

Abstract
Metabotropic glutamate receptor type 1 (mGluR1) is related with various neurological and psychiatric diseases, such as anxiety, depression, epilepsy, Parkinson’s disease, and neuropathic pain. Hence, mGluR1 is an important target for drug development and imaging. We synthesized [18F]cEFQ (3-ethyl-2-[18F]fluoroquinolin-6-yl cis-(4-methoxycyclohexyl)methanone) as a PET tracer for selective mGluR1 imaging and evaluated its properties in rodents. A chloroquinoline precursor was labeled by a nucleophilic substitution reaction, and the resulting [18F]cEFQ was obtained with high radiochemical purity (>99%) and specific activity (63-246 GBq/µmol). The log D value was 3.24, and the initial brain uptake at 10 min was over 4% of injected dose per gram in BALB/c mice. According to PET/CT and autoradiography in SD rats, [18F]cEFQ showed wide distribution in the whole brain and the highest uptake in the cerebellum. Pre-treatment with unlabeled cEFQ or the mGluR1-specific antagonist JNJ16259685 blocked the uptake of [18F]cEFQ. However, the uptake was not blocked by pre-treatment with the mGluR5-specific antagonist ABP688. The trans isomer [18F]tEFQ did not show high uptake in the mGluR1-rich region. [18F]cEFQ was straightforwardly prepared using a chloro-derivative precursor. Its feasibility as a specific and selective PET agent for imaging mGluR1 was proved by in vitro and in vivo experiments using rodents.
Keywords: Autoradiography, metabotropic glutamate receptor subtype 1 (mGluR1), positron emission tomography (PET), [18F]cEFQ
Introduction
Glutamic acid is an abundant and one of the most important excitatory neurotransmitters in the central nervous system (CNS) of mammals. It is stored in presynaptic vesicles and is released by calcium-dependent exocytosis. After being released from vesicles, it acts via membrane-bound glutamate receptors.
Glutamate receptors are categorized as ionotropic and metabotropic by their affinity to various ligands. Ionotropic glutamate receptors (iGluR) comprise N-methyl-D-aspartate (NMDA), α-amino-3-hydroxy-5-methyl-4-isoxazole propionate (AMPA), and kainate (KA) receptors.1,2 Metabotropic glutamate receptors (mGluRs) comprise G-protein-coupled receptors (GPCRs) that are classified into eight subtypes (mGluR1-8) based on their pharmacology, signal transduction mechanism, and sequence homology.3
Group I mGluRs include mGluR1 and mGluR5, which are coupled to Gq/11. Gq/11 stimulates phospholipase C, an enzyme that hydrolyzes phosphatidylinositol 4,5-bisphosphate to produce inositol triphosphate (IP3) and diacylglycerol. IP3 then binds to the IP3 receptor on the endoplasmic reticulum, triggering the release of Ca2+ from intracellular stores. The released Ca2+ and diacylglycerol activate protein kinase C.1–3
mGluR1 is expressed in the postsynaptic neurons of the CNS in mammals, especially in the cerebellum. mGluR1 is also found in the hippocampus, thalamus, and striatum but at lower levels than in the cerebellum.4 It is related to various neurological and psychiatric disorders, such as anxiety,5,6 depression,7–9 epilepsy,10 Parkinson’s disease,11–13 and neuropathic pain.14–16 Therefore, development of mGluR1-targeting drugs has been an attractive subject for decades.17
Quinoline derivatives that function as noncompetitive mGluR1 antagonists have been reported (Figure 1(a)).18 JNJ16259685 is a potent and selective mGluR1 antagonist that has been used as a standard for measuring the specific binding of new mGluR1 antagonist candidates.19 [11C]JNJ16567083 was the first tracer labeled with a positron emitter for use in mGluR1 imaging, and it showed high uptake in the rat cerebellum—the mGluR1-abundant region.20,21 However, no further evaluations or clinical investigations have been reported; moreover, 11C-labeled agents have some disadvantages for practical use due to a short half-life.
Figure 1.

Structures of quinoline derivatives for studying mGluR1 (a) and radiosynthetic scheme of [18F]cEFQ and [18F]tEFQ (b).
A fluorine-containing quinoline derivative, 3-ethyl-2-fluoroquinolin-6-yl cis-(4-methoxycyclohexyl)methanone (cEFQ), that acts as a noncompetitive mGluR1 antagonist and has high binding affinity to rat and human mGluR1 has been developed.18 A 18F-labeled compound, [18F]cEFQ, was developed as a PET agent for mGluR1 imaging (Figure 1(a)), and in vitro and in vivo studies using rats and baboons have demonstrated specific binding to mGluR1.21 However, brain images using [18F]cEFQ and analyses of its radioactive metabolites have not been reported. Furthermore, no study using a purified trans isomer, [18F]tEFQ, has been reported. Thus, in the present research, we aimed to prepare both cis and trans isomers ([18F]cEFQ and [18F]tEFQ) and examined their radiochemical properties, metabolites, and in vivo behavior using rats.
Materials and methods
General
The following ARRIVE guidelines were followed in the preparation of this manuscript: Title, Abstract, Background, Objectives, and Ethical statement; Study Design; Experimental procedures; Experimental animals, housing, and husbandry; Experimental outcomes; Statistical methods, Numbers analyzed; Interpretation and scientific implications; and Generalizability/translation and Funding. All animal studies were conducted with the approval of the Institutional Animal Care and Use Committee of Clinical Research Institute, Seoul National University Hospital. The facility was accredited by the Association for the Assessment and Accreditation of Laboratory Animal Care International. In addition, National Research Council guidelines for the care and use of laboratory animals (revised in 1996) were observed throughout the studies.
