

Abstract
Dimeric β-carbolines are cytotoxic against multiple NSCLC cell lines, and we report herein our preliminary studies on the mechanism of action of these dimeric structures. Dimeric β-carboline 1, which is more potent than the corresponding monomer in NSCLC cell lines, is a lysomotropic agent that inhibits autophagy and mediates cell death by apoptosis, upregulating the proapoptotic BH3-only protein PUMA (p53 upregulated modulator of apoptosis) in a dose dependent manner
Keywords: apoptosis, PUMA, β-carboline dimer, autophagy, lysosomotropic
Graphical abstract
According to the World Health Organization (WHO), lung cancer is one of the most 5 common types of cancer in both men and women and the leading cause of cancer death worldwide.1 Lung cancer is traditionally categorized into two major types; small cell (SCLC) and non-small cell (NSCLC) lung cancer. Most lung cancers are NSCLC (85%) and challenging to treat due to acquired resistance. With currently available drugs for the treatment of NSCLC, the 5-year survival rate is less than 15%. The development of novel and more effective chemotherapeutic agents is therefore an important goal.1
PUMA (p53 upregulated modulator of apoptosis), a proapoptotic BH3-only protein, mediates apoptosis by releasing proapoptotic proteins, Bax and/or Bak, from the anti-apoptotic Bcl-2 family members, or by directly activating Bax and/or Bak to cause mitochondrial dysfunction and induce apoptosis.2 PUMA activity is controlled by different transcription factors (e.g. p53, p73, c-Myc and E2F1), depending on cell type and stimuli.2 Loss of PUMA leads to impaired apoptosis, which can promote carcinogenesis.3 In addition to apoptosis, PUMA has also been reported to regulate autophagy through the autophagy regulator, Beclin-1,4 and to selectively induce mitochondrial autophagy, mitophagy, via a Bax-dependent mechanism.5 The upregulation of PUMA is therefore an attractive strategy for the development of new NSCLC chemotherapies.
Recently, Thorburn et.al. reported that autophagy modulates apoptosis by selective regulation of constitutive PUMA expression.6 Autophagy inhibition increases PUMA levels, leading to accelerated mitochondrial outer membrane permeabilization (MOMP) and apoptosis. Lysosomes, which have emerged as important targets for the development of cancer therapeutics,7 are key components in the autophagy pathway.8 The acidic environment of the lysosome is typically maintained by a vacuolar ATPase.9 Lysosomal dysfunction due to vacuolar ATPase inhibition leads to interior pH elevation, and thereby impairs autophagy and consequently activates apoptosis.10
Many lysosomotropic agents, for example, chloroquine11,12 and Lys05,13 have been reported as lysosome-targeting anticancer agents. Due to their weakly basic property, they are protonated and trapped in the acidic interior of lysosomes.12 The weak base accumulation enhances the interior pH, leading to inhibition of hydrolase activities and consequently to autophagy inhibition. In addition, the accumulation of the charged molecules in the lysosome can cause lysosomal leakage and/or lysis, mediating cell death due to the increase of an osmotic pressure across the lysosomal membrane.14 β-carbolines exhibit broad biological activity and exhibit antimalarial, anticancer, antiviral and anti-inflammatory properties.15,16 We have recently reported that dimeric β-carbolines17 exhibit significantly enhanced cytotoxicity relative to their monomeric counterparts against multiple cancer cell lines, including NSCLC, prompting us to study the mechanism of action of the cytotoxicity of these dimeric β-carbolines. We describe herein the preparation of a novel lysosomotropic agent, dimeric β-carboline 1 (Figure 1), its evaluation against six NSCLC cell lines, and preliminary results on the mechanism of action of 1.
Figure 1.
