSSEY Infrared Imaging System (Li-COR, Miami, FL, USA). The band intensities were measured with ImageJ (NIH). For details, see Gutierrez-Vargas et al.11
Cyclin-dependent kinase 5 assay
The animals (n = 5–6 per each experimental group) were sacrificed by decapitation, and their brains were quickly removed. Hippocampal area CA1 was dissected and rapidly frozen in liquid nitrogen. The brain tissues were kept at −70℃ to preserve enzymatic activity until the assay was performed. CDK5 was immunoprecipitated from 250 µg of total protein using 1 µg of a rabbit polyclonal anti-CDK5 IgG (C-8) antibody (Santa Cruz, CA, USA). The antibody and protein extract were incubated overnight at 4℃ in a rotator. CDK5 activity was measured according to the protocol described in Piedrahita et al.15
Calpain activation assay
The animals (n = 5–6 per experimental group) were sacrificed by decapitation, and their brains were quickly removed. Calpain activity was measured using a commercially available kit (Calpain-Glo protease assay, Promega) according to the manufacturer’s instructions. Human calpain-1 (Tocris-Bioscience, Bristol, UK) was used as a positive control, and the calpain activity of the hippocampal lysates was measured on a microplate luminometer using a luciferase-based assay (GlioMax, Promega).
BDNF immunoassay
The ipsilateral hippocampi were homogenized in 20 mM Tris HCl, 137 mM NaCl, 1% NP40, 10% glycerol, 1 mM PMSF, 10 µg/mL aprotinin, 1 µg/mL leupeptin, and 0.5 mM sodium vanadate. The homogenates were centrifuged at 14,000 g for 5 min, and the supernatants were collected for BDNF immunoassay. BDNF was quantified in all of the samples using a commercially available kit (BDNF Emax Immunoassay system; Promega, Madison, WI, USA) according to the manufacturer’s instructions. The samples were assayed in duplicate using 96-well plates that were coated with an anti-BDNF monoclonal antibody overnight. A BDNF standard dilution series was also applied to obtain the absorbance–concentration calibration curve. The concentration values were normalized to the total protein concentrations of the corresponding lysates.
Electrophysiology
The mice (C57BL/6) were anesthetized, and the brains were removed and placed in cold modified artificial cerebral spinal fluid (aCSF) bubbled with O2 and CO2, to maintain a pH near 7.4. Horizontal hippocampal slices were obtained as previously described.17 In short, the brains were sectioned through the ventral hippocampus into 400 µm thick slices. The slices were left at room temperature for a recovery period until the recording, which was performed at 34℃. The slices were treated with 10 μM glutamate (Sigma-Aldrich) for 30 min. The glutamate was then washed out, and the slices were left for 30 min in aCSF. For the next 15 min, a group of slices were treated with 20 μM roscovitine (CDK5 pharmacological inhibitor, Calbiochem) with/without 200 nM K252a (selective inhibitor of tyrosine protein kinase activity, Sigma-Aldrich) prepared in aCSF, as described in each experiment.
Field excitatory post-synaptic potentials (fEPSPs) were recorded in the CA1 stratum radiatum with glass electrodes filled with 150 mM NaCl (2–3 MΩ resistance). The fEPSPs were elicited by stimulating the Schaeffer collateral fibers with a bipolar electrode. Input–output curves were obtained, and a stimulus that evoked ∼40% of the maximum fEPSP was selected for the rest of the experiment. The same type of stimulus was used for the long-term potentiation (LTP) experiments, in which a baseline of test responses was obtained (15 min with an inter-stimulus interval of 30 s) before high-frequency stimulation (HFS) (one train of 100 stimuli at 100 Hz) was used to induce synaptic LTP. Responses were recorded for 60 min after HFS. The tungsten stimulating electrodes were connected to a stimulus isolation unit (Grass S88), and the recordings were made using an Axoclamp 2B amplifier (Molecular Devices) and then filtered (0.1 Hz to 10 kHz using −6 dB/octave). The voltage signals were digitized and stored on a PC using a DigiData 1200 A (Molecular Devices) for off-line analysis. The fEPSP slope was measured and expressed as a percentage of baseline. The data were analyzed using Axon™ pCLAMP™ software, and the results are expressed as the mean ± standard error of the mean (SEM).
Sample size, randomization, and statistical analysis
Upon arrival from the breeding colony, the animals were randomly allocated to the sham or ischemia group. After the surgery, we again randomly assigned the animals to receive SCRmiR or CDK5miR treatment. The sample size was determined based on previous ischemia studies in our laboratory,14 which showed that the effects of ischemia are reproducible with a minimal number of animals. Five to six rats per experimental group were histologically and biochemically analyzed, and 12–15 animals per group were evaluated with the water maze test. In this study, there was 10.13% mortality due to the cerebral ischemia procedure.
In an effort to reduce the variability of ischemic groups, we used the same intraluminal suture head diameter (1.5 mm). During the surgical phase, all ischemic animals had the same anesthetic parameters, and similar surgical time period (approx 30 ± 5 min, not including occlusion time). We had no surgical complications (v.gr., cardio-respiratory arrest). All animals were subjected to 60 min of occlusion and a subsequent reperfusion period. Post-mortem evaluation demonstrated that none of the subjects developed intracranial hemorrhage. Behavioral tests were all performed at the same post-surgical time. Animals did not present signs of distress or chronic pain. The animals that did not learn the water maze tasks (1 Isch-CDK5miR rat out of 16 rats) or that did not exhibit AAV GFP expression in the hippocampus by histological analyses (1 sham-CDK5miR rat out of 13 rats per group) were excluded from the study.