Unlabeled cEFQ and the precursor for 18F-labeling were prepared according to a previously reported procedure.18 ABP688 was synthesized as described previously.22 The other reagents and solvents were purchased from Sigma-Aldrich Korea Ltd. (Yongin, Korea), Fluka (Buchs, Switzerland), or TCI (Tokyo, Japan) and were used without further purification. All non-aqueous reactions were performed under an inert atmosphere of dry argon. Reactions were monitored by TLC analysis using glass-backed silica gel 60 F254 thin-layer plates (Merck, Darmstadt, Germany). The spots were detected under UV light (254 nm and 356 nm). Flash column chromatography for purification was carried out on silica gel 60 (230–400 mesh), and all chromatographic solvents used were HPLC grade and were purchased from Fisher Scientific (Fair Lawn, NJ, USA ). 1H and 13C nuclear magnetic resonance (NMR) spectra were recorded in δ units relative to a tetramethylsilane (TMS) as an internal reference using a 300 or 400 MHz NMR instrument (JEOL Ltd., Tokyo, Japan).
[18F]Fluoride was produced by the 18O(p,n)18F reaction using 18O-enriched (98%) water and a 16.5-MeV proton beam generated by a PETtrace 10 cyclotron (GE Healthcare, Uppsala, Sweden). A high-performance liquid chromatography (HPLC) system (Gilson Inc., Middleton, WI, USA ) equipped with a Gabi Star flow-through gamma radioactivity detector (Raytest GmbH, Straubenhardt, Germany) was used to confirm and purify the radiolabeled compounds. The radiochemical purity and specific activity were checked by analytical HPLC and radio-TLC scanning (AR-2000; Bioscan, Washington, DC, USA) of aluminum-backed silica gel 60 F254 TLC plates (Merck Co., Darmstadt, Germany). A dose calibrator (Atomlab 100; Biodex Medical Systems, Inc., New York, NY, USA) was used for measuring the radioactivity.
Two-tailed analysis of Student’s t-test was used to confirm the statistical significance of two group means.
Radiosynthesis
The cyclotron-produced [18F]fluoride was captured on a QMA light Sep-Pak cartridge (Waters) pre-washed with 5 mL of 0.5 M potassium bicarbonate and 10 mL of deionized water. [18F]Fluoride on the cartridge was eluted with a 1 mL solution of Kryptofix 2.2.2 (K222) (18.1 mg, 0.048 mmol)/K2CO3 (2.9 mg, 0.021 mmol) in 50% MeCN in H2O. Azeotropic evaporation was performed twice at 100℃ by purging with nitrogen gas after adding 1 mL of MeCN.
To the dried [18F]fluoride, 1.5 mg (4.5 µmol) of precursor in DMSO (0.5 mL) was added and heated at 100℃ for 5 min. The reaction mixture was loaded to a preparative HPLC (Xterra® preparative column RP18, 10 µm, 10 × 250 mm, Waters; 60% MeCN in H2O, isocratic; flow rate, 5 mL/min). The fractions of [18F]tEFQ and [18F]cEFQ appearing at 16.5 and 18.0 min, respectively, were collected separately (Figure 1(b)).
A pooled [18F]cEFQ or [18F]tEFQ fraction was diluted with 100 mL of water and passed through a Sep-Pak C18 light cartridge pre-washed with 5 mL of ethanol and 10 mL of deionized water successively. The adsorbed radioactive compounds were eluted with 0.8 mL of ethanol, passed through a sterile Millex®-FG filter (Merck Millipore, Billerica, MA, USA), and collected in a sterile vial. The final products were diluted with saline for the subsequent in vivo studies.
Radiochemical purities were checked using aluminum-backed silica gel 60 F254 TLC plates (hexane/EtOAc, 75:25) and analytical HPLC (Xterra® analytical column RP18, 3.5 µm, 4.6 × 100 mm, Waters; 45–50% MeCN gradient in H2O from 0 to 15 min; flow rate, 1 mL/min). Specific activities were also determined using the same analytical HPLC system.
Automated synthesis of [18F]cEFQ was performed by using 3 mg of precursor in 0.7 mL of DMSO in a TRACERLab FX-FN module (GE medical Systems, Germany). Reaction temperature and time were 110℃ and 6 min, respectively. All other conditions were the same as in the manual synthesis.
Distribution coefficient (log D)
The distribution coefficient log D value was measured according to a slight modification of a previous reported method.23 Briefly, 10 µL of ethanol solution containing [18F]cEFQ (radiochemical purity: 100%) was mixed with a mixture of 1-octanol (3.0 g) and 0.1 M phosphate buffer (3.0 g; pH 7.4), vortexed for 3 min at room temperature, and centrifuged at 3000 r/min for 5 min. The samples from the organic and aqueous phases were weighed and counted using a gamma scintillation counter (DREAM r-10, Shinjin Medics Inc., Korea). An aliquot of the 0.5 g organic layer was re-partitioned until log D values were consistent. The log D was calculated as the ratio of radioactivity in the organic phase and that in the aqueous phase.
Stability test
The in vitro stabilities of [18F]cEFQ in the prepared medium at room temperature and in human serum at 37℃ were measured. The prepared [18F]cEFQ was stored at room temperature for 4 h and was analyzed by instant TLC (ITLC) and analytical HPLC. For the stability test in human serum, the prepared [18F]cEFQ (25 µL) was mixed with human serum (325 µL) and incubated at 37℃ for 4 h. Absolute ethanol (775 µL) was added to the mixture to precipitate serum proteins followed by centrifugation (3000 r/min) for 5 min. The supernatant was analyzed using ITLC and analytical HPLC.