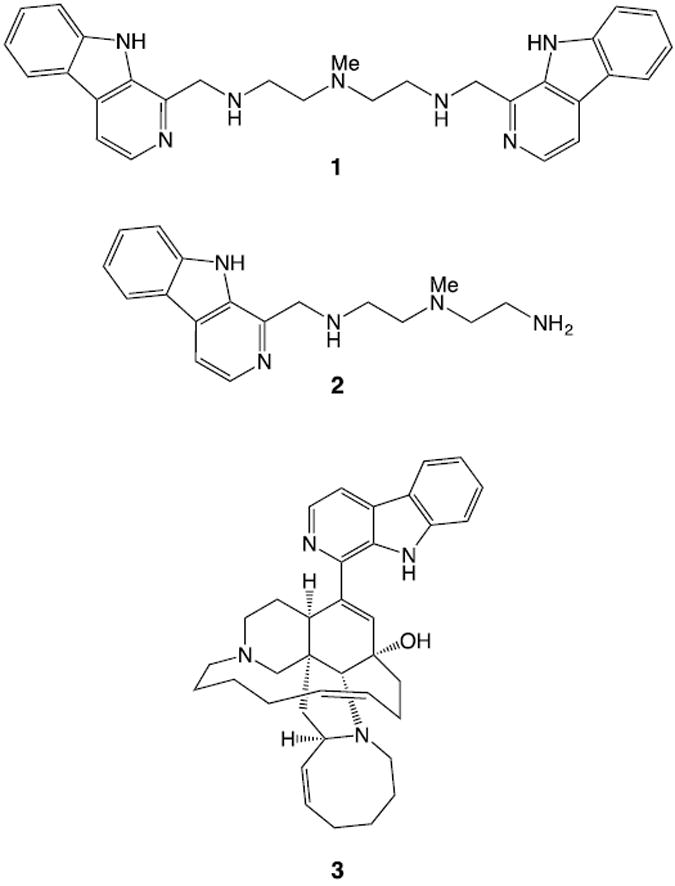
Synthetic dimeric β-carboline 1, monomer 2 and naturally occurring β-carboline manzamine A 3.
To determine the cytotoxicity of dimeric β-carboline 1 against NSCLC, we examined the biological activity of dimeric β-carboline 1, monomeric β-carboline 2, and the naturally occurring β-carboline alkaloid manzamine A 3, against six NSCLC cell lines (H1299, A549, H441, H1373, H1993 and H2009). The results are summarized in Table 1. Dimeric β-carboline 1 exhibited comparable potency to manzamine A 3 and significantly greater cytotoxicity than the corresponding monomer 2 in each of the NSCLC cell lines.17 The relative cytotoxicity of 1 was consistently greater than that of monomer 2 in NSCLC cells compared to IMR90 (human lung fibroblast) cells. These results support the importance of the dimeric structure of 1, and suggest that these dimeric structures could represent lead compounds for the development of new NSCLC therapeutics.
Table 1.
Cytotoxicity (IC50) for dimeric β-carboline 1, monomeric β-carboline 2 and manzamine A 3 against NSCLC and IMR90 (Human Caucasian fetal lung fibroblast)
| IC50(μM)
| |||||||
|---|---|---|---|---|---|---|---|
| H1299 | A549 | H441 | H1373 | H1993 | H2009 | IMR90 | |
| 1 | 1.6±0.7 | 2.9±0.8 | 0.7±0.3 | 3.3±2.1 | 1.2±1.1 | 1.4±0.2 | 5.6±0.6 |
| 2 | 15.1±0.4 | 20.5±9.2 | 4.3±2.8 | 13.2±0.3 | 9.3±7.1 | 8.9±0.1 | 12.9±1.2 |
| 3 | 1.5±0.5 | 2.3±0.5 | 1.1±0.1 | 2.1±0.1 | 3.9±1.7 | 1.6±0.2 | 6.8±0.7 |
We initiated our study of the mechanism of action of 1 by first determining the site(s) of its subcellular localization. Taking advantage of the inherent fluorescence of the β-carboline moiety (Ex/Em: 358/461 nm), we treated H1299 cells with compounds 1-3 for 1 hr before staining mitochondria with MitoTracker® Green FM (Invitrogen) and lysosomes with LysoTracker® Red DND-99. Live confocal microscopy established that each of these compounds localized in lysosomes (Figure 2a). These observations are similar to the findings seen previously for manzamine A 3, which targets vacuolar ATPases in lysosomes and inhibits autophagy by preventing autophagosome turnover in pancreatic cancer cells.10 To determine whether dimer 1 or monomer 2 show similar effect to 3, we performed western blot analysis for autophagy markers, LC3 and p62/SQSTM1 (Figure 2b).18
Figure 2.