The sample sizes (n) used for statistical analyses correspond to the number of animals per experimental group. The homogeneity of variance test was applied prior to statistical analysis. In the histological and biochemical analyses, the parametric data were compared using one-way analysis of variance (ANOVA) and Tukey’s post hoc tests were used for comparing several independent groups. ANOVA was also used to analyze the neurological test scores daily. The escape latencies during the training and transfer water maze tests were examined by repeated-measures ANOVA. The probe trial (water maze) results were analyzed by ANOVA followed by Fisher’s post hoc tests. The analyses were performed with SPSS 18.0. The data are expressed as the mean ± SEM, p < 0.05. To diminish inter-assay variation, all of the sample groups were processed in parallel.
For the electrophysiological experiments, the results were expressed as the mean ± SEM. The data were analyzed using repeated-measures ANOVA, and in specific cases, one-way ANOVA followed by Fisher's post hoc comparisons was used to analyze the differences between groups in the first and last 20 min of LTP. SPSS 18.0 software was used, and p < 0.05 was considered significant.
Results
CDK5miR prevents long-term post-ischemic CDK5 and calpain overactivation
In all of the experimental groups, AAV2.GFP was widely expressed in the injected area of the right CA1 (Figure 1(A – a and b)). The ischemic animals treated with SCRmiR exhibited a significant increase in CDK5 FI (p < 0.001), which was reduced by CDK5miR treatment (p < 0.001) (Figure 1(A – a and c)). CDK5 immunoreactivity (IR) was also significantly reduced by CDK5miR in the sham rats (p < 0.05; Figure 1(A – a and c)). These histological results were accompanied by an increase in the protein levels of CDK5 and p25, a coactivator associated with neurodegeneration, as well as by an increase in the p25/p35 ratio in the hippocampus of the ischemic animals (p < 0.05) compared with the control values. A significant reduction of p25 and CDK5 protein levels and the p25/p35 ratio were also observed in the CDK5miR-treated ischemic animals (p < 0.05) (Figure 1(B – a, c, and d)). However, the total p35 protein levels remained unchanged (Figure 1(B – b)). Interestingly, compared with the corresponding measures in the sham group, we observed higher calpain activation in the ischemic animals that were treated with SCRmiR (Figure 1(C); p < 0.05) as well as an increase in CDK5 activity (Figure 1(D – a and b); p < 0.05). Interestingly, calpain activation (Figure 1(C); p < 0.001) and CDK5 activity (Figure 1(C); p < 0.05) were significantly reduced by CDK5miR four months post-treatment.
Figure 1.

CDK5miR reduces CDK5 and p25 expression as well as prevents CDK5 and calpain activity at four months post-ischemia. (A) Representative photomicrographs of CDK5 expression in sections of the anterior portion of area CA1 (bregma −2.56). (a). The green fluorescent protein (GFP) signal represents the transduced CA1 cells in the ipsilateral hemisphere. The sections were photographed at 40 × , scale bars = 15 µm (40×). The animals were sacrificed four months after they received a unilateral injection with either CDK5miR or SCRmiR into the right CA1. (b) Identification of the injection site. (c). Quantification of the fluorescence intensity of CDK5 immunoreactivity in transduced neurons using Image Scope Pro software (Media Cybernetics). The fluorescence intensity was quantified over a total area of 1.2288 mm2 (40×); n = 4–6, **p < 0.001. (B) Representative Western blots of CDK5, p35, and p25 expression in CA1 samples collected from the animals treated with SCRmiR and CDK5miR (a, b, d). The p25/p35 ratio was determined (c). The densitometric quantification was expressed relative to the loading control (tubulin) and was normalized to the internal control (SCRmiR-treated sham rats). The values are presented as the mean ± SEM. (C) Calpain activity was measured in the ipsilateral CA1 region. n = 4 to 6, *p < 0.05, **p < 0.001. (D) The ipsilateral CA1 was dissected, and CDK5 kinase activity was detected using a phosphorylated histone antibody. A band corresponding to the IgG heavy chain was detected. Densitometric quantification was performed. pHISTONE expression was expressed relative to the total CDK5 from lysates (a), which was normalized to the loading control (tubulin) (b). n = 4 to 6, **p < 0.001. CDK5, cyclin-dependent kinase 5; RU, relative units; SCR, scrambled RNA sequence.
