

Abstract
Macrophage infiltration is a factor in most if not all inflammatory pathologies. Understanding molecular interactions that underlie this process is therefore important for our ability to modulate macrophage behavior for therapeutic purposes. Our studies show that cell surface expression of the nerveglial antigen 2 (NG2) proteoglycan is important for the ability of macrophages to colonize both brain tumors and sites of central nervous system (CNS) demyelination. Myeloid-specific ablation of NG2 using LysM-Cre deleter mice results in large decreases in macrophage abundance in both an intracranial melanoma model and a lysolecithin model of spinal cord demyelination. In the melanoma model, decreased macrophage recruitment in the NG2 null mice leads to diminished tumor growth. In line with observations in the literature, this phenomenon is based in part on deficits in tumor vascularization that result from loss of pericyte interaction with endothelial cells in the absence of a macrophage-derived factor(s). In the demyelination model, decreased macrophage infiltration in the NG2 null mice is associated with an initial reduction in lesion size, but nevertheless also with deficits in repair of the lesion. Diminished myelin repair is due not only to reduced clearance of myelin debris, but also to decreased proliferation/recruitment of oligodendrocyte progenitor cells in the absence of a macrophage-derived factor(s). Thus, in both models macrophages have secondary effects on other cell types that are important for progression of the specific pathology. Efforts are underway to identify mechanisms by which NG2 influences macrophage recruitment and by which macrophages signal to other cell types involved in the pathologies.
Keywords: NG2 proteoglycan, macrophages, pericytes, oligodendrocyte progenitor cells, CNS demyelination, inflammation, brain tumors
1. Introduction
Macrophages are an important part of the innate immune response, and as such, are vital components in the initial host response to pathogens. Thus, recruitment/infiltration of these myeloid cells is a prominent aspect of most, if not all, inflammatory pathologies. In cases of chronic inflammation, macrophage phenotypes can be subverted to perform functions that promote progression of the pathology. The actions of immune suppressive tumor-associated macrophages are a good example of this phenomenon [1, 2], but there are additional pathologies, such as atherosclerosis [3], in which the plasticity of macrophage phenotype and function are exploited to the advantage of the pathology. In our recent work, we have investigated the importance of the NG2 proteoglycan in macrophage recruitment and participation in two CNS pathologies: brain tumor progression and demyelination/remyelination of the spinal cord. NG2 expression in other types of immature and progenitor cells has been found to be important for various aspects of cell behavior, including proliferation, motility, and survival [4, 5]. NG2 therefore has the potential to participate in mechanisms that may help account for the plasticity of macrophage phenotypes.
2. Macrophage expression of NG2
Studies from several research groups have noted prominent expression of NG2 by macrophages [6-10]. This was initially puzzling to us, since in our hands, NG2 is expressed only at very low levels by quiescent CNS resident microglia, peritoneal macrophages, bone marrow-derived macrophages, RAW264.7 mouse macrophages, and THP-1 human monocytes. However, the work of Bu et al. (2001) [6] makes the important point that NG2 is expressed by activated, rather than quiescent, macrophages, in kainic acid-induced lesions in the CNS. This apparently is also true for microglia, which are induced to express NG2 following lipopolysaccharide (LPS) injection [11]. Consistent with these observations is our recent finding that NG2 expression can be induced in primary macrophages and in both RAW264.7 and THP-1 cells by treatment with tolllike receptor ligands such as poly-I:C or LPS [12].
Figure 1 shows an example of NG2 expression induced in RAW264.7 cells by treatment with LPS. These findings suggest that NG2 expression may be up-regulated on circulating monocytes in vivo in response to signals received from sites of inflammation, and that this event may be part of the transition from monocytes to macrophages. The report from Bu et al. (2001) [6] further notes the transience of NG2 expression by macrophages, a phenomenon that we have noted both in vivo and in vitro. This observation may have relevance to the functional role of NG2 in macrophage biology; i.e. NG2 could be more important for initial stages of macrophage recruitment than for subsequent macrophage activities at sites of inflammation.
Figure 1. Induction of NG2 expression in RAW264.7 macrophages.
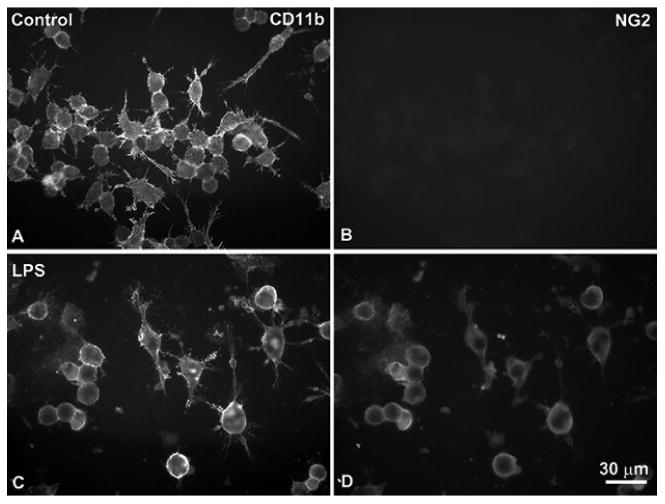
Double immunolabeling for the macrophage marker CD11b (A, C) and NG2 (B, D) was carried out at 4 °C on living RAW264.7 mouse macrophages under control conditions (A, B) and after a two-day stimulation with 2 μg/ml LPS (C, D). NG2 expression on cell surfaces is not detectable prior to stimulation, but is evident after activation with the toll-like receptor ligand. Bar in D = 30 μm.