Biodistribution study in mice
Male BALB/c mice (5 weeks old, n = 4 per group, 23 ± 1 g) were injected with 148 kBq (187 pg) of [18F]cEFQ in 0.1 mL of 10% ethanol through the tail vein. The injected mice were numbered randomly for blind test, and were sacrificed by decapitation at 10, 60, or 120 min after injection. Blood and other organs were rapidly separated, weighed, and counted using a gamma scintillation counter. Results are expressed as percentage of injected dose per gram of tissue (% ID/g). Values are expressed as the mean ± SD.
Analysis of metabolites in mice
After intravenous injection of [18F]cEFQ (72.5 MBq, 93.0 ng), male BALB/c mice (5 weeks old) were sacrificed by decapitation at 5, 15, or 30 min. Urine, blood, and the whole brain were removed and stored on ice. The urine sample was diluted with the same volume of distilled water and filtered through a 0.22 µm filter. The blood sample was centrifuged at 3300 r/min for 10 min at 4℃ to separate the plasma. The plasma sample was mixed with 0.4 mL of MeCN, and the mixture was then vortexed for 15 s and centrifuged at 3300 r/min for 10 min to precipitate protein. The supernatant was collected. The brain sample in ice-cooled MeCN (1.0 mL) was homogenized for 1 min using a Polytron homogenizer (Kinematica, Westbury, Canada). The homogenate was centrifuged at 3300 r/min for 10 min at 4℃, and the supernatant was collected. The brain precipitate was re-homogenenized after adding 1.0 mL of water, and the mixture was centrifuged at 3300 r/min for 10 min at 4℃. The supernatants were collected and centrifuged again at 3300 r/min for 10 min at 4℃ to remove the precipitate.
Samples from the urine, plasma, and brain homogenates were injected into the analytical HPLC system (Xterra® analytical column RP18, 3.5 µm, 4.6 × 100 mm, Waters; 0–45% MeCN gradient in H2O from 0 to 6 min; 45% MeCN in H2O, isocratic from 6 to 21 min; 100% MeCN, isocratic from 21 to 26 min; flow rate, 1 mL/min). Each 0.33-min eluent fraction was collected, and the radioactivity was measured using a gamma scintillation counter.
Animal PET
Male Sprague-Dawley rats (12 weeks old) anesthetized with 2% (v/v) isoflurane at 1 L/min oxygen flow were positioned in the imaging chamber and were intravenously administrated with 92.5 MBq of [18F]cEFQ (118.6 ng) or [18F]tEFQ (312.9 ng) prepared as described above. PET studies were performed with a dedicated small animal PET/CT scanner (eXplore VISTA, GE Healthcare, USA), and list-mode data were acquired for 60 min with an energy window of 400–700 keV. These list mode data were framed into a dynamic sequence of 10 × 30 s, 5 × 60 s, 4 × 300 s, and 3 × 600 s frames. These images were reconstructed to temporally framed sinograms using Fourier rebinning and an ordered subsets expectation maximization (OSEM) reconstruction algorithm without attenuation correction.24 All images were analyzed using parametric mapping software (SPM2, University College of London, London, UK).
To confirm the selectivity and specificity of [18F]cEFQ for mGluR1, blocking studies were performed. Rats were pre-administered through the tail vein with 3 mg/kg of cold cEFQ, the mGluR1 antagonist JNJ16259685, or the mGluR5 antagonist ABP688 10 min before the administration of [18F]cEFQ. After administration of [18F]cEFQ, PET images were obtained as described above.
Ex vivo autoradiography
Ex vivo autoradiography was performed to obtain higher resolution brain images than with animal PET. The prepared [18F]cEFQ (148 MBq, 190 ng, 0.5 mL) was intravenously injected into the tail vein of male Sprague-Dawley rats (12 weeks old), and the rats were sacrificed 10 min post-injection. The rats’ brains were dissected and rapidly frozen at −80℃. Sagittal brain sections of 20 µm thickness were cut using a cryostat microtome (Leica CM 1800, Leica Inc., Germany) and thaw-mounted on glass slides. The prepared brain sections were exposed to imaging plates, and autoradiograms were then obtained using a BAS-2000 system (FLA-2000, FUJIFILM Inc., Japan).25
Results
Radiochemistry
The radiolabeling was performed by the nucleophilic aromatic substitution reaction using a cis–trans mixture of precursor (Figure 1(b)). We found that both [18F]cEFQ and [18F]tEFQ were produced even if only a purified cis- or trans-precursor was used (Figure 1(b)). Thus, it was not necessary to use a purified cis-form precursor, which might be more cost-effective.
The optimum precursor quantity for rapid reaction was found to be 1.5 mg. When 1 mg of precursor was used for labeling, it took over 10 min to obtain enough labeled compounds at 100℃. However, no significant difference was found in the labeling time when using 1.5 mg and 2.0 mg of precursor. Labeled products appeared very rapidly after adding precursor in both cases. When the reaction temperature was 120℃, the labeling was very fast, but the product decomposed after 3 min of reaction. At 100℃, the decomposition rate was slow, and the maximum labeling efficiency was found after 5 min of reaction. From these results, we optimized the reaction temperature and time to 100℃ and 5 min, respectively.