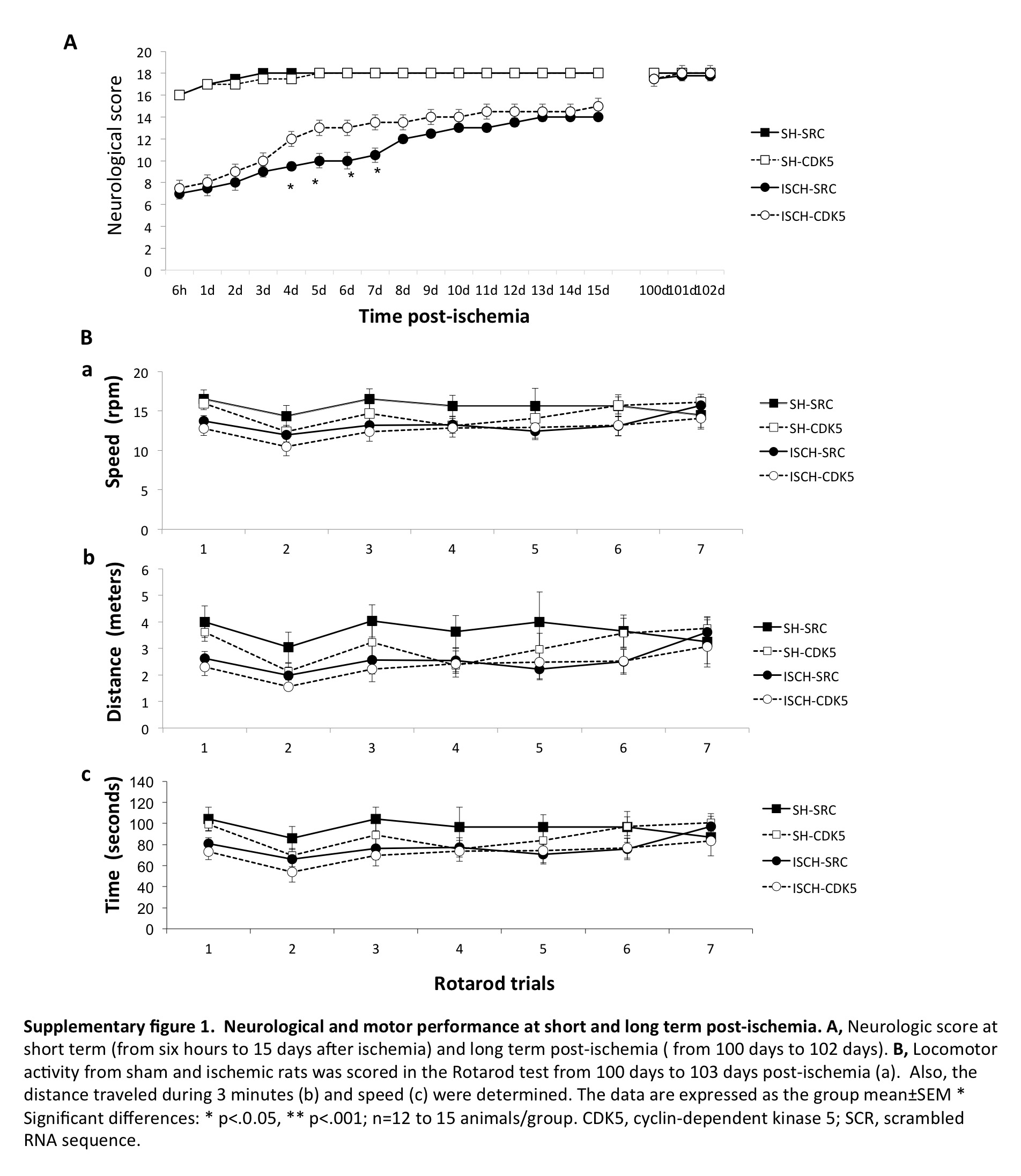
Neurological and motor deficits were not found four months post-ischemia
We evaluated the neurological scores at short- and long-term post-ischemia, as well as the rotarod test at the long-term time point (Supplementary Figure 1). Ischemic animals exhibited significantly lower neurological scores than the sham-operated animals during the first 15 days after surgery (p < 0.001; Supplementary Figure 1 A). During fourth to seventh day after ischemia, the CDK5miR-treated animals displayed neurological scores that were significantly higher than the SCRmiR-treated animals (p < 0.05; Supplementary Figure 1 A), which support our previous observations.11 However, we did not observe differences in the neurological scores (Supplementary Figure 1 A) or in the rotarod test (Supplementary Figure 1 B) between the groups at 100–102 days post-ischemia, indicating that untreated and treated ischemic rats had similar motor and sensory functions at long-term post-ischemia.
CDK5 down-regulation prevents reversal learning impairment at four months post-ischemia
The effects of CDK5 interference on the ischemia-induced impairment of hippocampus-dependent spatial navigation were examined using the Morris water maze. All groups were able to learn the hidden platform’s location during the six learning trials. Therefore, there were no differences between the groups in the learning test, indicating that neither the ischemia nor the treatment had an effect (Figure 2(A)). Forty-eight hours after the last training trial, the platform was removed from the maze. All groups displayed similar latencies to reach the original platform position (Figure 2(B)).
Figure 2.

CDK5miR prevents reversal learning deficits at four months post-ischemia. Spatial learning and memory function were assessed with the Morris water maze test. (A) Learning: average latency to find the hidden platform. (B) During the probe trial, the latency to reach the platform location was determined. (C) The reversal phase (reversal learning) was established, and the average latency to find the hidden platform (a), distance traveled to reach the platform (b), representative routes used to find the hidden platform in the last trial (c), and swimming speed (d) are shown. The data are expressed as the group mean ± SEM *Significant differences: p < 0.05; n = 12–15 animals/group. CDK5, cyclin-dependent kinase 5; SCR, scrambled RNA sequence.
Three additional complementary training trials were conducted with the hidden platform located in the quadrant opposite its original location. This test was conducted to evaluate the animals’ reversal learning abilities. In contrast to the CDK5miR-treated ischemic animals, the SCRmiR-treated ischemic rats displayed impaired performance on the last trial of reversal learning, taking a longer time (p < 0.05; Figure 2(C – a)) and traveling a greater distance (p < 0.05; Figure 2(C – b and c)) to find the new platform location, without significant differences in swimming speed (Figure 2(C – d)).
Reversal learning impairment was evaluated by analyzing the number of crossings over each quadrant during each reversal learning trial (Figure 3). We found that the SCRmiR ischemic animals exhibited fewer crossings over quadrant C (platform target) during the third trial (p < 0.05; Figure 3(B)) and instead presented more crossings over quadrants B and D (i.e. outside the platform target) (p < 0.05; Figure 3(B)), demonstrating that the ability of the SRC-miR-treated ischemic rats to find the platform in a new location (quadrant C) was impaired. However, they did not forget the platform’s first location (quadrant A). In contrast, the CDK5miR-treated ischemic animals persistently crossed quadrant C during the third trial (Figure 3(B); p < 0.05), suggesting that these animals were also able to learn the new location (quadrant C) without forgetting the first position learned (quadrant A). No significant differences among the groups were observed in the visible platform test (data not shown).