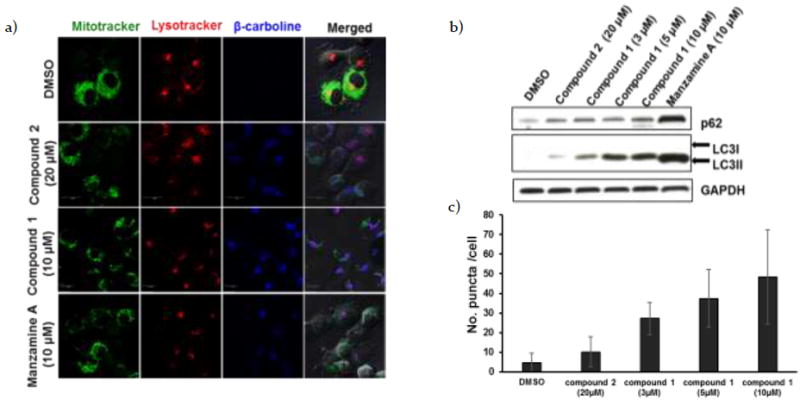
a) H1299 cells were treated with compounds 1-3 for 1 hr before staining mitochondria with MitoTracker® Green FM (Invitrogen) and lysosomes with LysoTracker® Red DND-99 (Invitrogen). Confocal analysis showed localization of the β-carbolines in lysosomes. b) Western blot analysis for LC3 and p62/SQSTM1 in H1299 cells incubated with DMSO, monomer 2 (20 μM), dimer 1 (3, 5 and 10 μM) or manzamine A 3(10 μM) for 24 hrs. c) GFP-LC3 transfected H1299 cells were treated with DMSO, monomer 2 (20 μM), dimer 1 (3, 5 and 10 μM) or manzamine A 3 (10 μM) for 24 hrs. The GFP-LC3 puncta were analyzed by confocal microscope. The number of GFP-LC3 puncta/cell was scored using ImageJ software (n=30; error bars, s.d.).
H1299 cells treated with dimeric β-carboline 1 showed an increase in autophagy adaptor protein, p62/SQSTM1, and a dose-dependent increase in LC3II levels, compared to the control DMSO. Since the LC3I signal, which is known to be less sensitive to detection by western blot analysis than LC3II,18 was not observed in this assay, we performed immunofluorescence staining of LC3II puncta in H1299 cells transfected with GFP-LC3 to confirm these results (Figure 2c; see also Supplementary Figure 1 (S1)). Consistent with the western blot analysis, the transfected cells treated with β-carbolines 1 and 2 and manzamine A 3 showed an increase in LC3II. These results suggest that dimeric β-carboline 1 functions similarly to manzamine A to inhibit autophagosome clearance and consequently inhibit autophagy.
Apoptosis and autophagy are mutually cross-regulatory,4,5,19 and autophagy inhibition has been reported to induce apoptotic cell death.6,20,21 We therefore examined whether 1, by inhibiting autophagy, induces cell death in H1299 cells through apoptosis. We examined Annexin V positive cells using the Guava Nexin reagent and Guava Personal Cell Analysis. H1299 cells were treated with either 1 or 2 for 48 hrs before staining with Annexin V-PE (Annexin V- phycoerythrin) and 7-AAD (7-Aminoactinomycin D). Flow cytometric analysis (Figure 3a) revealed that treatment with 1 (3 μM) led to a significant increase in apoptotic cell death (bottom right panel) relative to treatment with the monomer 2 at 5x concentration (15 μM). Similarly, treatment of H1299 cells with 1 induced a significant increase in cleaved caspase 3 expression in a dose-dependent manner as seen by western blot analysis (Figure 3b) and immunofluorescence (Figure 3c). Interestingly, other apoptotic markers, such as cleaved lamin A and cleaved PARP levels, were also increased following treatment with 1 relative to 2 (Figure 3b; For quantification, see Supplementary Figure 2 (S2)). These combined results indicate that 1 induces cell death through caspase-dependent apoptosis, and that it does so more efficiently than the corresponding monomer 2.