The reaction mixture was purified by semi-preparative HPLC, and the retention times of [18F]tEFQ and [18F]cEFQ were 16.5 and 18.0 min, respectively, that could be separated from the precursors. The radiolabeling efficiencies of [18F]cEFQ and [18F]tEFQ were 29% and 49%, respectively. The purified radioactive compounds were diluted with normal saline for intravenous injection. The total synthesis time from starting the reaction of precursor with [18F]fluoride to formulation was approximately 60 min. The purities and specific activities of prepared [18F]cEFQ and [18F]tEFQ were measured using analytical HPLC. The radiochemical purity of the prepared [18F]cEFQ was over 99%, and the specific activity was 63-246 GBq/µmol. The radiochemical purity and specific activity of [18F]tEFQ calculated from the chromatogram were over 99% and 93.2 GBq/µmol, respectively.
Distribution coefficient (log D)
The distribution coefficient log D value of [18F]cEFQ was 3.24 ± 0.09 (n = 6), representing an adequate lipophilicity for penetration of the blood–brain barrier (BBB).
Stability test
[18F]cEFQ showed no degradation over 4 h in the prepared medium at room temperature. However, in human serum at 37℃, 1.2% of fluoride and 3.0% of radioactive metabolite were detected at 1 h, which increased to 3.2% and 6.2% at 4 h, respectively. The radioactive metabolite was not a trans isomer, indicating that cis–trans isomerization did not occur.
Biodistribution study in mice
The biodistribution of [18F]cEFQ (148 kBq) in normal BALB/c mice at 10, 60, and 120 min was investigated in blood and 10 organs after intravenous injection through the tail vein (Table 1). At 10 min after injection, uptake in the liver (16.77 ± 1.42% ID/g) was the highest among the measured organs, followed by the kidney (10.54 ± 0.78% ID/g). However, these activities decreased over time, with increased intestinal uptake activities of 16.94 ± 1.87 and 26.11 ± 1.64% ID/g at 60 and 120 min, respectively, indicating rapid hepatobiliary excretion. Brain uptake was 4.42 ± 0.35% ID/g at 10 min, which decreased to 1.01 ± 0.11% ID/g at 60 min. The radioactivity in the bone was low at 10 min (2.39 ± 0.16% ID/g), but increased to 15.43 ± 3.76% ID/g at 120 min, indicating defluorination occurred in vivo.
Table 1.
Results of the biodistribution study of [18F]cEFQ in normal BALB/c mice (% ID/g tissue: mean ± SD).
| Tissue | 10 min | 60 min | 120 min |
|---|---|---|---|
| Blood | 2.51 ± 0.16 | 1.40 ± 0.07 | 0.71 ± 0.04 |
| Muscle | 2.76 ± 0.16 | 1.64 ± 0.17 | 1.32 ± 0.25 |
| Brain | 4.42 ± 0.35 | 1.01 ± 0.11 | 0.44 ± 0.04 |
| Bone | 2.39 ± 0.72 | 10.79 ± 0.39 | 15.43 ± 3.76 |
| Heart | 3.85 ± 0.23 | 1.76 ± 0.40 | 1.18 ± 0.46 |
| Lung | 6.61 ± 2.56 | 2.50 ± 0.15 | 1.08 ± 0.08 |
| Liver | 16.77 ± 1.42 | 12.98 ± 0.40 | 10.55 ± 1.39 |
| Spleen | 3.28 ± 0.42 | 2.43 ± 0.18 | 1.66 ± 0.16 |
| Stomach | 4.41 ± 0.92 | 21.14 ± 8.18 | 8.19 ± 3.88 |
| Intestine | 5.49 ± 0.58 | 16.94 ± 1.87 | 26.11 ± 1.64 |
| Kidney | 10.54 ± 0.78 | 7.68 ± 0.19 | 4.57 ± 0.33 |
Analysis of metabolites in mice
The metabolites of [18F]cEFQ in the plasma, brain, and urine of normal BALB/c mice were analyzed by HPLC (Figure 2, Table 2). The retention times of free [18F]fluoride, [18F]tEFQ, and [18F]cEFQ were 1.4, 18.0, and 19.0 min, respectively. After intravenous injection of [18F]cEFQ, plasma and brain homogenate samples showed six radiometabolites (metabolites 1–6) and free [18F]fluoride. No evidence of cis–trans isomerization was found, as [18F]tEFQ was not detected in the metabolites of any sample. Metabolites 1–4 were not separated well from each other in the HPLC condition that was used. All radiometabolites showed faster retention times than [18F]cEFQ, indicating that they became more hydrophilic after metabolism. The percentages of [18F]cEFQ in the brain were 88.9% and 59.5% at 5 and 30 min, respectively. Intact [18F]cEFQ in plasma was at 68.5% at 5 min, which decreased to 30.6% at 30 min. No intact [18F]cEFQ and metabolite 6 were found in urine at any time point. One the other hand, no metabolite 1 was found in the brain.
Figure 2.

HPLC profiles of metabolites in the plasma, brain and urine of BALB/c mice. The samples were obtained at 30 min after injection of [18F]cEFQ. Metabolites 1–6 were found in the plasma sample. Metabolite 1 was not found in brain while metabolites 5 and 6 were not found in urine.
Table 2.