Figure 3.

CDK5miR facilitates the learning of a new platform location without forgetting a prior one. (A) The platform was located in quadrant A during learning and quadrant B during reversal learning and it was never located in the quadrants B and D. (B) A representative depiction of the route traveled during the reversal learning test is shown, and the number of crossings over each quadrant during each trial in the reversal learning period was determined. The data are expressed as the group mean ± SEM *Significant differences: p < 0.05; n = 12–15 animals/group. CDK5, cyclin-dependent kinase 5; SCR, scrambled RNA sequence.
CDK5 targeting prevents long-term post-ischemic neurodegeneration and regulates survival signaling
At four months post-ischemia, neural damage was evident in the CA1 region of the hippocampus (Figure 4). We found a significant reduction of neuronal marker (NeuN) IR at the level of −2.56 from bregma in the SCRmiR-treated animals (p < 0.05; Figure 4(A) and (B – a)). In contrast, the CDK5miR-treated animals exhibited NeuN IR equivalent to that of the sham control animals (p < 0.05; Figure 4(A) and (B – a)). This result was supported by the detection of hyperphosphorylated tau with AT8 (a phospho-specific PHF-tau antibody). In sections from −2.56 to −3.60 from bregma, AT8 IR was absent in the sham groups but was observed in the ischemic animals treated with SCRmiR (Figure 4(A)) (p < 0.05; Figure 4(B – b)). When we analyzed the ischemic animals treated with CDK5miR, the taupathology was reduced to the control values (p < 0.05; Figure 3(A) and (B – b)).
Figure 4.

CDK5 silencing reduces neurodegenerative hallmarks in CA1 at four months post-ischemia. (A) Representative immunoreactivity levels in the ipsilateral CA1 region of the hippocampus (bregma −2.56) at four months post-ischemia. The micrographs were obtained at 10 × and 40×. Scale bars=50 µm (10×) and 25 µm (40×). (B) Intensity of immunoreactivity for (a) NeuN, (b) AT-8 (c) MAP-2, (d) GFAP, and (e) OX42. These values were quantified at bregma levels of −2.56, −3.60, and −5.20. RU: relative units. *Significant differences, p ≤ 0.05; n = 4–6 animals/group. (C) Total protein levels of pAKT (Ser 473), AKT (a), and phosphorylated p38 (b) were determined by Western blotting. Densitometric quantification was determined relative to the loading control (tubulin) and normalized to the internal control (SCRmiR-treated sham rats). The values are the mean ± SEM. *Significant differences, p ≤ 0.05; n = 4–6 animals/group. CDK5, cyclin-dependent kinase 5; SCR, scrambled RNA sequence.
To visualize the dendrites of the hippocampal neurons, we used an antibody against microtubule-associated protein 2 (MAP2), which is specifically expressed in neuronal perikarya and dendrites. Consistent with the loss of neurons and the tauopathy, MAP2 IR in the anterior CA1 region was altered in the SCRmiR-treated animals (Figure 4(A)). The distribution of MAP2 IR in the CDK5miR-treated ischemic animals was similar to that in the sham animals (Figure 4(A)). We observed significant between-group differences in MAP2 IR in area CA1 in sections −5.20 from bregma (Figure 4(B – c)).
When we evaluated the astrocytic response, the data showed that the astrocytes in the SCRmiR-treated ischemic animals were less branched than those in the sham groups (Figure 4(A)). Although an increase in GFAP IR was observed in the CDK5miR-treated ischemic animals at bregma levels of −2.56 to −3.60 (p < 0.05; Figure 4(B – d)), the astrocytic morphology in these animals was similar to that in the control groups and corresponded to non-hypertrophic astrocytes with more branches (Figure 4(A)). We also found that the SCRmiR-treated ischemic animals presented a notable increase in microglial hyperreactivity (Figure 4(A)) at bregma levels of −2.56 to −5.20 (p < 0.05; Figure 4(B – e)). CDK5 silencing prevented the inflammatory response, as revealed by expression of the OX42-IR indicator, which was less intense in the CDK5miR-treated animals compared with the SCRmiR-treated ischemic animals (p < 0.05; Figure 4(A) and (B – e)) and was similar to that in the sham groups. The CDK5miR treatment itself did not produce hippocampal tissue alterations.
These histological results were supported by changes in the phosphorylation state of the survival markers AKT and p38. We found reduced AKT phosphorylation (Ser 473) (p < 0.05; Figure 4(C – a)) and significantly increased p38 phosphorylation (p < 0.05; Figure 4(C – b)) in the ischemic animals treated with SCRmiR. In contrast, the CDK5miR-treated ischemic animals exhibited a significant increase in AKT phosphorylation (Ser 473) (p < 0.05; Figure 4(C – a)) and a clear tendency toward a reduction of p38 phosphorylation (Figure 4(C – b)), suggesting that the CDK5miR treatment protected the ischemic tissue.
CDK5 silencing enriched BDNF and pCREB long after ischemia
Previous studies have shown that an increase in BDNF levels is necessary to achieve significant post-stroke functional recovery in rats.14 We have previously shown that CDK5 silencing increases BDNF levels and improves the recovery of cognitive function at one month post-ischemia.11 Consequently, we evaluated BDNF levels by ELISA at four months post-ischemia. Our data showed that the CDK5miR-treated ischemic rats displayed increased BDNF levels (p < 0.05; Figure 5(A)) and activation of plasticity-related pathways, as revealed by CREB phosphorylation (p-Ser 133; p < 0.05; Figure 5(B – c)). Additionally, we found that in hippocampal area CA1, the cells that were immunoreactive for BDNF were also NeuN-immunoreactive (Figure 5(C – a)). However, these cells were not GFAP-immunoreactive (Figure 5(C – b)). Overall, these results suggest that the CDK5miR treatment induced neuronal BDNF production.