3. NG2-dependent macrophage contributions to CNS pathologies
Although NG2 expression by macrophages is not limited to CNS pathologies, several published studies have focused on macrophages that infiltrate damaged CNS areas. This is partly because of the need to distinguish between the contributions of macrophages and oligodendrocyte progenitor cells (OPCs), both of which express NG2 in CNS lesions. This is emphasized by our own work on brain tumor progression and on myelin damage and repair.
3.1. Intracranial melanoma progression
Several publications have noted the potentiating effects of NG2 expression on glioma and melanoma growth, establishing positive correlations between NG2 expression and glioma malignancy [13-19]. In almost all cases, the focus of the work has been on the importance of tumor cell NG2 for glioma malignancy and progression. To examine specifically the importance of NG2 in the stromal compartment, we have used intracranial microinjection of B16F10 melanoma cells [20] to investigate the role of NG2 in melanoma progression in the brain. Because B16F10 cells are NG2-negative, these studies have allowed us to focus on the importance of NG2 expression in the host stroma [21]. By comparing intracranial tumor progression in wild-type and germline NG2 null mice [22], we were able to demonstrate a significant role in tumor growth for NG2 expressed by pericytes and macrophages in the host microenvironment. Similar findings were made for growth of MMTV-PyMT mammary tumors in wild-type and germline NG2 null mice [23]. In the case of both brain and mammary tumors, a major factor underlying reduced tumor growth in germline NG2 null mice was a deficiency in tumor vascularization [21, 23].
In order to dissect the respective roles of NG2 in pericyte and macrophage contributions to tumor vascularization and progression, we used Cre-Lox technology for specific ablation of NG2 in these two cell types. NG2 floxed mice [24] were crossed with pdgfrb-Cre mice [25, 26] and with LysM-Cre mice [27, 28] to generate pericyte-specific (PC-NG2ko) and myeloid-specific (My-NG2ko) mice, respectively. Comparison of intracranial B16F10 tumor growth in control and PC-NG2ko mice revealed slower tumor growth in the PC-NG2ko animals [29, 30] (Figure 2C, D). The diminished tumor growth in PC-NG2ko mice appeared to be due at least in part to deficits in vascular function. Tumor vessels in PC-NG2ko mice exhibited decreased patency and increased leakiness compared to tumor vessels in control mice, resulting in higher levels of tumor hypoxia [29] (Table 1).
Figure 2. Intracranial melanoma growth in NG2 null mice.
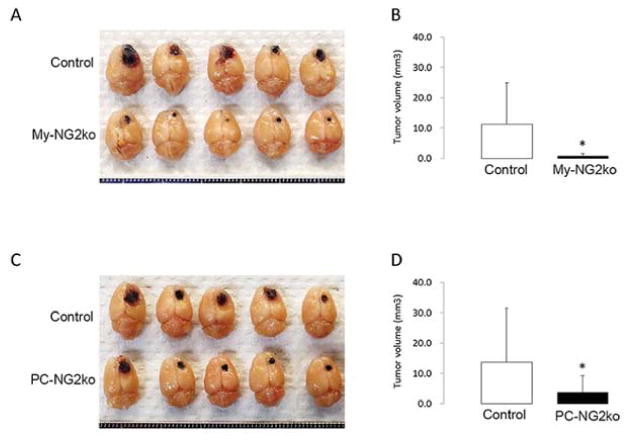
Brain tumors were initiated in control, PC-NG2ko, and My-NG2ko mice by intracranial microinjection of 2 × 104 pigmented B16F10 melanoma cells. Animals were euthanized after 10 days for determination of tumor volumes (mm3). (A, B): Control mice (n = 21) versus My-NG2ko mice (n = 20). (C, D): Control mice (n = 22) versus PC-NG2ko mice (n = 18). *p < 0.01. (Reproduced with permission from Yotsumoto, F., You, W. K., Cejudo-Martin, P., Kucharova, K., Sakimura, K. and Stallcup, W. B. 2015, Oncoimmunol., 4, e1001204.)
Table 1.
Differences in vessel structure and function between control and NG2 null mice#.
| Parameter | Change (versus control mice) | ||
|---|---|---|---|
| Germline-NG2ko mice | PC-NG2ko mice | Mac-NG2ko mice | |
| Vessel structure | |||
| Pericyte coverage of endothelial cells | 45% decrease | 33% decrease | 47% decrease |
| Detached pericyte | 200% increase | Unchanged | 181% increase |
| Vessel diameter | 20% decrease* | Unchanged | 53% decrease |
| Basal lamina assembly | 73% decrease | 31% decrease | 72% decrease |
| N-cadherin (endothelial cell) | N.D. | Unchanged | 74% decrease |
| N-cadherin (pericyte) | N.D. | Unchanged | 97% decrease |
| Vessel function | |||
| Vessel patency | 50% decrease | 43% decrease | 48% decrease |
| Vessel leakiness | 400% increase | 270% increase | 416% increase |
| Intratumoral hypoxia | 2000% increase | 560% increase | 1580% increase |
Change in vessel diameter of spontaneous mammary tumors between control and global-NG2 ko mice [23].
Reproduced with permission from Yotsumoto, F., You, W. K., Cejudo-Martin, P., Kucharova, K., Sakimura, K. and Stallcup, W. B. 2015, Oncoimmunol., 4, e1001204.