Analysis of metabolites in the plasma, brain, and urine of BALB/c mice.
| Rt (min) | Plasma |
Brain |
Urine |
|||||||
|---|---|---|---|---|---|---|---|---|---|---|
| 5 min | 15 min | 30 min | 5 min | 15 min | 30 min | 5 min | 15 min | 30 min | ||
| [18F]fluoride | 1.4 | 4.2 | 19.0 | 16.8 | 0.2 | 1.6 | 4.6 | 9.5 | 52.4 | 11.3 |
| Metabolite 1 | 6.7 | 0 | 6.6 | 11.1 | 0 | 0 | 0 | 4.0 | 4.1 | 5.9 |
| Metabolite 2 | 7.3 | 4.8 | 10.6 | 16.6 | 0.2 | 1.3 | 5.2 | 26.1 | 17.7 | 11.3 |
| Metabolite 3 | 8.4 | 9.3 | 11.5 | 14.0 | 1.5 | 3.3 | 5.5 | 23.1 | 18.9 | 15.4 |
| Metabolite 4 | 9.0 | 2.1 | 5.5 | 4.0 | 0.8 | 2.7 | 4.4 | 4.6 | 1.0 | 7.1 |
| Metabolite 5 | 10.9 | 9.9 | 11.5 | 13.1 | 5.6 | 14.9 | 20.0 | 2.5 | 3.0 | 0 |
| Metabolite 6 | 13.2 | 1.0 | 0.6 | 0.4 | 1.4 | 1.1 | 0.8 | 0 | 0 | 0 |
| [18F]cEFQ | 19.0 | 68.5 | 36.7 | 30.6 | 88.9 | 74.8 | 59.5 | 0 | 0 | 0 |
The values represent the percentage of each radioactive material.
Animal PET
Dynamic animal-PET/CT images of the rat brain for 1 h were obtained after intravenous injection of [18F]cEFQ through the tail vein. [18F]cEFQ showed the highest uptake in the cerebellum. Other mGluR1-rich regions, such as the hippocampus, thalamus, and striatum, also showed some uptake (Figure 3). As shown in the time–activity curves (TACs), radioactivity uptake in the cerebellum remained the highest among all regions for all investigated times after injection, peaking at 2 min and then decreasing (Figure 4(a)). Although the affinity of cEFQ was high (IC50 = 5 nM in rat18), cerebellum uptake decreased faster than expected. Blocking tests using unlabeled cEFQ, JNJ16259685, and ABP688 were performed to investigate the selectivity and specificity of [18F]cEFQ binding to mGluR1. JNJ16259685 and ABP688 are proven specific antagonists of mGluR1 and mGluR5, respectively. When the rats were pre-injected with unlabeled cEFQ or JNJ16259685 10 min before the injection of [18F]cEFQ, there was decreased homogeneous distribution of radioactivity in all brain regions, indicating non-specific uptake due to mGluR1 blocking (Figure 3(c) and (d) and Figure 4(c) and (d)). However, pre-injection of ABP688 did not result in blocked uptake of [18F]cEFQ (Figures 3(e) and (e)).
Figure 3.

PET and CT images of normal SD rat brains. PET images of [18F]cEFQ (a), [18F]tEFQ (b), [18F]cEFQ 10 min after pretreatment with 3 mg/kg cEFQ (c), [18F]cEFQ 10 min after pretreatment with 3 mg/kg JNJ16259685 (d), [18F]cEFQ 10 min after pretreatment with 3 mg/kg ABP688 (e) and CT image (f). All PET images are sum images of 2–10 min after injection.
Figure 4.

Time–activity curves of [18F]cEFQ (a), [18F]tEFQ (b), [18F]cEFQ 10 min after pretreatment with 3 mg/kg cEFQ (c), [18F]cEFQ 10 min after pretreatment with 3 mg/kg JNJ16259685 (d), and [18F]cEFQ 10 min after pretreatment with 3 mg/kg ABP688 (e) in SD rat brains. These TACs are representative results from a rat.
[18F]tEFQ did not show specific uptake in any brain region; this pattern was similar to the blocked non-specific uptake patterns of [18F]cEFQ (Figures 3(b) and 4(b)). The TAC showed that the trans isomer [18F]tEFQ could enter into the brain rapidly through the blood flow and was washed out quickly without binding to mGluR1 (Figure 4(b)).
Ex vivo autoradiography
We examined the binding of [18F]cEFQ in the rat brain using ex vivo autoradiography (Figure 5). Similar to the results of the PET studies, the autoradiograms also showed a wide distribution of radioactivity in almost all brain regions. The highest radioactivity level was found in the cerebellum, followed by the thalamus, hippocampus, and striatum (Figure 5(a)). Slightly increased radioactivity uptakes also were found in mGluR1 localized regions such as olfactory tubercle and substantia nigra pars reticulate (Figure 5(a)). [18F]cEFQ uptakes in the rat brain were blocked by the presence of JNJ16259685 (Figure 5(b)), which was consistent with the animal PET study.
Figure 5.
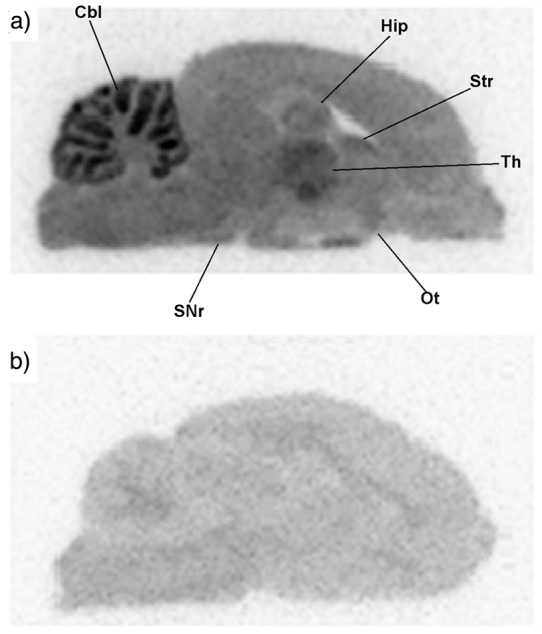
Autoradiograms of [18F]cEFQ in the SD rat brain (a) and with co-injection of JNJ16259685 (b). Cbl, cerebellum; Hip, hippocampus; Str, striatum; Th, thalamus; Ot, olfactory tubercle; SNr, substantia nigra pars reticulata.
Discussion
In this study, we synthesized [18F]cEFQ and [18F]tEFQ for imaging mGluR1 in the brain and evaluated their chemical and biological properties. [18F]cEFQ was labeled with 18F by a nucleophilic substitution reaction at high temperature. In this labeling condition, both cis- and trans isomers [18F]cEFQ and [18F]tEFQ were produced when using either a pure cis-form precursor or a cis–trans precursor mixture. The ratio of produced [18F]cEFQ to [18F]tEFQ was approximately 2:3 due to the more stable thermodynamic nature of the trans product.
It has been reported that compounds having a trans configuration on the cyclohexyl ring of chloroquinoline derivatives have no affinity to mGluR1 but that cis-compounds have high affinity (IC50 = 9 and 4 nM to human and rat mGluR1, respectively).18 Thus, it was essential to separate [18F]cEFQ from [18F]tEFQ by HPLC before use.
Lipophilicity (log D) is an important parameter for brain receptor imaging agents. High lipophilicity is required for BBB penetration; however, too high lipophilicity might increase nonspecific binding to albumin or other tissues. The log D of [18F]cEFQ was measured to be 3.24, which was within the proposed optimal range for BBB penetration of 2.0–3.5,26 and a high initial brain uptake (4.24% ID/g) at 10 min after injection was obtained due to the optimal lipophilicity (Table 1).
In vitro stability tests were performed in prepared medium at room temperature and in human serum at 37℃. [18F]cEFQ did not show any degradation in the prepared medium over 4 h at room temperature, indicating that it can be stored at room temperature for at least 4 h before use. However, it slowly degraded into free 18F and other radioactive metabolites over time in human serum at body temperature, indicating a somewhat unstable metabolic nature in vivo. No [18F]tEFQ peak was found among the radioactive metabolite peaks in the metabolite analysis, which is evidence that cis–trans isomerization did not occur. The isomerization of [18F]cEFQ in vivo could be a serious undesirable reaction for brain PET imaging because the trans isomer has no affinity to the mGluR1 receptor.
Most quinoline derivatives used as mGluR1 antagonists have been reported to have low metabolic stability.18 [11C]JNJ16567083 (Figure 1(a)) showed high affinity to mGluR1; its IC50 values to rat and human mGluR1 were 3 and 8 nM, respectively. However, only 8% of the parent compound remained after a 30-min incubation in human liver microsome.18 Compared with [11C]JNJ16567083, [18F]cEFQ showed a similar affinity to mGluR1 and higher stability (25% of the parent compound remained in human liver microsomes after 30 min) in the same experimental condition.18 Radiometabolites in the brain are problematic for PET imaging because it is not possible to determine which chemical is the source of the detected radioactivity.27 From the metabolism analysis study, we found that approximately 90% of the intact compound was detected in the brain at 5 min post-injection and that 74% was detected at 15 min post-injection (Table 2). If the PET image is obtained at 10 min, more than 80% of the radioactivity will be from the intact compound. Thus, correction using metabolism modeling might be required for quantitative PET studies.
The biodistribution study showed rapid uptake and wash out of [18F]cEFQ into and from the brain. According to the results, brain PET images should be obtained no later than 10 min in mice. For clinical imaging, a further PET TAC study might be necessary to obtain the best quality image. The major excretion route was found to be a hepatobiliary tract according to a biodistribution study. Increased radioactivity in the bone over time represents the in vivo defluorination of [18F]cEFQ in mice. Generally, higher levels of defluorination are found in mice than in humans.28,29
PET images and TACs of rats with [18F]cEFQ showed the distribution of mGluR1 in the brain (Figures 3 and 4). The expression pattern of mGluR1 in the autoradiography study was similar but clearer than in the PET images (Figure 5). It has been reported that although mGluR1 is distributed in most parts of the brain, the distribution in the cerebellum is over nine times higher than in the cortex and approximately eight times higher than in the hippocampus and striatum in the normal rat brain.4,21,30,31 As expected, the highest and most distinct radioactivity uptake was in the cerebellum, and slightly higher radioactivities were found in the thalamus, hippocampus, and striatum than in the cortex. Pretreatment with unlabeled cEFQ or the known mGluR1 antagonist JNJ16259685 resulted in a marked decrease in radioactivity in the whole brain compared to the [18F]cEFQ baseline PET image. However, pretreatment with the mGluR5 selective antagonist ABP688 had no influence on [18F]cEFQ binding. These results confirmed the specificity and selectivity of [18F]cEFQ as a PET tracer for mGluR1 imaging in vivo. Although bone uptake was demonstrated in the biodistribution study, there was no skull uptake in the 10-min PET image.