Figure 5.

CDK5 silencing increases BDNF levels in neurons at four months post-ischemia. (A) BDNF levels were measured in the hippocampus at four months post-ischemia. *Significant differences, p ≤ 0.05; n = 4–6 animals/group. (B) Total protein levels of TRKB (a, b), pCREB (Ser 133) (c), and CREB (d) were determined by Western blotting. Densitometric quantification was determined relative to the loading control (tubulin) and normalized to the internal control (SCRmiR-treated sham rats). The values are the mean ± SEM. *Significant differences, p ≤ 0.05; n = 4–6 animals/group. (C) Immunofluorescence in the ipsilateral CA1 region of the hippocampus (bregma−2.56) at four months post-ischemia is shown for NeuN/BDNF (a) and GFAP/NeuN (b). The photomicrographs were obtained at 40 x. Scale bars=15 µm, n = 4–6 animals/group. CDK5, cyclin-dependent kinase 5; SCR, scrambled RNA sequence; GFP, green fluorescent protein.
The LTP deficits produced by excitotoxic exposure are rescued by roscovitine in a TRKB-dependent manner
In light of the significant BDNF production observed in the context of CDK5 RNAi-induced neuroprotection (see above and Figure 5), we used K252a, a compound that inhibits the tyrosine kinase activity of the TRKB receptor, in brain slices treated with glutamate ± roscovitine (a CDK5 pharmacological inhibitor) and determined its effects on a form of hippocampal synaptic plasticity (LTP) (Figure 6(A)).
Figure 6.

Inhibition of TRKB blocks the CDK5 inhibition-mediated recovery of LTP after excitotoxic glutamate stimulation. (A) Slices (400 µm) were pre-treated with glutamate (10 μM) for 30 min and then maintained in aCSF for 30 min. Subsequently, the slices were treated with roscovitine (20 μM) ± K252a (200 nM) for 15 min. (a) Schematic diagram of a horizontal hippocampal slice showing the arrangement of the recording and stimulating electrodes. (b) LTP was induced in the CA1 region by high-frequency stimulation (HFS, 1 train, 100 Hz, indicated by the arrow) of the Schaffer collateral–CA1 pathway. The data represent the mean ± SEM of the fEPSP slope at the indicated times, expressed as a percentage of the baseline value. (c) Representative traces of fEPSPs before and after HFS and the overlap between the experimental groups. LTP was calculated between 0 and 20 min after HFS stimulation (d) and between 40 and 60 min after HFS stimulation (d). (n = 6–7 slices). The error bars indicate the SEM. (e) fEPSP, field excitatory postsynaptic potential; aCSF, artificial cerebrospinal fluid. (B) Total protein levels of pERK1/2, ERK1/2 (a, b), pCAMKII (T286) (c), and NR2B (d) in brains treated with SCRmiR or CDK5miR were determined by Western blotting. Densitometric quantification was expressed relative to the loading control (tubulin) and normalized to the internal control (SCRmiR-treated sham rats). The values are the mean ± SEM. *Significant differences, p ≤ 0.05; n = 4–6 animals/group. CDK5, cyclin-dependent kinase 5; SCR, scrambled RNA sequence.
A growing body of evidence suggests that perturbations in systems that utilize the excitatory amino acid l-glutamate (l-Glu) may underlie several of the pathogenic mechanisms of hypoxia-ischemia. Transient exposure of hippocampal slices to extracellular glutamate (μM concentrations) has been shown to produce spontaneous epileptiform discharges.18 Glutamate is thus considered a long-lasting excitotoxic stimulus that partially replicates the increased neuronal excitability that is observed post-stroke. We therefore evaluated whether extracellular glutamate also affected synaptic plasticity. In slices of the ventral hippocampus that were pre-treated for 30 min with 10 μM glutamate and recorded in normal aCSF, hippocampal LTP was impaired during the last 20 min of recording as compared to vehicle-treated slices (114 ± 3.87% vs 224 ± 3.77%; n = 7; p < 0.05) (Figure 6(A – b, c, and e)). Subsequently, we tested whether the pharmacological inhibitor of CDK5 was able to restore LTP deficits induced by glutamate exposure. Pre-treatment with roscovitine (20 μM) for 15 min increased the LTP in the first 20 min (235.53 ± 1.18% vs 288.38 ± 2.23%; n = 6; p < 0.05) until the last 20 min of recording (288.39 ± 2.71%; vs 224 ± 3.7%; n = 6; p < 0.05) (Figure 6(A – b to e)). Glutamate-induced LTP deficits were rescued when 20 μM roscovitine was applied 30 min after the excitotoxic stimulus for 15 min, which resulted in potentiation of 287.47% ± 2.07% vs 236.37 ± 2.97% at 20 min (Figure 6(A – d)) and 234.13 ± 2.01% vs 114.12 ± 4.01 at 60 min post-HFS (Figure 6(A – b, c, and e)).
The application of roscovitine and K252a (200 nM) to glutamate-pretreated slices did not recover LTP deficits, as demonstrated by comparing LTP during the last 20 min of recording between glutamate/roscovitine/K252a and glutamate-/roscovitine-treated slices (121.96 ± 1.28% vs 234.13 ± 2.01%; n = 7; p < 0.05) (Figure 6(A – e)). This result suggests that the effect of roscovitine on LTP recovery requires normal TRKB signaling. Additionally, we evaluated candidate proteins related to LTP14,19 (Figure 6(B)) as molecular correlates of the recovery of synaptic plasticity in brain lysates from the CDK5 RNAi-treated ischemic rats. We found a significant increase in CAMKII (T286) phosphorylation (p < 0.05; Figure 6(B – c)) as well as in GluN2B levels (p < 0.05; Figure 6(B – d)) in the CDK5miR-treated ischemic rats, although markers of other pathways involved in long-term synaptic plasticity, such as pERK, were not modified by the treatments (Figure 6(B – a and b)).