Tumor growth in My-NG2ko mice was retarded to an even greater degree than in PC-NG2ko mice [30] (Figure 2A, B). Interestingly, vascular deficits in tumors in My-NG2ko mice were more pronounced than in PC-NG2ko mice, providing a possible explanation for the greater reduction in tumor growth. In addition to reduced vessel patency and greater vessel leakiness, the diameter of tumor vessels in My-NG2ko mice was reduced by half, a phenomenon not seen in PC-NG2ko mice (Table 1; Figure 5). As a result of these vascular changes, tumor hypoxia was increased in My-NG2ko mice to a greater extent than in PC-NG2ko mice [30].
Figure 5. Pericyte ensheathment of endothelial cells in tumors in NG2 null mice.
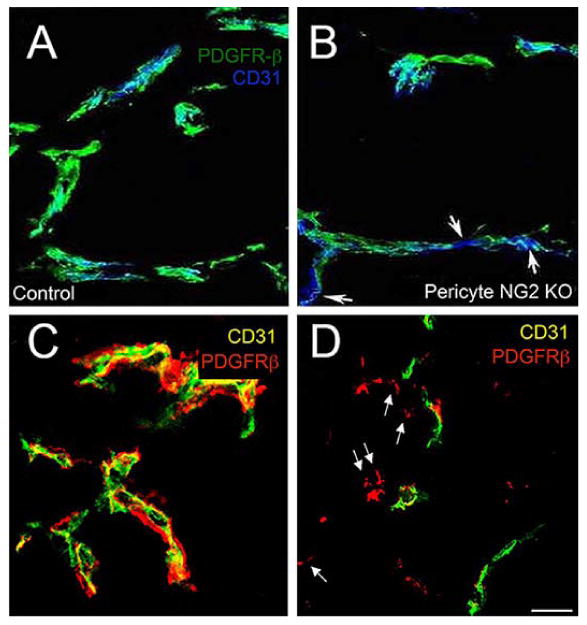
Vessels in intracranial B16F10 tumors were compared at 10 days in control (A) versus PC-NG2ko (B) mice, and in control (C) versus My-NG2ko (D) mice by immunolabeling sections for CD31 (endothelial cells) and PDGFRβ (pericytes). Pericyte (green) ensheathment of endothelial cells (blue) is incomplete in PC-NG2ko vessels (B), as judged by gaps in coverage (arrows). These gaps are not present in control vessels (A). In My-NG2ko vessels (D), many pericytes (red, arrows) fail to associate with endothelial cells (green). These detached pericytes are not seen in control vessels (C). These images also highlight the reduced diameter of vessels in My-NG2ko mice, a phenotype that is not observed in PC-NG2ko mice. Bar in D = 20 μm. (Reproduced with permission from You, W. K., Yotsumoto, F., Sakimura, K., Adams, R. H. and Stallcup, W. B. 2014, Angiogenesis, 17, 61 and from Yotsumoto, F., You, W. K., Cejudo-Martin, P., Kucharova, K., Sakimura, K. and Stallcup, W. B. 2015, Oncoimmunol., 4, e1001204.)
3.2. Spinal cord demyelination and remyelination
NG2 is frequently used as a marker for OPCs in studies of myelin damage and repair [31-34]. Importantly, our studies on lysolecithin-induced myelin damage in the spinal cord reveal that in addition to OPCs, NG2 is also expressed by macrophages and pericytes in demyelinated lesions [35]. It is therefore of importance to investigate the overall significance of NG2 expression in this model, for the sake of both NG2 utility as a marker and understanding NG2 functions in the various cell types. Our initial studies used microinjection of lysolecithin into the dorsal spinal cord white matter to compare myelin damage and repair in wild-type mice and germline NG2 null mice. Lesions in NG2 null mice were smaller at 1-week post-lysolecithin injection than lesions in wild-type mice. Nevertheless, myelin repair was retarded in the NG2 null animals, such that at 6-weeks post-injury remyelination was still incomplete. Germline ablation of NG2 resulted in reduced proliferation of OPCs, macrophages, and pericytes in demyelinated lesions at 1-week post-injury, such that the abundance of all 3 cell types was decreased in lesion sites.
Because OPCs and macrophages were the most abundant NG2-expressing cell types in lysolecithin-induced lesions, we used Cre-Lox methods to ablate NG2 in these two cell types. The My-NG2ko mice have been described above. OPC-specific NG2 null mice were generated by crossing NG2 floxed mice [24] with Olig2-Cre mice [36]. While lysolecithin-induced spinal cord lesions in OPC-NG2ko mice were similar in size at 1-week post-injury to lesions in control mice, repair of these lesions was retarded, due to reduced OPC proliferation that limited the production of myelinating oligodendrocytes (Table 2 lines 4, 5, 6). At 6-weeks post-injury, unmyelinated axons were still abundant in OPC-NG2ko mice, as judged by immunolabeling for myelin basic protein (MBP) and by high-resolution quantification in semi-thin sections of unmyelinated axons and myelin in lesions in OPC-NG2ko mice at 6-weeks post-thickness [37] (Figure 3B, E; Table 2, lines 7, 8). injury Table 2, line 9), emphasizing the importance Numbers of surviving neurons were also diminished in lesions in OPC-NG2ko mice at 6-weeks post-injury Table 2, line 9), emphasizing the importance of myelin for neuronal health.
Table 2.