The low signal in the brain with [18F]tEFQ in the PET study was expected because of its low affinity to mGluR1 and the results of the study by Mabire et al.18 PET has received much attention as a cutting edge technology for drug development. With radiolabeled drug candidates and PET, it is possible to estimate the suitability of new drug molecules in preclinical studies in vivo quantitatively without sacrificing animals. It is a cost-effective technology and helps minimize the risks of drug development.32
Huang et al. verified the binding affinity of [18F]cEFQ to mGluR1 in rodent and non-human primates in a previous study.21 The affinity of [18F]cEFQ changed with temperature, as the affinity at body temperature was lower than at low temperature. The binding affinity of [18F]cEFQ to mGluR1 also differed by species. Generally, lower affinity was found for cloned human mGluR1 than for rat mGluR1. Furthermore, non-human primates and humans have relatively lower mGluR1 receptor densities than rodents.21,30,33 Although the discrepancy between receptor density, binding affinity, and metabolism by species is a barrier for the clinical application of [18F]cEFQ,34 this PET tracer is still useful for preclinical studies.
Conclusions
We described the production and physicochemical and biological properties of [18F]cEFQ as a PET tracer for mGluR1 imaging in the brain. We successfully synthesized [18F]cEFQ both manually and using an automatic module with high specific activity and radiochemical purity with a short preparation time. [18F]cEFQ had an appropriate lipophilicity for a brain receptor imaging agent, and it showed a rapid initial brain uptake. In PET and autoradiography studies, we demonstrated that [18F]cEFQ specifically and selectively bound to mGluR1-abundant regions, especially the cerebellum. In conclusion, [18F]cEFQ can be considered a suitable PET tracer for imaging and quantification of mGluR1 in rats.
Funding
The author(s) disclosed receipt of the following financial supports for the research, authorship, and/or publication of this article: This work was supported by a National Research Foundation of Korea (NRF) grant funded by the Korean government (MEST) (grant nos NRF-2013R1A2A1A05006227 and 1711026888).
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Authors’ contributions
BL performed the synthesis and evaluation of radiotracers, and wrote the manuscript; YKK performed PET/CT data analysis and supervised this study; JYL performed biodistribution, animal PET/CT and autoradiographic experiments; YJK performed biodistribution experiment; Y-SL supervised this study; DSL supervised this study; J-KC supervised this study; and JMJ supervised and funded this study and wrote the manuscript. All authors provided final approval of the present version for publication.
References
- 1.Monaghan DT, Bridges RJ, Cotman CW. The excitatory amino acid receptors: their classes, pharmacology, and distinct properties in the function of the central nervous system. Ann Rev Pharmacol Toxicol 1989; 29: 365–402. [DOI] [PubMed] [Google Scholar]
- 2.Hollmann M, Heinemann S. Cloned glutamate receptors. Ann Rev Neurosci 1994; 17: 31–108. [DOI] [PubMed] [Google Scholar]
- 3.Schoepp DD. Unveiling the functions of presynaptic metabotropic glutamate receptors in the central nervous system. J Pharmacol Exp Therapeut 2001; 299: 12–20. [PubMed] [Google Scholar]
- 4.Lavreysen H, Pereira SN, Leysen JE, et al. Metabotropic glutamate 1 receptor distribution and occupancy in the rat brain: a quantitative autoradiographic study using [3H]R214127. Neuropharmacology 2004; 46: 609–619. [DOI] [PubMed] [Google Scholar]
- 5.Steckler T, Lavreysen H, Oliveira AM, et al. Effects of mGlu1 receptor blockade on anxiety-related behaviour in the rat lick suppression test. Psychopharmacology 2005; 179: 198–206. [DOI] [PubMed] [Google Scholar]
- 6.Swanson CJ, Bures M, Johnson MP, et al. Metabotropic glutamate receptors as novel targets for anxiety and stress disorders. Nature Rev Drug Discov 2005; 4: 131–144. [DOI] [PubMed] [Google Scholar]
- 7.Belozertseva IV, Kos T, Popik P, et al. Antidepressant-like effects of mGluR1 and mGluR5 antagonists in the rat forced swim and the mouse tail suspension tests. Eur Neuropsychopharmacol: J Eur Coll Neuropsychopharmacol 2007; 17: 172–179. [DOI] [PubMed] [Google Scholar]
- 8.Luscher C, Huber KM. Group 1 mGluR-dependent synaptic long-term depression: mechanisms and implications for circuitry and disease. Neuron 2010; 65: 445–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kang SJ, Liu MG, Chen T, et al. Plasticity of metabotropic glutamate receptor-dependent long-term depression in the anterior cingulate cortex after amputation. J Neurosci 2012; 32: 11318–11329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tang FR, Lee WL, Yeo TT. Expression of the group I metabotropic glutamate receptor in the hippocampus of patients with mesial temporal lobe epilepsy. J Neurocytol 2001; 30: 403–411. [DOI] [PubMed] [Google Scholar]
- 11.Senkowska A, Ossowska K. Role of metabotropic glutamate receptors in animal models of Parkinson's disease. Polish J Pharmacol 2003; 55: 935–950. [PubMed] [Google Scholar]