Discussion
One of the major difficulties shared by the different strategies to provide neuroprotection against cerebral ischemia is the limited therapeutic window and the poor long-term effectiveness post-ischemia; to date, such strategies have failed to prevent the neurological sequelae and general cognitive problems that appear several months or even years after ischemia. Here, we showed that silencing CDK5 during the ischemia/reperfusion phase prevented reversal learning impairment at four months post-ischemia as well as prevented the neurodegenerative hallmarks of tauopathy and neuronal population loss. Furthermore, we showed that these effects were associated with increased BDNF expression and the activation of survival- and plasticity-related pathways.
CDK5 plays a critical role in glutamate excitotoxicity after ischemia/reperfusion.20,21 Our results showed that the levels of CDK5 as well as its neurodegeneration-associated target proteins (p25 and calpain)22 were increased in hippocampal area CA1 long after ischemia. These effects were also associated with tauopathy in ischemic animals, likely because CDK5 is a major kinase that contributes to tau pathology in cerebral ischemia.23 Interestingly, CDK5 and p25 protein levels as well as CDK5 and calpain activity were reduced by CDK5 silencing at four months post-ischemia. These biochemical findings were associated with a reduction in the histopathological hallmarks of neurodegeneration, especially tauopathy, and the recovery of cognitive dysfunction.
The behavioral changes that take place following cerebral ischemia include cognitive impairments that can be detected by the “Morris” water maze test.24 However, most of the studies have been developed no more than a month post-injury, making novel the analyses of learning, memory and reversal learning at four months post-ischemia in the current study. In our study, the ischemic animals displayed no alterations in performance on the learning and memory tasks but showed deficits in reversal learning. This test reveals if animals are able to extinguish the memory of the initial platform position and learn a new location, which requires reorganization of hippocampal-to-cortical circuits,25 the consolidation of previous learning, and the establishment of new connections to integrate the newly learned information. Interestingly, our findings showed that the CDK5 RNAi-treated ischemic animals displayed better reversal learning than the untreated rats, suggesting that CDK5 reduction in the hippocampus might also might protect the neuronal circuit to the cerebral cortex, similar to what we previously observed at one month post-injury.11 These data are supported by the observation of enhanced re-learning skills in CDK5 conditional knockout mice26 and in other neurodegenerative disease animal models,27 which also showed a reduction of tauopathy, a hallmark of cognitive disorders.15
At four months post-ischemia, the ischemic animals presented tauopathy as well as neuronal and dendritic loss, an increase in the expression of inflammatory indicators, such as OX42 and alterations in astrocyte morphology. Large mineralized calcium deposits, which are associated with neuroinflammatory and astroglial reactions, have been found in hippocampal area CA1 and the thalamus 250 days post-lesion.6 This phenomenon is called “tertiary cell death” and can appear between 90 days and 125 days after ischemia.28 These calcium deposits have been attributed to delayed neuronal death, probably as a consequence of calcium overload.29,30 CA1 neurons that are born after cerebral ischemia have difficulty maintaining calcium homeostasis, either because of deficiencies in intracellular homeostatic mechanisms or because of poor regional calcium regulation in the extracellular space.29 As a result, these cells may experience a continual excess of intracellular calcium, which is stored as calcium hydroxyl-apatite.29 Interestingly, our data showed a reduction of calpain activation and of the p35/p25 ratio, suggesting control over the Ca2+ levels in the CDK5 knockdown ischemic environment. This hypothesis was supported by an increase in AKT phosphorylation (Ser 473) and the prevention of p38 up-regulation, which is predominantly observed in a survival context.31
However, we also unexpectedly found that the reduction in CDK5 correlated with increased GFAP IR at four months post-ischemia. We found that the astrocytes in the CDK5 RNAi-treated ischemic animals displayed a more healthy morphology than those in the ischemic animal. The astrocytes in the CDK5 RNAi-treated ischemic animals were also more stellate than those in the sham animals. Some studies have demonstrated a protective role of astrocytes in in vitro ischemia models.32 Additionally, we previously observed that astrocytes provide neuroprotection under conditions of CDK5 knockdown33 and that this neuroprotection was associated with astrocytic morphology changes, such as lamellipodia protrusions, increased cell area, and stellation, as well as with Rac1 activation and BDNF release.33 In this study, however, we found that astrocytes were not the primary producers of BDNF, at least in the transduced CA1 zone.
In the present study, we found that at four months post-treatment in the ischemic animals, BDNF was mainly located in neurons in hippocampal area CA1. We also observed increased CREB phosphorylation at Ser 133, suggesting the induction of neuronal plasticity. To support these findings, we evaluated LTP, which is a form of synaptic plasticity, in the hippocampus.34 Pharmacological inhibition of CDK5 with roscovitine rescued the LTP deficits induced by glutamate exposure. This recovery was blocked by pharmacological inhibition of TRKB with K252a, suggesting an important role of BDNF/TRKB signaling in the neuroprotection and plasticity mediated by CDK5 down-regulation within an excitotoxic context. Several lines of evidence have shown that BDNF plays a crucial role in stimulating LTP, a synaptic model of memory storage. The application of exogenous BDNF can trigger a long-lasting increase in synaptic efficacy (BDNF-LTP) in neurons in the hippocampus, dentate gyrus, visual cortex, and insular cortex.35 BDNF activates Ras, which triggers mitogen-activated protein kinase (MAPK) activity and is part of the CREB pathway; this pathway can lead to synaptic restructuring in support of LTP.36,37 We previously reported that CDK5 silencing increased the phosphorylation of ERK1, CREB, and BDNF one month after ischemia; however, we did not detect ERK activation at four months, although the CDK5 RNAi-mediated upregulation of pCREB was maintained at this timepoint in the sham and ischemic rats.