Figure 3.

a) H1299 cells were incubated with DMSO (control experiment), monomer 2 (15 μM) or dimer 1 (3 μM) for 48 hrs. After incubation, cells were stained with Guava Nexin Reagent, and data were acquired on the PCA-96 System. The results demonstrated 4%, 11% and 97% apoptotic cells, respectively (n=3; error bars, s.d.). b) Western blot analysis of apoptotic proteins in H1299 cells treated with DMSO, monomer 2 (20 μM) or dimer 1 (3, 5 and 10 μM) for 24 hrs incubation. c) Confocal immunofluorescent analysis for active caspase 3 in H1299 cells treated with DMSO, monomer 2 (20 μM) or dimer 1 (5 μM) for 24 hrs. Apoptotic cells were quantified from the ratio of number of cell expressing active caspase to total cell number (n=3; error bars, s.d.).
We have established that 1 accumulates in lysosomes (Figure 2a), inhibits autophagy (Figure 2b and 2c) and mediates caspase-dependent apoptotic cell death (Figure 3). We next explored the connection between lysosomal dysfunction and caspase-mediated cell death. Several BH3-only proteins are known to play a dual role in regulating apoptosis and autophagy.5,19 PUMA, in particular, mediates caspase cleavage by either activation of Bax/Bak or inhibition of anti-apoptotic Bcl-2 family proteins.2 Autophagy inhibition has been shown to cause upregulation of PUMA expression.6 Consistent with this premise, Western blot analysis of H1299 cells treated with different concentrations of dimer 1 show a dose-dependent increase in PUMA levels (Figure 4a). To determine whether 1 induces PUMA expression at the transcription level, we performed qRT-PCR analysis and found that the PUMA mRNA levels in H1299 cells treated with 1 increase in both a dose- and time-dependent manner, which correlates with the observed increase in PUMA protein levels (Figure 4b). Depending on stimuli and cell type, the expression of PUMA can be regulated by several different transcription factors (p53, p73 and E2F1).2 H1299 cells do not express p5322. Western blot analysis showed no alteration of p73 and E2F1, compared to the DMSO control (see Supplementary Figure 3 (S3), although this does not rule out the possibility that these proteins may still be involved. Our data suggest that 1, possibly by accumulation in lysosomes, inhibits autophagy and causes PUMA upregulation in a p53-independent manner, and that this consequently leads to caspase-dependent apoptosis. 6
Figure 4.
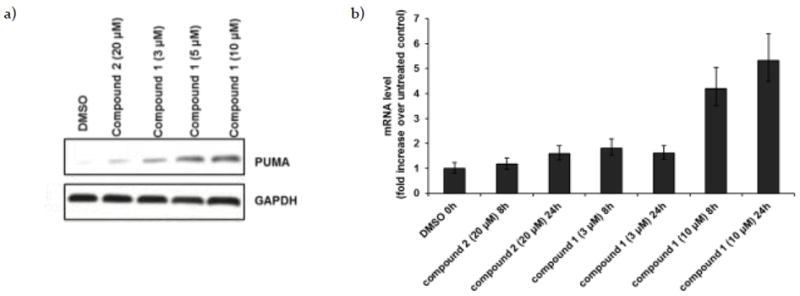
a) Western blot analysis of PUMA in H1299 cells treated with DMSO, monomer 2 (20 μM) or dimer 1 (3, 5 and 10 μM) for 24 hrs. b) RNA was harvested 8 and 24 hrs, respectively, after treatment with DMSO, monomer 2 (20 μM), or dimer 1 (3 and 10 μM). PUMA expression was analyzed by qRT-PCR. Expression was normalized to 18S levels and is reported as x-fold increase over untreated control.
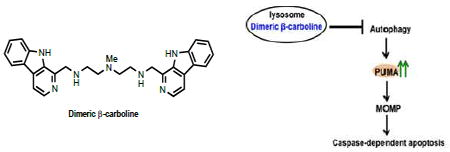
The cytotoxicity of dimeric β-carboline 1 against various NSCLC cell lines prompted us to further study its mechanism of action. Our studies indicate that dimeric β-carboline 1 induces apoptosis in H1299 cells significantly more efficiently than the corresponding monomer 2, accompanied by an increase in caspase-dependent apoptosis markers and in the expression of the proapoptotic protein, PUMA. These data support our proposal that dimeric β-carboline 1 induces cell death through caspase-dependent apoptosis mediated by PUMA (Figure 5).