- 12.Ossowska K, Wardas J, Pietraszek M, et al. The striopallidal pathway is involved in antiparkinsonian-like effects of the blockade of group I metabotropic glutamate receptors in rats. Neurosci Lett 2003; 342: 21–24. [DOI] [PubMed] [Google Scholar]
- 13.Hodgson RA, Hyde LA, Guthrie DH, et al. Characterization of the selective mGluR1 antagonist, JNJ16259685, in rodent models of movement and coordination. Pharmacol Biochem Behavior 2011; 98: 181–187. [DOI] [PubMed] [Google Scholar]
- 14.Wu WL, Burnett DA, Domalski M, et al. Discovery of orally efficacious tetracyclic metabotropic glutamate receptor 1 (mGluR1) antagonists for the treatment of chronic pain. J Med Chem 2007; 50: 5550–5553. [DOI] [PubMed] [Google Scholar]
- 15.Schkeryantz JM, Kingston AE, Johnson MP. Prospects for metabotropic glutamate 1 receptor antagonists in the treatment of neuropathic pain. J Med Chem 2007; 50: 2563–2568. [DOI] [PubMed] [Google Scholar]
- 16.Tappe-Theodor A, Fu Y, Kuner R, et al. Homer1a signaling in the amygdala counteracts pain-related synaptic plasticity, mGluR1 function and pain behaviors. Mol Pain 2011; 7: 38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Owen DR. Recent advances in the medicinal chemistry of the metabotropic glutamate receptor 1 (mGlu(1)). ACS Chem Neurosci 2011; 2: 394–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mabire D, Coupa S, Adelinet C, et al. Synthesis, structure-activity relationship, and receptor pharmacology of a new series of quinoline derivatives acting as selective, noncompetitive mGlu1 antagonists. J Med Chem 2005; 48: 2134–2153. [DOI] [PubMed] [Google Scholar]
- 19.Lavreysen H, Wouters R, Bischoff F, et al. JNJ16259685, a highly potent, selective and systemically active mGlu1 receptor antagonist. Neuropharmacol 2004; 47: 961–972. [DOI] [PubMed] [Google Scholar]
- 20.Huang Y, Narendran R, Bischoff F, et al. A positron emission tomography radioligand for the in vivo labeling of metabotropic glutamate 1 receptor: (3-ethyl-2-[11C]methyl-6-quinolinyl)(cis- 4-methoxycyclohexyl)methanone. J Med Chem 2005; 48: 5096–5099. [DOI] [PubMed] [Google Scholar]
- 21.Huang Y, Narendran R, Bischoff F, et al. Synthesis and characterization of two PET radioligands for the metabotropic glutamate 1 (mGlu1) receptor. Synapse 2012; 66: 1002–1014. [DOI] [PubMed] [Google Scholar]
- 22.Hintermann S, Vranesic I, Allgeier H, et al. ABP688, a novel selective and high affinity ligand for the labeling of mGlu5 receptors: identification, in vitro pharmacology, pharmacokinetic and biodistribution studies. Bioorg Med Chem 2007; 15: 903–914. [DOI] [PubMed] [Google Scholar]
- 23.Chang YS, Jeong JM, Lee YS, et al. Synthesis and evaluation of benzothiophene derivatives as ligands for imaging beta-amyloid plaques in Alzheimer's disease. Nucl Med Biol 2006; 33: 811–820. [DOI] [PubMed] [Google Scholar]
- 24.Yao R, Lecomte R, Crawford ES. Small-animal PET: what is it, and why do we need it? J Nucl Med Technol 2012; 40: 157–165. [DOI] [PubMed] [Google Scholar]
- 25.Chang YS, Jeong JM, Yoon YH, et al. Biological properties of 2'-[18F]fluoroflumazenil for central benzodiazepine receptor imaging. Nucl Med Biol 2005; 32: 263–268. [DOI] [PubMed] [Google Scholar]
- 26.Waterhouse RN. Determination of lipophilicity and its use as a predictor of blood-brain barrier penetration of molecular imaging agents. Mol Imag Biol 2003; 5: 376–389. [DOI] [PubMed] [Google Scholar]
- 27.Pike VW. PET radiotracers: crossing the blood-brain barrier and surviving metabolism. Trends Pharmacol Sci 2009; 30: 431–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ouyang Y, Tinianow JN, Cherry SR, et al. Evaluation of 2-[(1)(8)F]fluoroacetate kinetics in rodent models of cerebral hypoxia-ischemia. J Cereb Blood Flow Metab 2014; 34: 836–844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ponde DE, Dence CS, Oyama N, et al. 18F-fluoroacetate: a potential acetate analog for prostate tumor imaging – in vivo evaluation of 18F-fluoroacetate versus 11C-acetate. J Nucl Med 2007; 48: 420–428. [PubMed] [Google Scholar]
- 30.Mutel V, Ellis GJ, Adam G, et al. Characterization of [(3)H]Quisqualate binding to recombinant rat metabotropic glutamate 1a and 5a receptors and to rat and human brain sections. J Neurochem 2000; 75: 2590–2601. [DOI] [PubMed] [Google Scholar]
- 31.Lavreysen H, Janssen C, Bischoff F, et al. [3H]R214127: a novel high-affinity radioligand for the mGlu1 receptor reveals a common binding site shared by multiple allosteric antagonists. Mol Pharmacol 2003; 65: 1082–1093. [DOI] [PubMed] [Google Scholar]
- 32.Wang J, Maurer L. Positron emission tomography: applications in drug discovery and drug development. Curr Topic Med Chem 2005; 5: 1053–1075. [DOI] [PubMed] [Google Scholar]
- 33.Hostetler ED, Eng W, Joshi AD, et al. Synthesis, characterization, and monkey PET studies of [18F]MK-1312, a PET tracer for quantification of mGluR1 receptor occupancy by MK-5435. Synapse 2011; 65: 125–135. [DOI] [PubMed] [Google Scholar]
- 34.Li S, Huang Y. In vivo imaging of the metabotropic glutamate receptor 1 (mGluR1) with positron emission tomography: recent advance and perspective. Curr Med Chem 2014; 21: 113–123. [DOI] [PubMed] [Google Scholar]