Furthermore, other proteins involved in synaptic plasticity, such as p-CaMKII and the NR2 subunit of the GluN2B NMDA receptor, which are also associated with the induction of LTP,14,19 were upregulated under the neuroprotective conditions of CDK5 RNAi, during which BDNF also remained increased. Previous work has shown that BDNF activates CaMKII and that activated CaMKII binds GluN2B and rapidly increases its lateral diffusion to the post-synaptic density, inducing LTP.38 Other reports have indicated that CAMKII activation is upstream of the activity of Rho GTPases, which facilitate spinaptogenesis and neurotransmission.39 Furthermore, some GTPase-activating proteins (GAPs) are blocked by CDK5 overactivation.40 Complementarily, in previous studies, we found that CDK5 down-regulation induced Rac activation, and this event was associated with neuroprotection and better cognitive performance.27
Based on our present results, CDK5 silencing could be a therapeutic strategy for reducing long-term post-ischemic neuropathological hallmarks and cognitive deficits. This type of CDK5 silencing gene therapy could also become an important tool to prevent relapse in successive ischemia episodes as well as cognitive sequelae that are not detected early but that can have devastating long-term consequences.
Supplementary Material
Acknowledgements
We would like to thank Dr Beverly Davidson and Dr Maria Scheel at the Viral Vector Core and Davidson Laboratory, University of Iowa, USA for their expert viral vector advice.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: The research reported in this publication was supported by Colciencias Projects #111551928905 and #111554531400 (GPC-G).
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Authors’ contributions
JAGV designed the study, did the experiments and wrote the paper. HM coordinated and advised the LTP registers. He also drafted and edited the manuscript. GPCG designed and directed the study. She also drafted and edited the manuscript. All authors read and approved the final manuscript.
Supplementary material
Supplementary material for this paper can be found at http://jcbfm.sagepub.com/content/by/supplemental-data
References
- 1.Moskowitz MA, Lo EH, Iadecola C. The science of stroke: mechanisms in search of treatments. Neuron 2010; 67: 181–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Desmond DW, Moroney JT, Sano M, Stern Y. Mortality in patients with dementia after ischemic stroke. Neurology 2002; 59: 537–543. [DOI] [PubMed] [Google Scholar]
- 3.Tatemichi TK, Desmond DW, Stern Y, et al. Cognitive impairment after stroke: frequency, patterns, and relationship to functional abilities. J Neurol Neurosurg Psychiatry 1994; 57: 202–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bowler JV, Hadar U, Wade JP. Cognition in stroke. Acta Neurol Scand 1994; 90: 424–429. [DOI] [PubMed] [Google Scholar]
- 5.Glosser G, Goodglass H. Disorders in executive control functions among aphasic and other brain-damaged patients. J Clin Exp Neuropsychol 1990; 12: 485–501. [DOI] [PubMed] [Google Scholar]
- 6.Bueters T, von Euler M, Bendel O, et al. Degeneration of newly formed CA1 neurons following global ischemia in the rat. Exp Neurol 2008; 209: 114–124. [DOI] [PubMed] [Google Scholar]
- 7.Tatemichi TK, Desmond DW, Mayeux R, et al. Dementia after stroke: baseline frequency, risks, and clinical features in a hospitalized cohort. Neurology 1992; 42: 1185–1193. [DOI] [PubMed] [Google Scholar]
- 8.Loeb C, Gandolfo C, Croce R, et al. Dementia associated with lacunar infarction. Stroke 1992; 23: 1225–1229. [DOI] [PubMed] [Google Scholar]
- 9.Tatemichi TK, Paik M, Bagiella E, et al. Dementia after stroke is a predictor of long-term survival. Stroke 1994; 25: 1915–1919. [DOI] [PubMed] [Google Scholar]
- 10.Slevin M, Krupinski J. Cyclin-dependent kinase-5 targeting for ischaemic stroke. Curr Opin Pharmacol 2009; 9: 119–124. [DOI] [PubMed] [Google Scholar]
- 11.Gutierrez-Vargas JA, Munera A, Cardona-Gomez GP. CDK5 knockdown prevents hippocampal degeneration and cognitive dysfunction produced by cerebral ischemia. J Cereb Blood Flow Metab 2015; 35: 1937–1949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Barnett DG, Bibb JA. The role of Cdk5 in cognition and neuropsychiatric and neurological pathology. Brain Res Bull 2011; 85: 9–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Camins A, Verdaguer E, Folch J, et al. The role of CDK5/P25 formation/inhibition in neurodegeneration. Drug News Perspect 2006; 19: 453–460. [DOI] [PubMed] [Google Scholar]
- 14.Gutierrez-Vargas JA, Munoz-Manco JI, Garcia-Segura LM, et al. GluN2B N-methyl-D-aspartic acid receptor subunit mediates atorvastatin-Induced neuroprotection after focal cerebral ischemia. J Neurosci Res 2014; 92: 1529–1548. [DOI] [PubMed] [Google Scholar]
- 15.Piedrahita D, Hernandez I, Lopez-Tobon A, et al. Silencing of CDK5 reduces neurofibrillary tangles in transgenic alzheimer's mice. J Neurosci 2010; 30: 13966–13976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Morris R. Developments of a water-maze procedure for studying spatial learning in the rat. J Neurosci Meth 1984; 11: 47–60. [DOI] [PubMed] [Google Scholar]