Figure 5.
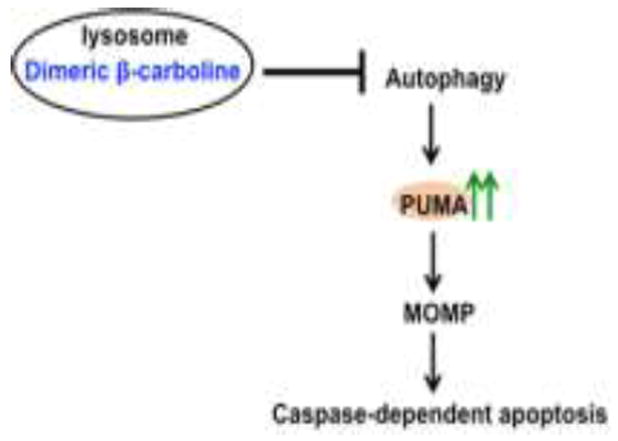
Proposed mechanism of action of dimeric β-carbolines.
The subcellular co-localization analysis demonstrate that both dimeric β-carboline 1 and monomeric 2 predominantly localize in the lysosome, in analogy to other weakly basic agents such as chloroquine, Lys0513 and manzamine A10, which were all reported to cause lysosomal dysfunction and consequently autophagy inhibition. H1299 cells treated with 1 showed a dose-dependent increase in LC3II levels, indicating increased autophagosome accumulation. Many studies have shown that autophagy and apoptosis regulate each other.5, 19, 23 In particular, autophagy has been reported to regulate apoptosis via modulation of constitutive PUMA expression, which controls the rate of mitochondrial outer membrane permeabilization (MOMP).6 These findings support the hypothesis that dimeric β-carboline 1 accumulates in lysosomes and inhibits autophagy, thereby inducing PUMA expression and caspase-dependent apoptosis.
In conclusion, we have demonstrated that the dimeric β-carboline 1 targets lysosomes in H1299 NSCLC cells and mediates caspase-dependent apoptosis, in part through PUMA induction.
These data suggest that these simple dimeric structures can serve as leads for the development of new NSCLC therapeutic strategies.
Supplementary Material
Acknowledgments
We would like to thank James Hayden and Frederick Keeney (Wistar Imaging Facility) for their assistance, Professor Lewis Cantley (Weill Cornell) for the NSCLC cell lines, and the Royal Thai Government for a scholarship (to J. C.). Financial support from the NIH (P01-CA114046) is gratefully acknowledged.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and notes
- 1.Nascimento AV, Bousbaa H, Ferreira D, Sarmento B. Curr Drug Targets. 2015;16:1448–63. doi: 10.2174/1389450115666140528151649. [DOI] [PubMed] [Google Scholar]
- 2.Yu J, Zhang L. Oncogene. 2008;27:S71–S83. doi: 10.1038/onc.2009.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Michalak EM, Vandenberg CJ, Delbridge ARD, Wu L, Scott CL, Adams JM, Strasser A. Genes Dev. 2010;24:1608–1613. doi: 10.1101/gad.1940110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Marino G, Niso-Santano M, Baehrecke EH, Kroemer G. Nat Rev Mol Cell Biol. 2014;15:81–94. doi: 10.1038/nrm3735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yee KS, Wilkinson S, James J, Ryan KM, Vousden KH. Cell Death Differ. 2009;16:1135–45. doi: 10.1038/cdd.2009.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Thorburn J, Andrysik Z, Staskiewicz L, Gump J, Maycotte P, Oberst A, Green DR, Espinosa JM, Thorburn A. Cell Rep. 2014;7:45–52. doi: 10.1016/j.celrep.2014.02.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rebecca VW, Amaravadi RK. Oncogene. 2015;35:1–11. doi: 10.1038/onc.2015.99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Glick D, Barth S, Macleod KF. J Pathol. 2010;221:3–12. doi: 10.1002/path.2697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Boya P, Kroemer G. Oncogene. 2008;27:6434–6451. doi: 10.1038/onc.2008.310. [DOI] [PubMed] [Google Scholar]
- 10.Kallifatidis G, Hoepfner D, Jaeg T, Guzmán EA, Wright AE. Mar Drugs. 2013;11:3500–3516. doi: 10.3390/md11093500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lakhter AJ, Sahu RP, Sun Y, Kaufmann WK, Androphy EJ, Travers JB, Naidu SR. J Invest Dermatol. 2013;133:2247–2254. doi: 10.1038/jid.2013.56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Giraldo AMV, Appelqvist H, Ederth T, Ollinger K. Biochem Soc Trans. 2014;42:1460–1464. doi: 10.1042/BST20140145. [DOI] [PubMed] [Google Scholar]
- 13.McAfee Q, Zhang Z, Samanta A, Levi SM, Ma X, Piao S, Lynch JP, Uehara T, Sepulveda AR, Davis LE, Winkler JD, Amaravadi RK. Proc Natl Acad Sci U S A. 2012;109:8253–8258. doi: 10.1073/pnas.1118193109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Turk B, Stoka V. Protease signalling in cell death: caspases versus cysteine cathepsins. FEBS Lett. 2007;581:2761–2767. doi: 10.1016/j.febslet.2007.05.038. [DOI] [PubMed] [Google Scholar]
- 15.Cao R, Peng W, Wang Z, Xu A. Curr Med Chem. 2007;14:479–500. doi: 10.2174/092986707779940998. [DOI] [PubMed] [Google Scholar]
- 16.Ashok P, Ganguly S, Murugesan S. Drug Discov Today. 2014;19:1781–1791. doi: 10.1016/j.drudis.2014.06.010. [DOI] [PubMed] [Google Scholar]
- 17.Chatwichien J, Basu S, Murphy ME, Hamann MT, Winkler JD. Tetrahedron Lett. 2015;56:3515–3517. doi: 10.1016/j.tetlet.2015.01.142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Klionsky DJ, Abeliovich H, Agostinis P, Agrawal DK, Aliev G, Askew DS, Baba M, Baehrecke EH, Bahr BA, Ballabio A, Bamber BA, Bassham DC, Bergamini E, Kroemer G, Kuan C, Kumar R, Kundu M, Landry J, Laporte M, Le W, Lei H, Michael J, Levine B, Lieberman A, Lim K, Lin F, Liou W, Liu LF, Lopez-berestein G, López-otín C, Macleod KF, Malorni W, Martinet W, Matsuoka K, Mistiaen WP, Mizushima N, Mograbi B, Münz C, Murphy LO, Naqvi NI, Thomas P, Ogawa M, Oleinick NL, Olsen LJ, Ozpolat B, Perry G, Piacentini M, Pinkas-kramarski R. Autophagy. 2008;4:151–175. doi: 10.4161/auto.5338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nishida K, Yamaguchi O, Otsu K. Circ Res. 2008;103:343–351. doi: 10.1161/CIRCRESAHA.108.175448. [DOI] [PubMed] [Google Scholar]
- 20.Boya P, Gonzalez-Polo, Rosa-Ana Casares N, Perfettini J-L, Dessen P, Larochette N, Métivier D, Meley D, Souquere S, Yoshimori T, Pierron G, Codogno P, Kroemer G. Mol Cell Biol. 2005;25:1025–1040. doi: 10.1128/MCB.25.3.1025-1040.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Amaravadi RK, Yu D, Lum JJ, Bui T, Christophorou MA, Evan GI, Thomas-Tikhonenko A, Thompson CB. J Clin Invest. 2007;117:326–336. doi: 10.1172/JCI28833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang Y, Blandino G, Oren M, Givol D. Oncogene. 1998;17:1923–1930. doi: 10.1038/sj.onc.1202113. [DOI] [PubMed] [Google Scholar]
- 23.Ruan Y, Hu K, Chen H. Mol Med Rep. 2015;12:5796–5806. doi: 10.3892/mmr.2015.4148. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.