Effects of cell type-specific ablation of NG2 on cell and axon biology.
| Control | OPC-NG2ko | My-NG2ko | ||
|---|---|---|---|---|
| Sham-operated animals | ||||
| 1 | Number of Olig2+ cells | 115.27 ± 3.8 | 111.02 ± 5.7 | 117.63 ± 5.4 |
| 2 | Number of Olig2+/PDGF-Rα+ OPCs | 14.55 ± 1.7 | 12.31 ± 2 | 14.46 ± 3.5 |
| 1 week after demyelination | ||||
| 3 | Number of Iba1+ macrophages/ microglia | 117.19 ± 22.3 | 120.1 ± 28.1 | 39.64 ± 19.8** |
| 4 | Number of Olig2+/PDGF-Rα+ OPCs | 42.92 ± 1.5 | 32.4 ± 1.7*** | 23.02 ± 7.2*** |
| 5 | Mitotic index of Olig2+/PDGF-Rα+ OPCs (%) | 22.91 ± 3.5 | 13.49 ± 1.5** | 11.93 ± 2.1*** |
| 6 weeks after demyelination | ||||
| 6 | Number of APC+ oligodendrocytes | 62.14 ± 6.7 | 44.48 ± 8.6* | 43.17 ± 4.1** |
| 7 | Well-myelinated axons | 1056 ± 133 | 480 ± 128** | 499 ± 80** |
| 8 | Myelin thickness (G-value) | 0.745 ± 0.089 | 0.861 ± 0.099*** | 0.87 ± 0.065*** |
| 9 | Number of NF-positive axons | |||
| Small: diameter < 1 μm | 1103 ± 60 | 660 ± 66*** | 812 ± 85** | |
| Medium: diameter = 1-2.5 μm | 201 ± 29 | 135 ± 10* | 118 ± 14* | |
| Large: diameter > 2.5 μm | 18 ± 3 | 6 ± 1** | 8 ± 2* | |
The key parameters for Olig2/PDGFRα-positive OPCs, Iba1-positive macrophages/microglia, and APC-positive oligodendrocytes were quantified by immunolabeling in lesions in control, OPC-NG2ko, and My-NG2ko mice (n = 4) for each group) from 1 to 6 weeks after lysolecithin microinjection into the dorsal spinal cord white matter (lines 1-6). Myelination parameters and axon diameters were determined in toluidine blue-stained semi-thin sections (lines 7-9). G-value = diameter of axon only/diameter of axon + myelin sheath. Cell and axon numbers are determined in volumes of 100,000 μm3. Values represent means ± SD. Statistically significant values evaluated by ANOVA and t-test are indicated by
P < 0.05,
P < 0.01,
P < 0.001.
(Reproduced with permission from Kucharova, K. and Stallcup, W. B. 2015, J. Neuroinflamm., 12, 161.)
Figure 3. Myelin repair in NG2 null mice.
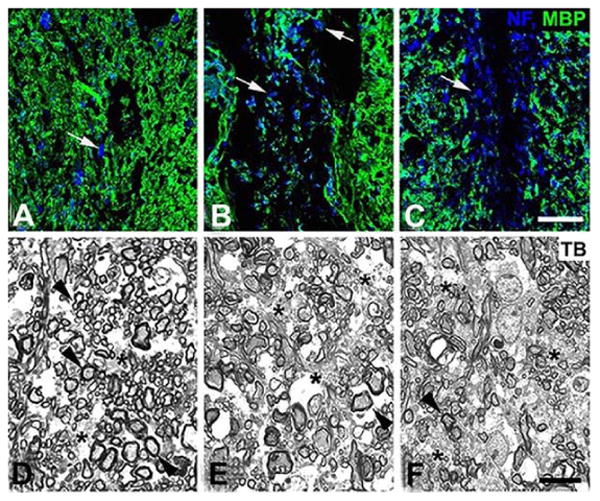
Six weeks after lysolecithin microinjection (1.5 μl of 1% lysolecithin) into the dorsal spinal cord white matter of control, OPC-NG2ko and My-NG2ko mice, animals were euthanized for determination of the extent of myelin repair. (A-C): Tissue sections were immunolabeled for myelin basic protein (MBP; green) and neurofilament protein (NF; blue), allowing visualization of myelinated axons and axons lacking association with myelin (arrows). (D-F): Toluidine blue-stained (TB) semi-thin sections enabled identification of well-myelinated axons (arrowheads) and unmyelinated axons (asterisks), along with determination of g-values for evaluating myelin thickness. Controls: A, D; OPC-NG2ko: B, E; My-NG2ko: C, F. Bar in C = 30 μm. Bar in F = 10 μm. (Reproduced with permission from Kucharova, K. and Stallcup, W. B. 2015, J. Neuroinflamm., 12, 161.)
In contrast to OPC-NG2ko mice, demyelinated lesions in My-NG2ko mice were reduced in size by almost 50% at 1-week post-injury compared to lesions in control mice. In spite of this reduced size, lesion repair was retarded in My-NG2ko mice, such that remyelination was still incomplete at 6-weeks post-injury. Surprisingly, OPC abundance was diminished in My-NG2ko mice to an extent at least as great as seen in OPC-NG2ko mice, even though OPCs still retain NG2 expression in the myeloid-specific knockouts (Table 2, lines 4, 5). This offers a possible explanation for diminished myelin repair in the My-NG2ko animals. As in OPC-NG2ko mice, immunolabeling for MBP and examination of semi-thin sections from 6-week My-NG2ko lesions revealed more unmyelinated axons, reduced myelin thickness, and fewer surviving axons than in control mice [37] (Figure 3C, F; Table 2, lines 7, 8, 9).
4. NG2-dependent macrophage recruitment
In a number of other cell types, NG2 promotes cell proliferation and/or motility [5, 29, 38-41]. It has therefore been of interest to investigate the effects of NG2 ablation on macrophage abundance in CNS lesions. While the decreased abundance of macrophages we observe in both brain tumors and demyelinated spinal cord lesions in My-NG2ko mice (described below) could be due to reduced proliferation, it could also be due to increased apoptosis, reduced responsiveness to macrophage recruitment signals such as M-CSF, CCL2, or CXCL12, or to diminished endothelial attachment and extravasation from the circulation into tissues. Our finding of reduced β1 and β2 integrin activation in NG2 knockdown THP-1 macrophages might be relevant to any of these mechanisms [12].
4.1. Intracranial melanomas
Pericyte-specific ablation of NG2 had no effect on either pericyte abundance or macrophage abundance in intracranial melanomas [29, 30]. In contrast, macrophage abundance in these melanomas was greatly diminished in My-NG2ko mice [30], as judged by immunolabeling for the macrophage markers F4/80, CD11b, and CD18. Figure 4A, B shows that a similar loss of macrophage recruitment was observed in intracranial melanomas in wild-type mice that had been previously gamma-irradiated and reconstituted with enhanced green fluorescent protein (EGFP)-labeled bone marrow from β-actin EGFP/germline NG2 null mice [30], demonstrating that loss of macrophage recruitment is independent of the method of NG2 ablation. The fact that the large majority of CD11b expressing cells in tumors in mice receiving wild-type bone marrow are positive for the EGFP marker further indicates that these tumor myeloid cells are macrophages rather than resident microglia. The fact that My-NG2ko mice do not exhibit decreased abundance of circulating monocytes [30] suggests that the reduction in tumor macrophage number is not due to deficits in generating monocytes.
Figure 4. Macrophage recruitment in NG2 null mice.
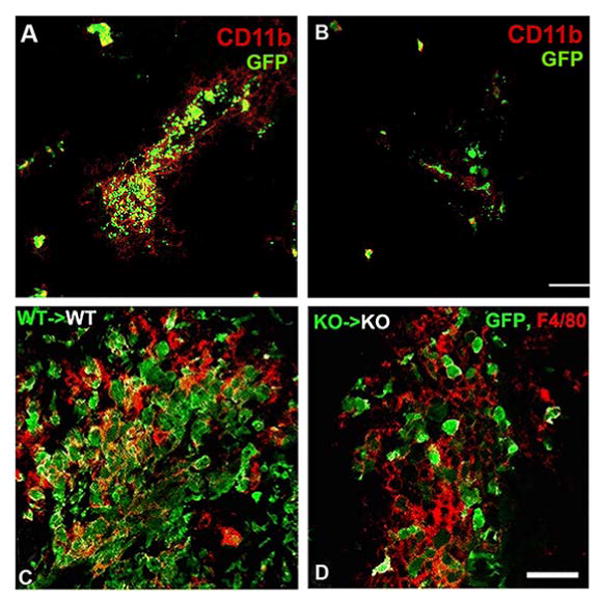
Gamma-irradiated wild-type mice were engrafted with bone marrow from wild-type β-actin-EGFP donors or with bone marrow from germline NG2 null β-actin-EGFP donors. After a 6-week recovery, chimeric mice were used for establishment of intracranial B16F10 tumors (A, B) and for lysolecithin-induced spinal cord demyelination (C, D). (A, B): At 10 days post-tumor initiation, animals were euthanized for analysis of macrophage infiltration into tumors. Immunolabeling for CD11b (red) was compared to the EGFP marker for identification of bone marrow-derived macrophages in tumors in mice receiving wild-type bone marrow (A) and mice receiving NG2 null bone marrow (B). Bar in B = 40 μm. (C, D): After a 6-week recovery, chimeric mice were used for lysolecithin induction of spinal cord demyelination. At 1-week post-injury, animals were euthanized for analysis of macrophage infiltration of demyelinated lesions. Immunolabeling for F4/80 (red) was compared to the EGFP marker for identification of bone marrow-derived macrophages in lesions in mice receiving wild-type bone marrow (C) and mice receiving NG2 null bone marrow (D). Bar in D = 20 μm. (Reproduced with permission from Yotsumoto, F., You, W. K., Cejudo-Martin, P., Kucharova, K., Sakimura, K. and Stallcup, W. B. 2015, Oncoimmunol., 4, e1001204 and from Kucharova, K. and Stallcup, W. B. 2015, J. Neuroinflamm., 12, 161.)
4.2. Demyelinated lesions in spinal cord
While OPC-specific ablation of NG2 had no effect on macrophage abundance in demyelinated spinal cord lesions, macrophage numbers were greatly reduced in lesions in My-NG2ko mice at 1-week post-injury [37] (Table 2, line 3). Demyelinated lesions in wild-type mice that had been previously gamma-irradiated and reconstituted with NG2 null EGFP-labeled bone marrow also exhibited this loss of macrophage abundance (Figure 4C, D), compared to wild-type mice that received wild-type EGFP-labeled bone marrow [37]. As in the case of brain tumors, the presence of the EGFP marker in more than 80% of F4/80-positive myeloid cells suggests that most of these lesional cells are bone marrow-derived macrophages rather than resident microglia.
5. Secondary effects of macrophages on other cell types
Macrophages are widely acknowledged to be an important source of factors that can affect other cells in the microenvironment, including tumor cells, OPCs, and vascular cells [42-48]. Our studies have implicated macrophage-derived signals in the biology of both pericytes (in brain tumors) and OPCs (in myelin repair).
5.1. Intracranial melanomas
It was noted above that vascular defects in tumors in PC-NG2ko mice are reflected by increased tumor hypoxia and reduced tumor growth. These vascular deficits appear to arise from a decrease in pericyte ensheathment of endothelial cells (Figure 5A, B), due to loss of pericyte NG2 activation of β1 integrin signaling in the endothelial cells [29]. This signaling deficit leads to reduced formation of intercellular junctions between endothelial cells and diminished assembly of the vascular basement membrane (Table 1). Pericyte maturation is also impaired by the loss of pericyte-endothelial cell interaction. These structural defects give rise to the deficits in patency and leakiness that are detected in tumor vessels.
Surprisingly, vascular defects are more pronounced in tumors in My-NG2ko mice than in PC-NG2ko mice [30]. Even though pericytes still retain NG2 expression in My-NG2ko mice, pericyte ensheathment of endothelial cells is nevertheless reduced to a greater degree than in PC-NG2ko mice. In fact, even though pericyte abundance is unaffected, many pericytes fail to interact at all with endothelial cells in the macrophage-deficient tumors [30] (Figure 5C, D). Significantly, expression of N-cadherin is reduced in these mice on both pericytes and endothelial cells, providing a possible explanation for decreased pericyte-endothelial cell interaction. Formation of endothelial junctions, assembly of the vascular basal lamina, and pericyte maturation are reduced to a greater extent in My-NG2ko mice than in PC-NG2ko mice (Table 1), suggesting mechanisms that underlie the functional deficits in these tumor vessels. We hypothesize that the reduced macrophage abundance in My-NG2ko tumors results in loss of a macrophage-derived signal that is required for promoting pericyte interaction with endothelial cells. This putative signal might be responsible for triggering N-cadherin expression in the two vascular cell populations as a means of promoting vessel formation. These observations are relevant to a growing number of reports on the importance of tumor macrophages for tumor vascularization [49, 50].
5.2. Demyelinated lesions in spinal cord
Based on previously known mechanisms of NG2 action [4, 5], it was predictable to us that OPC-specific ablation of NG2 would lead to reduced OPC proliferation, and thus to decreased generation of mature oligodendrocytes and diminished myelination [37]. Much more surprising, however, was the finding that myeloid-specific ablation of NG2 led to an equivalent or even greater decrease in OPC proliferation, and to comparable deficits in myelin repair. Two possible explanations for this effect are that reduced macrophage abundance in My-NG2ko lesions (a) diminishes clearance of myelin debris that is inhibitory to OPC proliferation, and/or (b) reduces production of macrophage-derived factors that promote OPC proliferation. Decreased macrophage abundance is most likely also responsible for the reduced myelin damage seen at 1-week post-injury in My-NG2ko mice. It will be of interest to investigate whether these two contrary aspects of macrophage impact on both myelin damage and repair are mediated by the same or distinct populations of macrophages.
6. Prospects and perspectives
Our work so far adds macrophages to the diverse list of cell types that express the NG2 proteoglycan. Our findings are consistent with the overall view that NG2 is not a marker for a specific cell lineage, but is expressed by cells that have been activated in some way, whether as part of a normal developmental program or as part of a response to pathological stimuli [5]. Macrophages appear to be an extreme example of the transience of NG2 expression, which is observed in many NG2-positive progenitors as these cells differentiate. The work reviewed here identifies NG2 as a marker for activated macrophages, and more importantly, establishes NG2 as an important player in macrophage colonization of sites of inflammation in the CNS. More work will be required to determine mechanisms by which NG2 promotes macrophage abundance in these sites (recruitment, extravasation, proliferation, survival) and to establish whether these findings can be generalized to include sites of inflammation in other pathologies. An additional topic of importance will be determining whether NG2 continues to influence macrophage function subsequent to infiltration into tissues. In our CNS models, these functions would include the production of factors that are important for OPC proliferation and for pericyte interaction with endothelial cells, but it can be imagined that NG2 might also be important for determining macrophage polarization toward immune-suppressive versus inflammatory phenotypes. Identification of factors that control the extreme phenotypic plasticity of macrophages is a key aspect of working with these myeloid cells.
Acknowledgments
We thank Regina Kapono for her help in preparing the manuscript. This work was supported by NIH grant RO1 CA95287 and by Sanford Burnham Prebys Lab Funding Initiative to WBS.
Abbreviations
- CCL2
chemokine ligand 2
- CNS
central nervous system
- Cre
Cre recombinase
- CXCL12
chemokine ligand 12
- EGFP
enhanced green fluorescent protein
- Flox
flanked by LoxP sites
- LPS
lipopolysaccharide
- Lys
lysozyme
- MBP
myelin basic protein
- M-CSF
macrophage colony stimulating factor
- MMTV-PyMT
polyoma middle T driven by mouse mammary tumor virus promoter
- My
myeloid
- NG2
nerve-glial antigen 2
- Olig2
oligodendrocyte transcription factor 2
- PC
pericyte
- PDGFRα
platelet-derived growth factor receptor alpha
- PDGFRβ
platelet-derived growth factor receptor beta
- Poly-I:C
poly-inosinic:polycytidylic acid
Footnotes
Conflict of Interest Statement: The authors declare that they have no conflicts of interest in this work.
References
- 1.Qian BZ, Pollard JW. Cell. 2010;141:39. doi: 10.1016/j.cell.2010.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Noy R, Pollard JW. Immunity. 2014;41:49. doi: 10.1016/j.immuni.2014.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.She ZG, Chang Y, Pang HB, Han W, Chen HZ, Smith JW, Stallcup WB. Arterioscler Thromb Vasc Biol. 2016;36:49. doi: 10.1161/ATVBAHA.115.306074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Stallcup WB. J Neurocytol. 2002;31:423. doi: 10.1023/a:1025731428581. [DOI] [PubMed] [Google Scholar]
- 5.Stallcup WB, Huang FJ. Cell Adh Migr. 2008;2:192. doi: 10.4161/cam.2.3.6279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bu J, Akhtar N, Nishiyama A. Glia. 2001;34:296. doi: 10.1002/glia.1063. [DOI] [PubMed] [Google Scholar]
- 7.de Castro R, Jr, Tajrishi R, Claros J, Stallcup WB. Exp Neurol. 2005;192:299. doi: 10.1016/j.expneurol.2004.11.027. [DOI] [PubMed] [Google Scholar]
- 8.Jones LL, Yamaguchi Y, Stallcup WB, Tuszynski MH. J Neurosci. 2002;22:2792. doi: 10.1523/JNEUROSCI.22-07-02792.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Larsen PH, Wells JE, Stallcup WB, Opdenakker G, Yong VW. J Neurosci. 2003;23:11127. doi: 10.1523/JNEUROSCI.23-35-11127.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tigges U, Hyer EG, Scharf J, Stallcup WB. Development. 2008;135:523. doi: 10.1242/dev.002071. [DOI] [PubMed] [Google Scholar]
- 11.Gao Q, Lu J, Huo Y, Baby N, Ling EA, Dheen ST. Neurosci. 2010;165:386. doi: 10.1016/j.neuroscience.2009.10.022. 2010. [DOI] [PubMed] [Google Scholar]
- 12.Stallcup WB, You WK, Kucharova K, Cejudo-Martin P, Yotsumoto F. Microcirc. 2016;23:122. doi: 10.1111/micc.12251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ligon KL, Alberta JA, Kho AT, Weiss J, Kwaan MR, Nutt CL, Louis DN, Stiles CD, Rowitch DH. J Neuropathol Exp Neurol. 2004;63:509. doi: 10.1093/jnen/63.5.499. [DOI] [PubMed] [Google Scholar]
- 14.Al-Mayhani MT, Grenfell R, Narita M, Piccirillo S, Kenney-Herbert E, Fawcett JW, Collins VP, Ichimura K, Watts C. Neuro-oncol. 2011;13:830. doi: 10.1093/neuonc/nor088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chekenya M, Krakstad C, Svendsen A, Netland IA, Staalesen V, Tysnes BB, Selheim F, Wang J, Sakariassen PO, Sandal T, Lonning PE, Flatmark T, Enger PO, Bjerkvig R, Sioud M, Stallcup WB. Oncogene. 2008;27:5182. doi: 10.1038/onc.2008.157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Persson AI, Petritsch C, Swartling FJ, Itsara M, Sim FJ, Auvergne R, Goldenberg DD, Vandenberg SR, Nguyen KN, Yakovenko S, Ayers-Ringler J, Nishiyama A, Stallcup WB, Berger MS, Bergers G, McKnight TR, Goldman SA, Weiss WA. Cancer Cell. 2010;18:669. doi: 10.1016/j.ccr.2010.10.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Price MA, Colvin Wanshura LE, Yang J, Carlson J, Xiang B, Li G, Ferrone S, Dudek AZ, Turley EA, McCarthy JB. Pigment Cell Melanoma Res. 2011;24:1148. doi: 10.1111/j.1755-148X.2011.00929.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang J, Svendsen A, Kmiecik J, Immervoll H, Skaftnesmo KO, Planaguma J, Reed RK, Bjerkvig R, Miletic H, Enger PO, Rygh CB, Chekenya M. PloS ONE. 2011;6:e23062. doi: 10.1371/journal.pone.0023062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yadavilli S, Hwang EI, Packer RJ, Nazarian J. Trans Oncol. 2016;9:63. doi: 10.1016/j.tranon.2015.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fidler IJ. Cancer Res. 1975;35:21. 1975. [PubMed] [Google Scholar]
- 21.Huang FJ, You WK, Bonaldo P, Seyfried TN, Pasquale EB, Stallcup WB. Dev Biol. 2010;344:1035. doi: 10.1016/j.ydbio.2010.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Grako KA, Ochiya T, Barritt D, Nishiyama A, Stallcup WB. J Cell Sci. 1999;112:905. doi: 10.1242/jcs.112.6.905. [DOI] [PubMed] [Google Scholar]
- 23.Gibby K, You WK, Kadoya K, Helgadottir H, Young LJ, Ellies LG, Chang Y, Cardiff RD, Stallcup WB. Breast Cancer Res. 2012;14:R67. doi: 10.1186/bcr3174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chang Y, She ZG, Sakimura K, Roberts A, Kucharova K, Rowitch DH, Stallcup WB. PloS ONE. 2012;7:e30637. doi: 10.1371/journal.pone.0030637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Foo SS, Turner CJ, Adams S, Compagni A, Aubyn D, Kogata N, Lindblom P, Shani M, Zicha D, Adams RH. Cell. 2006;124:161. doi: 10.1016/j.cell.2005.10.034. [DOI] [PubMed] [Google Scholar]
- 26.Stenzel D, Nye E, Nisancioglu M, Adams RH, Yamaguchi Y, Gerhardt H. Blood. 2009;114:915. doi: 10.1182/blood-2008-10-186239. [DOI] [PubMed] [Google Scholar]
- 27.Clausen BE, Burkhardt C, Reith W, Renkawitz R, Forster I. Transgenic Res. 1999;8:265. doi: 10.1023/a:1008942828960. [DOI] [PubMed] [Google Scholar]
- 28.Stockmann C, Doedens A, Weidemann A, Zhang N, Takeda N, Greenberg JI, Cheresh DA, Johnson RS. Nature. 2008;456:814. doi: 10.1038/nature07445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.You WK, Yotsumoto F, Sakimura K, Adams RH, Stallcup WB. Angiogenesis. 2014;17:61. doi: 10.1007/s10456-013-9378-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yotsumoto F, You WK, Cejudo-Martin P, Kucharova K, Sakimura K, Stallcup WB. Oncoimmunol. 2015;4:e1001204. doi: 10.1080/2162402X.2014.1001204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chang A, Nishiyama A, Peterson J, Prineas J, Trapp BD. J Neurosci. 2000;20:6404. doi: 10.1523/JNEUROSCI.20-17-06404.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Watanabe M, Toyama Y, Nishiyama A. J Neurosci Res. 2002;69:826. doi: 10.1002/jnr.10338. [DOI] [PubMed] [Google Scholar]
- 33.Boda E, Buffo A. Frontiers Neurosci. 2014;8:122. doi: 10.3389/fnins.2014.00122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kang Z, Wang C, Zepp J, Wu L, Sun K, Zhao J, Chandrasekharan U, DiCorleto PE, Trapp BD, Ransohoff RM, Li X. Nature Neurosci. 2013;16:1401. doi: 10.1038/nn.3505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kucharova K, Chang Y, Boor A, Yong VW, Stallcup WB. J Neuroinflamm. 2011;8:158. doi: 10.1186/1742-2094-8-158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Schuller U, Heine VM, Mao J, Kho AT, Dillon AK, Han YG, Huillard E, Sun T, Ligon AH, Qian Y, Ma Q, Alvarez-Buylla A, McMahon AP, Rowitch DH, Ligon KL. Cancer Cell. 2008;14:123. doi: 10.1016/j.ccr.2008.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kucharova K, Stallcup WB. J Neuroinflamm. 2015;12:161. doi: 10.1186/s12974-015-0385-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kucharova K, Stallcup WB. Neurosci. 2010;166:185. doi: 10.1016/j.neuroscience.2009.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kadoya K, Fukushi J, Matsumoto Y, Yamaguchi Y, Stallcup WB. J Histochem Cytochem. 2008;56:295. doi: 10.1369/jhc.7A7349.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Makagiansar IT, Williams S, Mustelin T, Stallcup WB. J Cell Biol. 2007;178:155. doi: 10.1083/jcb.200612084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Makagiansar IT, Williams S, Dahlin-Huppe K, Fukushi J, Mustelin T, Stallcup WB. J Biol Chem. 2004;279:55262. doi: 10.1074/jbc.M411045200. [DOI] [PubMed] [Google Scholar]
- 42.Franklin RJ, Hinks GL. J Neurosci Res. 1999;58:207. [PubMed] [Google Scholar]
- 43.Hinks GL, Franklin RJ. Mol Cell Neurosci. 1999;14:153. doi: 10.1006/mcne.1999.0771. [DOI] [PubMed] [Google Scholar]
- 44.Mason JL, Ye P, Suzuki K, D’Ercole AJ, Matsushima GK. J Neurosci. 2000;20:5703. doi: 10.1523/JNEUROSCI.20-15-05703.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Miron VE, Boyd A, Zhao JW, Yuen TJ, Ruckh JM, Shadrach JL, van Wijngaarden P, Wagers AJ, Williams A, Franklin RJ, Ffrench-Constant C. Nature Neurosci. 2013;16:1211. doi: 10.1038/nn.3469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bonde AK, Tischler V, Kumar S, Soltermann A, Schwendener RA. BMC Cancer. 2012;12:35. doi: 10.1186/1471-2407-12-35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Fan QM, Jing YY, Yu GF, Kou XR, Ye F, Gao L, Li R, Zhao QD, Yang Y, Lu ZH, Wei LX. Cancer Lett. 2014;352:160. doi: 10.1016/j.canlet.2014.05.008. [DOI] [PubMed] [Google Scholar]
- 48.Rymo SF, Gerhardt H, Wolfhagen Sand F, Lang R, Uv A, Betsholtz C. PloS One. 2011;6:e15846. doi: 10.1371/journal.pone.0015846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Coffelt SB, Lewis CE, Naldini L, Brown JM, Ferrara N, De Palma M. Am J Pathol. 2010;176:1564. doi: 10.2353/ajpath.2010.090786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.De Palma M, Venneri MA, Galli R, Sergi L, Politi LS, Sampaolesi M, Naldini L. Cancer Cell. 2005;8:211. doi: 10.1016/j.ccr.2005.08.002. [DOI] [PubMed] [Google Scholar]