- 17.Cepeda-Prado E, Popp S, Khan U, et al. R6/2 Huntington's disease mice develop early and progressive abnormal brain metabolism and seizures. J Neurosci 2012; 32: 6456–6467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wong M, Guo D. Dendritic spine pathology in epilepsy: cause or consequence? Neuroscience 2013; 251: 141–150. [DOI] [PubMed] [Google Scholar]
- 19.Wang N, Chen L, Cheng N, et al. Active calcium/calmodulin-dependent protein kinase II (CaMKII) regulates NMDA receptor mediated postischemic long-term potentiation (i-LTP) by promoting the interaction between CaMKII and NMDA receptors in ischemia. Neural Plast 2014; 2014: 827161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Meyer DA, Torres-Altoro MI, Tan Z, et al. Ischemic stroke injury is mediated by aberrant Cdk5. J Neurosci 2014; 34: 8259–8267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Menn B, Bach S, Blevins TL, et al. Delayed treatment with systemic (S)-roscovitine provides neuroprotection and inhibits in vivo CDK5 activity increase in animal stroke models. PloS ONE 2010; 5: e12117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ahlijanian MK, Barrezueta NX, Williams RD, et al. Hyperphosphorylated tau and neurofilament and cytoskeletal disruptions in mice overexpressing human p25, an activator of cdk5. Proc Natl Acad Sci U S A 2000; 97: 2910–2915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wen Y, Yang S, Liu R, et al. Transient cerebral ischemia induces site-specific hyperphosphorylation of tau protein. Brain Res 2004; 1022: 30–38. [DOI] [PubMed] [Google Scholar]
- 24.Nunn JA, LePeillet E, Netto CA, et al. Global ischaemia: hippocampal pathology and spatial deficits in the water maze. Behav Brain Res 1994; 62: 41–54. [DOI] [PubMed] [Google Scholar]
- 25.McKenzie S, Eichenbaum H. Consolidation and reconsolidation: two lives of memories? Neuron 2011; 71: 224–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hawasli AH, Benavides DR, Nguyen C, et al. Cyclin-dependent kinase 5 governs learning and synaptic plasticity via control of NMDAR degradation. Nature Neurosci 2007; 10: 880–886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Posada-Duque RA, Lopez-Tobon A, Piedrahita D, et al. p35 and Rac1 underlie the neuroprotection and cognitive improvement induced by CDK5 silencing. J Neurochem 2015; 134: 354–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.von Euler M, Bendel O, Bueters T, et al. Profound but transient deficits in learning and memory after global ischemia using a novel water maze test. Behav Brain Res 2006; 166: 204–210. [DOI] [PubMed] [Google Scholar]
- 29.Herrmann G, Stunitz H, Nitsch C. Composition of ibotenic acid-induced calcifications in rat substantia nigra. Brain Res 1998; 786: 205–214. [DOI] [PubMed] [Google Scholar]
- 30.Kato H, Araki T, Itoyama Y, et al. Calcium deposits in the thalamus following repeated cerebral ischemia and long-term survival in the gerbil. Brain Res Bull 1995; 38: 25–30. [DOI] [PubMed] [Google Scholar]
- 31.Lai TW, Zhang S, Wang YT. Excitotoxicity and stroke: identifying novel targets for neuroprotection. Prog Neurobiol 2014; 115: 157–188. [DOI] [PubMed] [Google Scholar]
- 32.Shinotsuka T, Yasui M, Nuriya M. Astrocytic gap junctional networks suppress cellular damage in an in vitro model of ischemia. Biochem Biophys Res Commun 2014; 444: 171–176. [DOI] [PubMed] [Google Scholar]
- 33.Posada-Duque RA, Palacio-Castaneda V, Cardona-Gomez GP. CDK5 knockdown in astrocytes provide neuroprotection as a trophic source via Rac1. Mol Cell Neurosci 2015; 68: 151–166. [DOI] [PubMed] [Google Scholar]
- 34.Larkman AU, Jack JJ. Synaptic plasticity: hippocampal LTP. Curr Opin Neurobiol 1995; 5: 324–334. [DOI] [PubMed] [Google Scholar]
- 35.Escobar ML, Figueroa-Guzman Y, Gomez-Palacio-Schjetnan A. In vivo insular cortex LTP induced by brain-derived neurotrophic factor. Brain Res 2003; 991: 274–279. [DOI] [PubMed] [Google Scholar]
- 36.English JD, Sweatt JD. Activation of p42 mitogen-activated protein kinase in hippocampal long term potentiation. J Biol Chem 1996; 271: 24329–24332. [DOI] [PubMed] [Google Scholar]
- 37.Wang J, Chen YB, Zhu XN, et al. Activation of p42/44 mitogen-activated protein kinase pathway in long-term potentiation induced by nicotine in hippocampal CA1 region in rats. Acta Pharmacol Sin 2001; 22: 685–690. [PubMed] [Google Scholar]
- 38.Dupuis JP, Ladepeche L, Seth H, et al. Surface dynamics of GluN2B-NMDA receptors controls plasticity of maturing glutamate synapses. EMBO J 2014; 33: 842–861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Murakoshi H, Wang H, Yasuda R. Local, persistent activation of Rho GTPases during plasticity of single dendritic spines. Nature 2011; 472: 100–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Walkup WGt, Washburn L, Sweredoski MJ, et al. Phosphorylation of synaptic GTPase-activating protein (synGAP) by Ca2+/calmodulin-dependent protein kinase II (CaMKII) and cyclin-dependent kinase 5 (CDK5) alters the ratio of its GAP activity toward Ras and Rap GTPases. J Biol Chem 2015; 290: 4908–4927. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials