

Abstract
Background
The interplay between viral infection and alloimmunity is known to influence the fate of transplanted organs. Clarifying how local virus-associated inflammation/injury and antiviral immunity can alter host alloimmune responses in transplantation remains a critical question.
Methods
We used a mouse model of polyomavirus (PyV) infection and kidney transplantation to investigate the roles of direct viral pathology, the antiviral immune response, and alloimmunity in the pathogenesis of PyV-associated allograft injury. We have previously shown that an effective primary T cell response is required in PyV-associated graft injury.
Results
Here we show that the transfer of primed antidonor, but not antiviral, T cells results in PyV-associated allograft injury. In further studies, we use a surrogate minor antigen model (ovalbumin) and show that only antidonor specific T cells and not antiviral specific T cells are sufficient to mediate injury. Lastly, we demonstrate that local but not systemic virus-mediated inflammation and injury within the graft itself are required.
Conclusions
These data suggest that in this mouse model, the predominant mechanism of allograft injury in PyV-associated injury is due to an augmented alloimmune T cell response driven by virus-induced inflammation/injury within the graft. These studies highlight the important interplay between viral infection and alloimmunity in a model system.
The concept of heterologous immunity and the interplay between viral infection, whether acute or persistent, and the initiation of a distinct antigen-specific immune response has generated considerable interest in transplantation. There is substantial literature outlining the relationship between previous viral infection and the subsequent immune response to transplanted tissues.1-9 Heterologous immunity poses a significant barrier to transplant tolerance,10-14 and a further understanding of the mechanisms by which previous and concurrent viral infections give rise to transplant rejection remains a critical area of study.1,15-19 The tissue tropism of a given viral infection in the host is also an interesting facet of this interplay, because recent evidence has shown that the tropic nature of certain viral infections plays a crucial role in the development of clinical disease.20
BK virus is a human polyomavirus (PyV) that is primarily and increasingly associated with renal transplantation and its attendant requirement for immunosuppression.21-24 BKV-associated nephropathy (BKVN), or more generally speaking, polyomavirus-associated allograft nephropathy (PVAN), affects up to 10% of renal transplant recipients and has been implicated in resultant graft loss in up to 7% of cases.19,25-28 Despite its growing recognition as an important clinical problem, much of the mechanistic pathogenesis remains unknown.29,30 We have previously described a mouse model of PyV-associated allograft injury, in which acute infection with mouse polyomavirus (MPyV) and transplantation of an allogeneic kidney resulted in allograft loss.31 Further studies in this model revealed that the adaptive immune response, and not viral cytopathology, was responsible for rejection.32 These studies established the relationship between alloimmunity and local PyV-mediated allograft injury.
In this study, we further investigate the role of the adaptive immune response in the context of viral infection for PyV-mediated rejection of kidney allografts in the mouse model. Using adoptive transfer of primed T cells into mice incapable of generating a primary T cell response as well as an antigen-specific transplant model, we demonstrate that donor-reactive T cells, and not antiviral T cells, are necessary and sufficient to mediate rejection in the presence of local viral infection. We also find that this is not a consequence of generalized viral inflammation or acute infection, but rather infection of the graft itself. Furthermore, graft-infiltrating alloantigen-specific T cells in infected mice were more likely to have an effector phenotype compared with T cells in uninfected recipients. These data suggest a mechanism in which viral-induced local inflammation within the kidney augments the alloimmune response, leading to PyV-mediated rejection of the allograft. These studies describe a key role for tissue-localized viral infection promoting antidonor T cell activation and effector function that results in rejection.
MATERIALS AND METHODS
Mice and Kidney Transplantation
C3H/HeJ (H-2k), C57BL/6 (H-2b), and B6C3F1 mice were purchased from The Jackson Laboratory (Bar Harbor, Maine). Alymphoplasia (aly/aly) mice on the B6 background were obtained from F. Lakkis (University of Pittsburgh). C57BL/6-Tg(CAG-OVA)916Jen/J mice were used33 (University of Minnesota). Kidney transplants were performed in 8- to 12-week-old male mice as previously described.32 There were no significant differences in perioperative mortality between groups, and overall surgical mortality was less than 10%. All transplanted mice were observed for 48 hours after transplant to monitor for immediate complications from surgical technique. All transplantation and procedures were performed in accordance with the Institutional Animal Care and Use Committee of Emory University.
MPyV Infection
MPyV was prepared as previously described.34 On day 1 posttransplantation, mice received 1.5 × 106 plaque-forming units of MPyV in hind footpads, unless noted otherwise. The construction of recombinant MPyV virus carrying the SIINFEKL epitope embedded in the middle T open reading frame (MPyV.OVAI) has been described elsewhere.35
Murine Herpesvirus 68 Infection
Murine herpesvirus 68 (MHV-68) was obtained from S. Speck (Emory University), prepared and administered as previously described.36 Briefly, B6 mice received 1.0 × 105 plaque-forming units of MHV-68 intraperitoneally on day 1 posttransplantation.
T Cell Purification and Transfer
For adoptive transfer experiments using antiviral T cells, splenocytes were harvested from C57BL/6 mice 8 days after MPyV infection. For those using allospecific T cells, spleens or draining lymph nodes were isolated from C57BL/6 mice 14 days after placement of a C3H/HeJ skin graft. T cells were purified by positive selection using an AutoMACS (anti-CD90.1) (Thy1.1)-coated microbeads; Miltenyi Biotec). A total of 1 × 107 T cells were transferred on day 1 or 2 days before transplantation.
OT-I T Cell Transfer
Bulk splenocytes from OT-I mice were assayed for CD8 and Valpha2 expression via flow cytometry.37 A total of 3.5 × 106 OT-I T cells (transgenic CD8+ T cells specific for chicken ovalbumin [OVA]) were transferred 1 or 2 days before transplantation.
Creatinine Measurements
To assess renal function, the creatinine (Cr) concentration in plasma was measured using the modified kinetic Jaffe reaction, as previously reported. The baseline level of mouse serum creatinine is approximately 0.2 mg/dL, as reported by our group and others.31,38
Real-Time Polymerase Chain Reaction for MPyV DNA
Taqman real-time polymerase chain reaction was used to quantify genome copies of MPyV, as previously described.39 The following primers were used: forward primer 5′-CGC ACA TAC TGC TGG AAG AAG A-3′ corresponding to nt 1040 to 1061 of the PyV A2 strain genomic sequence; reverse primer 5′-TCT TGG TCG CTT TCT GGA TAC AG-3′ corresponding to nt 1120 to 1142. The limit of detection is 10 copies of genomic viral DNA per mg of tissue.
IFNγ Production Measurement
Intracellular fixation and staining for IFNγ was performed on cells per the provided instructions from an available Intracellular Cytokine kit (BD Biosciences, San Jose, CA). SIINFEKL peptide was used for stimulation.
Histologic Evaluation
Kidney sections were fixed in 10% neutral-buffered formalin and embedded in paraffin. Serial 4-μm sections were stained with Harris hematoxylin and eosin (H&E).
Statistical Analysis
For survival, significance was evaluated using the log-rank test. For viral loads and serum Cr levels, significance was evaluated using either a 2-tailed Mann-Whitney U test or the Kruskal-Wallis test and Dunn multiple-comparison procedure, if applicable. These calculations were performed with Prism statistical software (GraphPad, La Jolla, CA). A P value less than 0.05 was considered significant.
RESULTS
Primed Allospecific But Not Antiviral T Cells Mediate PyV-Associated Allograft Injury
We have previously demonstrated the requirement of a primary T cell response in PyV-associated graft injury/rejection.32 One limitation with this model was the inability to distinguish the contributions of the alloimmune versus the antiviral immune responses. To further delineate which T cell population was responsible for the allograft injury, we performed adoptive transfer experiments in which splenectomized aly/aly mice were transplanted with C3B6F1 kidneys and infected by MPyV on day 1 posttransplantation (Figure 1A). One day before transplant, we transferred 1 × 107 T cells from a WT B6 that 14 days earlier had received a C3H skin graft (allospecific T cells) or 1 × 107 T cells from a WT B6 mouse that was inoculated with MPyV 2 weeks earlier (antiviral T cells).
FIGURE 1.
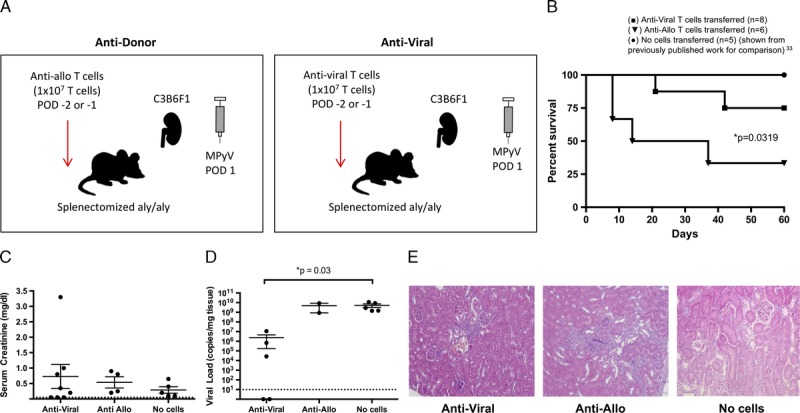
An augmented alloimmune response, but not an augmented antiviral response, promotes PyV-associated allograft injury. A, Experimental model for antidonor and antiviral adoptive T cell transfer in the splenectomized aly/aly mouse. C3B6F1 donor kidneys were transplanted into B6 aly/aly recipients with splenectomy and bilateral nephrectomy. Mice were infected by MPyV on day 1 posttransplantation. T cells were transferred on days −2 or −1 pretransplantation. B, Survival of transplanted mice receiving no cells (n = 5), anti-allo cells (n = 6), or antiviral cells (n = 8). C, Serum creatinine at day 60 or time of death and (D) viral load in kidneys at day 60 or time of death. (The number of data points per group is decreased from the initial survival curves due to mouse death before samples could be obtained). Dots represent individual mice. Dashed lines indicate limits of detection. E, Representative histology at day 60 or time or death. Although the transfer of antiviral T cell shows expected infiltration, the transfer of anti-allo T cells shows the greatest degree of histologic damage (H&E staining; original magnification, ×400).
Compared with a control cohort where splenectomized aly/aly mice were transplanted and inoculated with MPyV alone, only the transfer of allospecific T cells, and not the transfer of antiviral T cells, caused a significant increase in PyV-associated injury and death as evidenced by percent survival by day 60 (Figure 1B). The PyV-associated injury was graded by histologic analysis (% infiltrate and tubulitis); however, due to the difficulty in predicting death in mice, serial creatinine values in the preterminal period were not possible. The transfer of antiviral T cells did result in a significant decrease in viral load (indicating that the antiviral cells were functional) with a mild increase in serum creatinine levels, indicative of nonlethal kidney dysfunction (Figure 1C). Correspondingly, the transfer of allospecific cells resulted in uncontrolled high viral loads similar in number to inoculated mice who did not receive any T cell transfer (Figure 1D). The transfer of allospecific T cells also resulted in a more severe degree of injury as assessed by histology (Figure 1E). These data suggest that the alloimmune response and not the antiviral response is required for PyV-associated injury in this mouse model and confirm our previous finding that high viral loads themselves are not associated with allograft injury, excluding a role for directly mediated viral injury in the absence of other effector mechanisms.
Antigen-Specific Antidonor T Cells Are Required for PyV-Associated Allograft Rejection
Although the experiments described above using aly/aly mice allowed us to distinguish the impact of the allospecific and antiviral T cell response, the transfer of bulk T cell populations limited our ability to normalize both numbers and activation status between the allospecific and antiviral groups. More importantly, with the adoptive transfer of the antiviral T cells, there may have been introduction of some allo-cross-reactive T cells limiting our ability to isolate the effects of the antiviral response.40 To better control for variables in the transferred cell population, we performed adoptive transfer experiments using a well-characterized surrogate minor antigen transplant model system that uses tissue obtained from mice that express chicken OVA and OVA-specific OT-I T cell receptor-transgenic CD8+ T cells.41 In 1 set of experiments, donor kidneys were harvested from mice genetically engineered to express membrane-bound OVA (mOVA) under the β-actin promoter such that it is expressed on the surface of all tissues including the transplanted kidney. In this group of experiments, the transferred OT-I T cells were effectively antidonor T cells. In the other set of experiments, a recombinant MPyV in which the gene for OVA was inserted in the middle T reading frame was used so that all virally infected cells express OVA (MPyV.OVAI). In this latter experiment, the transferred OT-I cells were functionally antiviral. Therefore, although the actual antigenic peptide (SIINFEKL) remains the same, the method of antigen delivery differs—donor kidney tissue versus viral infection. In this model, we can finely control the number of transferred antigen-specific T cells, all primed by the same antigen, to ensure uniformity between the 2 groups.
To assess the contribution of antidonor OT-I T cells to PyV-associated allograft injury in this model, an mOVA kidney on the B6 background was transplanted into a B6 mouse that had undergone bilateral native nephrectomies (Figure 2A). Mice were infected with MPyV on day 1 posttransplantation and the OT-I T cells were transferred a day before transplant. In the presence of both OT-I T cells and virus (but not either one alone), there was PyV-associated rejection of the mOVA kidneys (Figure 2B). Correspondingly, these mice had elevated serum creatinines and histologic evidence of increased allograft damage at the time of death (Figures 2C-E). In agreement with previously published reports,32 we saw no correlation between viral load and survival.
FIGURE 2.
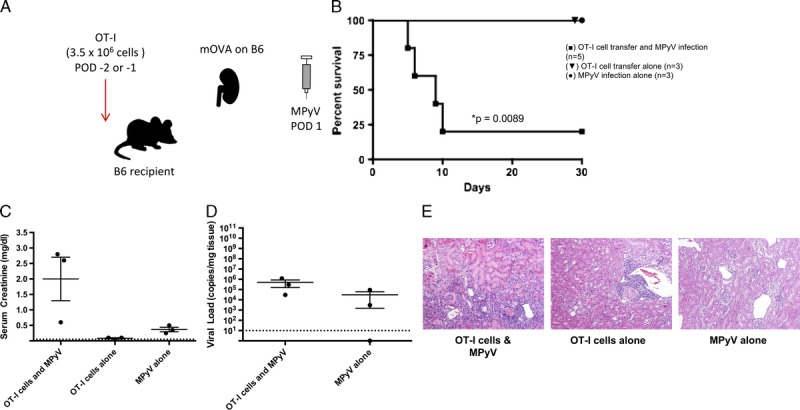
Antidonor CD8+ T cells and MPyV are both required to cause rejection. A, Experimental model for functional antidonor adoptive OT-I T cell transfer in the B6 mouse. B6 mOVA donor kidneys were transplanted into B6 recipients with bilateral nephrectomy. Mice were infected by MPyV on day 1 posttransplantation. OT-I T cells were transferred on days −2 or −1 pretransplantation. B, Survival of mice receiving MPyV infection alone (n = 3), OT-I cell transfer alone (n = 3), or both MPyV infection and OT-I cell transfer (n = 5). C, Serum creatinine at day 30 or time of death and (D) viral load in kidneys at day 30 or time of death. (The number of data points per group is decreased from initial survival curves due to mouse death before samples could be obtained). Dots represent individual mice. Dashed lines indicate limits of detection. E, Representative histology. OT-I T cell transfer and viral infection together result in a greater degree of histologic damage than either condition alone (H&E staining; original magnification, ×400).
To assess the contribution of antiviral T cells to allograft injury, B6 kidneys from donors previously infected with a recombinant MPyV.OVAI were transplanted into B6 mice passively immunized with a neutralizing VP1 mAb,42 with OT-I T cell transfer the day before transplant (Figure 3A). Passive immunization of the recipients was necessary to avoid systemic dissemination of viral infection, but such immunization does not prevent allograft dysfunction and rejection in this mouse model (data not shown). Transfer of functional antiviral OT-I T cells resulted in 100% survival, low serum creatinines, and less histologic evidence of allograft damage (Figures 3B-D). Taken together, these data support the conclusions drawn from the previous experiments in aly/aly mice; namely, that the mechanism of allograft dysfunction and rejection in the mouse model of PyV-associated allograft injury is a consequence of the antidonor, and not the antiviral, immune response.
FIGURE 3.
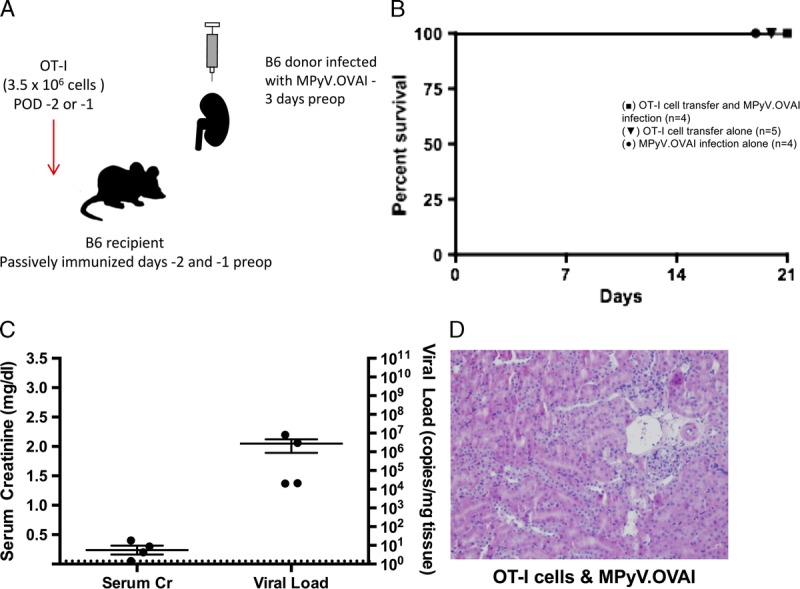
Antiviral OT-I T cells and MPyV are insufficient to cause rejection. A, Experimental model for functional antiviral adoptive OT-I T cell transfer in the B6 mouse. B6 donors were infected with a recombinant version of MPyV recognized by OT-I T cells (MPyV.OVAI), and the recipients passively immunized with VP-1 before transplant to prevent the dissemination of viral infection confounding the study. OT-I T cells were transferred to B6 recipients before transplant with the MPyV.OVAI B6 kidney. B, Survival of mice receiving MPyV.OVAI infection and OT-I cell transfer (n = 4), OT-I cell transfer alone (n = 5), or MPyV.OVAI infection alone (n = 4). C, Serum creatinine and viral load in kidneys at day 21. Dots represent individual mice. Dashed lines indicate limits of detection. D, Representative histology. OT-I T cell transfer and viral infection together result in infiltration and subclinical histologic damage (H&E staining; original magnification, ×400).
MPyV Infection Enhances the Effector Function of Antidonor T Cells Within the Graft and Spleen
To explore the effect of viral infection on the functional phenotype of antidonor T cells, mOVA kidneys were transplanted into B6 recipients and either infected on postoperative day 1 with MPyV or left uninfected. As with previous experiments, OT-I T cells were transferred to the recipients the day before transplant. On post-operative day 7, recipients were sacrificed and kidney and spleen tissue were processed for flow cytometric analysis. We found that in the presence of MPyV infection, the number of these antidonor T cells that exhibited effector function was significantly higher in the infected graft (Figure 4). The number of antidonor effector T cells was higher in the infected spleens as well (Figure 4), perhaps because of ongoing allograft rejection induced by tropic infection. These data confirm that the allospecific cells that are present in MPyV infected transplant recipients have significantly higher effector function as evidenced by their increased production of IFNγ after peptide stimulation. Future studies could include additional cytotoxicity assays and intracellular staining for increased production of other cytokines.
FIGURE 4.
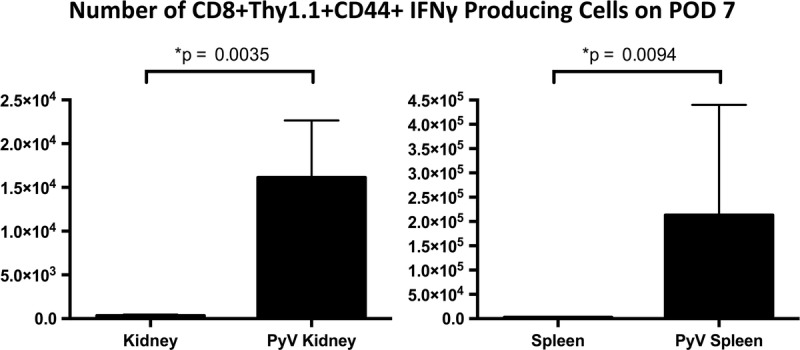
Infection with MPyV enhances the effector function of anti–alloantigen-specific OT-I T cells. mOVA kidneys were transplanted into B6 recipient mice which had been given adoptive transfers of OT-I T cells pretransplant. One group of transplanted mice were infected on POD 1 with MPyV; the other group was left uninfected. The number of effector allospecific cells present in the MPyV-infected transplant recipients is significantly higher as evidenced by their increased production of IFNγ with SIINFEKL stimulation. Median values ± SEM shown.
Systemic Viral Infection Without Local Graft Involvement Fails to Promote Rejection
Acute viral infections can affect local and systemic environments and result in altered pathology. In both humans and mice, it has been demonstrated that acute infections result in decreased graft survival.43-45 To determine whether the kidney allograft rejection we observed was not due to generalized inflammation or another consequence of acute viral infection, we substituted MHV-68 for MPyV. MHV-68, like MPyV, is endemic in mice, but lacks tropism for the kidney, initially replicating in lung tissue and then establishing latency in B cells.46 For these experiments, we transplanted a B6 mOVA kidney into a nephrectomized B6 mouse. OT-I cells were adoptively transferred approximately 1 day before transplantation, and mice were infected with MHV-68 1 day posttransplantation.
Under these same conditions, we found that 80% of mice infected with MPyV suffered rejection (Figure 2). In sharp contrast to the high rejection rate observed with MPyV, the recipient mice infected with MHV-68 exhibited 100% allograft survival (Figure 5A). Correspondingly, these mice showed serum creatinine levels consistent with uninfected recipients (Figure 5B), and the allografts displayed less histologic evidence of injury compared with those that received MPyV infection (Figure 5C). Although we cannot rule out the possibility that another viral infection may recapitulate the results seen with MPyV, these data indicate that a generalized acute viral infection without allograft involvement is insufficient to provoke immunologic injury of the renal allograft.
FIGURE 5.
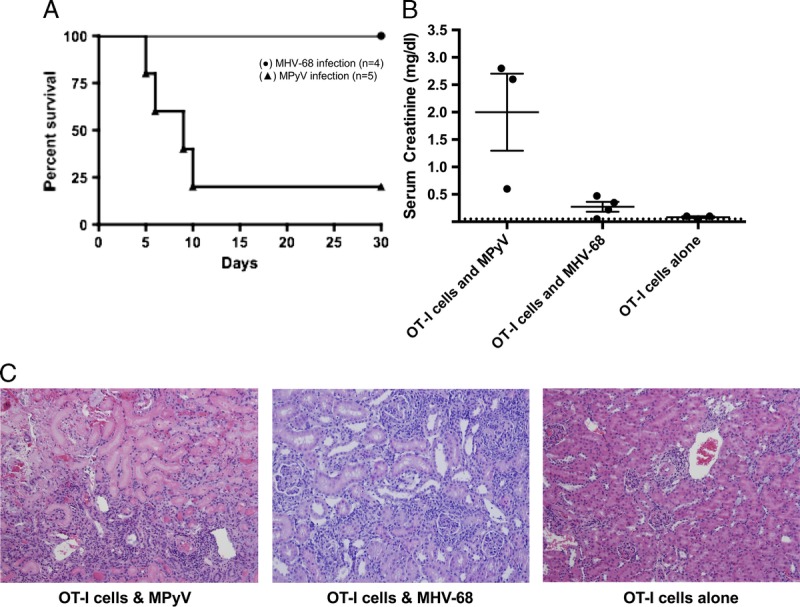
Viral infection lacking kidney tropism does not recapitulate the PyV-associated allograft injury phenotype seen in MPyV infection. B6 mOVA donor kidneys were transplanted into B6 recipients with bilateral nephrectomy. Mice were preoperatively given OT-I cells and infected on day 1 posttransplantation. Mice were infected with either MHV-68 or MPyV on day 1 posttransplantation. MPyV data are from Figure 2 and are shown for comparison. A, Survival. (●) MHV-68 infection (n = 4), (▲) MPyV infection (n = 5). B, Serum creatinine at time of death or day 30. Dots represent individual mice. Dashed line indicates limit of detection. C, Representative histology at day 30. OT-I T cell transfer and MHV-68 infection result is subclinical damage with minimal infiltration (H&E staining; original magnification, ×400).
DISCUSSION
The interplay between PyV infection, alloimmunity, and the fate of transplanted kidneys remains an area of interest to investigators and importance to clinicians. In the clinical realm, PyV infection in kidney transplant recipients continues to pose significant problems related to morbidity, premature loss of transplanted kidneys, and issues related to the expense and logistics of patient monitoring.47 It has been reported that polyomavirus-associated allograft nephropathy affects a significant proportion (1-10%) of renal transplant patients and may lead to allograft loss if it is not recognized and treated early.28,48 Despite the observed clinical impact of PVAN, the mechanisms by which it contributes to allograft injury are poorly understood.
Previous work from our group established a mouse model of PyV-associated allograft injury and used that model to demonstrate that the adaptive immune response (and not viral mediated injury alone) was essential for PyV-mediated allograft injury. The studies reported herein extend these findings, implicating the predominant role of the antidonor immune response as opposed to the antiviral immune response in the pathogenesis of PyV-associated injury. Support for this conclusion comes from the use of alymphoplasia mice, where only transfer of antidonor T cells is sufficient to cause allograft rejection. In addition, we used a more elegant surrogate minor antigen mOVA system, where OT-I T cell transfer recapitulates allograft injury if and only if the cells are directed against antigens expressed directly by the kidney allograft. Finally, the percentage of antidonor cells with an effector phenotype was significantly higher in the presence of PyV infection.
Because this model requires acute infection with MPyV, we wanted to rule out the possibility that the observed phenotype was simply a general consequence of an acute systemic infection at the time of transplantation. To investigate this possibility, we chose MHV-68, a mouse EBV homologue, because it lacks tropism for the kidney but still results in a systemic infection and inflammation.49-51 As seen in Figure 5, infection with MHV-68, in place of MPyV, does not result in substantial functional or histologic evidence of allograft injury. These data suggest that circulating inflammatory molecules and other systemic changes associated with acute infection do not contribute significantly to the pathobiology of PyV-associated allograft injury; rather, it is the local environment of the kidney that must be compromised to promote disease. In support of this conclusion, it has been reported that renal transplant patients with PVAN demonstrate an extremely high level of proinflammatory transcripts in their allografts, greater in magnitude than what is normally seen in cases of acute rejection.52-55
Taken together, these data allow us to propose a mechanism for the pathogenesis of PyV-associated allograft injury (Figure 6). The PyV-mediated rejection of a transplanted kidney requires both viral infection and an antidonor T cell response. It is well established that mice tolerate an allogeneic kidney transplant in the absence of infection,56 so the unmodified alloimmune response generated against the allograft is insufficient to cause rejection. Our previously published data also indicate that direct viral cytopathology alone is insufficient for irreversible graft injury in mice. This is demonstrated by the aly/aly experiment, in which the absence of the adaptive immune response results in survival despite high viral loads. With support from our MHV-68 data, we therefore conclude that the mechanism of action by which MPyV contributes to allograft injury involves localized inflammation driven by viral infection in the kidney. This environment potentiates the alloimmune response such that it becomes sufficiently potent to cause irreversible kidney injury.
FIGURE 6.
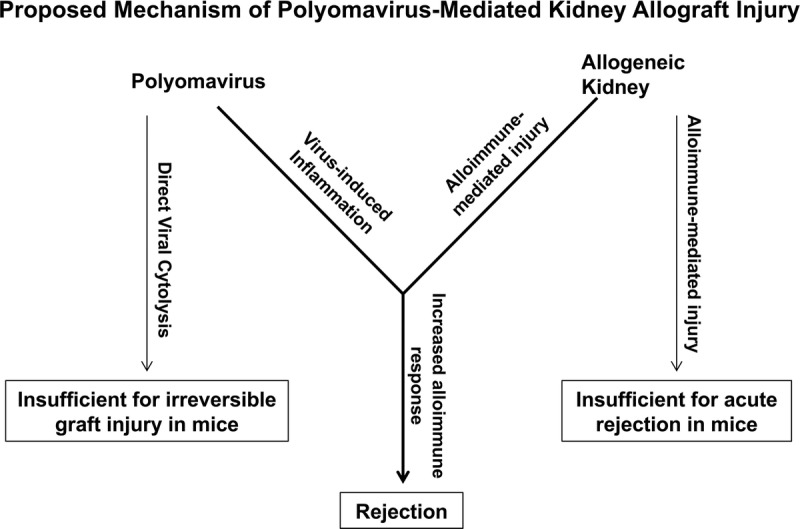
A proposed model describing the mechanism of PyV-mediated kidney allograft injury. As we have demonstrated, direct viral cytopathology of PyV infection alone is insufficient to cause irreversible graft injury in mice. Likewise, it is well established in the literature that mice tolerate an allogeneic kidney transplant, suggesting that alloimmune-mediated injury alone is also insufficient for acute kidney rejection. However, when this alloimmune injury encounters the PyV-induced inflammation of the kidney, the response is boosted, and the kidney is now rejected.
The observations from these experiments underscore important dynamics between virally mediated immunopathology and alloimmune responses. However, it is necessary to highlight some of the limitations of the model used in this study in relation to its clinical counterpart, BKVN. As mentioned, BK virus is a human PyV which is associated with renal transplantation and its attendant requirement for immunosuppression.21-24,57 Although polyomavirus-associated nephropathy has been linked to the family of DNA viruses that include BK virus and JC virus, BK virus's latent infection in the genitourinary system poses a specific challenge in caring for newly transplanted kidney allograft recipients on immunosuppression.58,59 Modeling the pathogenesis of BK viral infection and its resultant associated nephropathy is difficult due to the species specificity of Polyomaviridae family members. MPyV, genetically and structurally similar to human polyomaviruses, resembles human BK virus in terms of its prevalence, infectivity, and tropism, but it does not recapitulate all aspects of BKVN in humans.60,61 The lack of significant antigen exposure and immune challenges in specific pathogen-free mice limits the mouse model’s ability to recapitulate human allospecific responses in vivo. Many groups have demonstrated the importance of virus-specific immunologic memory as a barrier to transplantation.1,4,14,16,62 Furthermore, other viruses in the PyV family have failed to demonstrate clinical progression to interstitial nephritis and resultant allograft failure in humans,63,64 so the modeling of clinical PVAN with an infectious agent other than BK virus may pose key differences in disease progression or even manifestation. In light of these differences between the mouse model used in this study and the clinical BKVN seen in newly transplanted patients, direct translation of these findings to the pathogenesis and mechanisms of BKVN may not be appropriate. However, the insights provided by these experiments using MPyV-associated allograft rejection and injury remain informative in furthering our understanding of the role that tropic viral infection plays in allospecific injury.
In conclusion, we provide novel data from the mouse model of PyV-associated allograft injury, indicating that an augmented alloimmune response is the primary immunologic mechanism of disease. Importantly, these data highlight a previously undervalued target for therapeutic intervention—the interplay between viral inflammation and host alloimmune response. Many important questions remain: how does an otherwise well-controlled viral infection lead to a subset of activated, pathogenic alloreactive T cells infiltrating the graft? What biochemical signals underlie this trafficking and activation? Although this study provides important data into the nature of virally mediated allograft rejection, it also supports investigation into new studies exploring the compromise of the local kidney environment leading to augmented alloimmune response—an area of interest that explores the broader question of how viral infection interplays with immune-mediated disease progression. By better understanding the pathogenesis of PyV-associated allograft injury and nephropathy, we will be able to improve posttransplant outcomes and extend long-term survival for kidney transplant patients.
Footnotes
Published online 15 May, 2017.
This work was supported by NIH grants R01AI078426 (KAN), R01CA071971(AEL), and R01 AI102543 (AEL).
The authors declare no conflicts of interest.
S.C.K. and J.W. contributed equally.
S.C.K. substantially contributed to the analysis and interpretation of data for the work; and drafted and revised it critically for important intellectual content. J.W. substantially contributed to mouse kidney transplants, adoptive transfers, flow cytometry; and drafted and revised it critically for important intellectual content. Y.D. substantially contributed to mouse kidney transplants, adoptive transfers, flow cytometry; and drafted and revised it critically for important intellectual content. D.M. substantially contributed to the analysis and interpretation of data for the work; and drafted and revised it critically for important intellectual content. J.A. substantially contributed to the design and interpretation of data for the work; and drafted and revised it critically for important intellectual content. C.B. substantially contributed to the acquisition and analysis of data; and drafted and revised it critically for important intellectual content. A.F. substantially contributed to graft biopsy grading and histology interpretation; and drafted and revised it critically for important intellectual content. A.L. substantially contributed to the conception, design, and interpretation of data for the work; and drafted and revised it critically for important intellectual content. M.F. substantially contributed to the conception, design, and interpretation of data for the work; and drafted and revised it critically for important intellectual content. K.N. substantially contributed to the conception, design, and interpretation of data for the work; and drafted and revised it critically for important intellectual content. A.A. substantially contributed to the conception, design, and interpretation of data for the work; and drafted and revised it critically for important intellectual content.
All authors gave final approval of the version to be published; and agreed to be accountable for all aspects of the work.
REFERENCES
- 1.Adams A, Williams M, Jones T, et al. Heterologous immunity provides a potent barrier to transplantation tolerance. J Clin Invest. 2003;111:1887–1895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ford M. Virally-induced heterologous immunity in renal transplant recipients: important or inconsequential? Am J Transplant. 2016;16:1348–1349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Heutinck K, Yong S, Tonneijck L, et al. Virus-specific CD8+ T cells cross-reactive to donor-alloantigen are transiently present in the circulation of kidney transplant recipients infected with CMV and/or EBV. Am J Transplant. 2016;16:1480–1491. [DOI] [PubMed] [Google Scholar]
- 4.Pantenburg B, Heinzel F, Das L, et al. T cells primed by Leishmania major infection cross-react with alloantigens and alter the course of allograft rejection. J Immunol. 2002;169:3686–3693. [DOI] [PubMed] [Google Scholar]
- 5.Rist M, Hibbert K, Croft N, et al. T cell cross-reactivity between a highly immunogenic EBV epitope and a self-peptide naturally presented by HLA-B*18:01+ cells. J Immunol. 2015;194:4668–4675. [DOI] [PubMed] [Google Scholar]
- 6.Nguyen T, Rowntree L, Pellicci D, et al. Recognition of distinct cross-reactive virus-specific CD8+ T cells reveals a unique TCR signature in a clinical setting. J Immunol. 2014;192:5039–5049. [DOI] [PubMed] [Google Scholar]
- 7.Nickel P, Bold G, Presber F, et al. High levels of CMV-IE-1-specific memory T cells are associated with less alloimmunity and improved renal allograft function. Transpl Immunol. 2009;20:238–242. [DOI] [PubMed] [Google Scholar]
- 8.Selin L, Welsh R. Plasticity of T cell memory responses to viruses. Immunity. 2004;20:5–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mou D, Espinosa J, Stempora L, et al. Viral-induced CD28 loss evokes costimulation independent alloimmunity. J Surg Res. 2015;196:241–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sharma S, Thomas PG. The two faces of heterologous immunity: protection or immunopathology. J Leukoc Biol. 2014;95:405–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Welsh R, Che J, Brehm M, et al. Heterologous immunity between viruses. Immunol Rev. 2010;235:244–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Clute S, Naumov Y, Watkn L, et al. Broad cross-reactive TCR repertoires recognizing dissimilar Epstein-Barr and influenza A virus epitopes. J Immunol. 2010;185:6753–6764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Barton E, White D, Cathelyn J, et al. Herpesvirus latency confers symbiotic protection from bacterial infection. Nature. 2007;447:326–329. [DOI] [PubMed] [Google Scholar]
- 14.Smithey M, Li G, Venturi V, et al. Lifelong persistent viral infection alters the naive T cell pool, impairing CD8 T cell immunity in late life. J Immunol. 2012;189:5356–5366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sheil JM, Bevan MJ, Lefrancois L. Characterization of dual-reactive H-2Kb-restricted anti-vesicular stomatitis virus and alloreactive cytotoxic T cells. J Immunol. 1987;138:3654–3660. [PubMed] [Google Scholar]
- 16.Yang H, Welsh R. Induction of alloreactive cytotoxic T cells by acute virus infection of mice. J Immunol. 1986;136:1186–1193. [PubMed] [Google Scholar]
- 17.Braciale T, Andrew M, Braciale V. Simultaneous expression of H-2-restricted and alloreactive recognition by a cloned line of influenza virus-specific cytotoxic T lymphocytes. J Exp Med. 1981;153:1371–1376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Patenburg B, Heinzel F, Das L, et al. T cells primed by Leishmania major infection cross-react with alloantigens and alter the course of allograft rejection. J Immunol. 2002;169:3686–3693. [DOI] [PubMed] [Google Scholar]
- 19.Smith JM, Dharnidharka VR, Talley L, et al. BK virus nephropathy in pediatric renal transplant recipients: an analysis of the North American Pediatric Renal Trials and Collaborative Studies (NAPRTCS) registry. Clin J Am Soc Nephrol. 2007;2:1037–1042. [DOI] [PubMed] [Google Scholar]
- 20.Boothpur R, Brennan DC. Human polyoma viruses and disease with emphasis on clinical BK and JC. J Clin Virol. 2010;47:306–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hirsch HH, Suthanthiran M. The natural history, risk factors and outcomes of polyomavirus BK-associated nephropathy after renal transplantation. Nat Clin Pract Nephrol. 2006;2:240–241. [DOI] [PubMed] [Google Scholar]
- 22.Mbianda C, El-Meanawy A, Sorokin A. Mechanisms of BK virus infection of renal cells and therapeutic implications. J Clin Virol. 2015;71:59–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Comoli P, Hirsch H, Ginevri F. Cellular immune responses to BK virus. Curr Opin Organ Transplant. 2008;13:569–574. [DOI] [PubMed] [Google Scholar]
- 24.Egli A, Binggeli S, Bodaghi S, et al. Cytomegalovirus and polyomavirus BK posttransplant. Nephrol Dial Transplant. 2007;22:viii72–viii82. [DOI] [PubMed] [Google Scholar]
- 25.Ramos E, Drachenberg CB, Wali R, et al. The decade of polyomavirus BK-associated nephropathy: state of affairs. Transplantation. 2009;87:621–630. [DOI] [PubMed] [Google Scholar]
- 26.Comoli P, Binggeli S, Ginevri F, et al. Polyomavirus-associated nephropathy: update on BK virus-specific immunity. Transpl Infect Dis. 2006;8:86–94. [DOI] [PubMed] [Google Scholar]
- 27.Sawinski D, Forde KA, Trofe-Clark J, et al. Persistent BK viremia does not increase intermediate-term graft loss but is associated with de novo donor-specific antibodies. J Am Soc Nephrol. 2015;26:966–975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kuypers D. Management of polyomavirus-associated nephropathy in renal transplant recipients. Nat Rev Nephrol. 2012;8:390–402. [DOI] [PubMed] [Google Scholar]
- 29.Schachtner T, Stein M, Babel N, et al. The loss of BKV-specific immunity from pretransplantation to posttransplantation identifies kidney transplant recipients at increased risk of BKV replication. Am J Transplant. 2015;15:2159–2169. [DOI] [PubMed] [Google Scholar]
- 30.Bohl DL, Brennan DC. BK virus nephropathy and kidney transplantation. Clin J Am Soc Nephrol. 2007;2(Suppl 1):S36–S46. [DOI] [PubMed] [Google Scholar]
- 31.Han Lee ED, Kemball CC, Wang J, et al. A mouse model for polyomavirus-associated nephropathy of kidney transplants. Am J Transplant. 2006;6(5 Pt 1):913–922. [DOI] [PubMed] [Google Scholar]
- 32.Albrecht J, Dong Y, Wang J, et al. Adaptive immunity rather than viral cytopathology mediates polyomavirus-associated nephropathy in mice. Am J Transplant. 2012;12:1419–1428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ehst B, Ingulli E, Jenkins M. Development of a novel transgenic mouse for the study of interactions between CD4 and CD8 T cells during graft rejection. Am J Transplant. 2003;3:1355–1362. [DOI] [PubMed] [Google Scholar]
- 34.Lukacher AE, Wilson CS. Resistance to polyoma virus-induced tumors correlates with CTL recognition of an immunodominant H-2Dk-restricted epitope in the middle T protein. J Immunol. 1998;160:1724–1734. [PubMed] [Google Scholar]
- 35.Andrews NP, Pack CD, Lukacher AE. Generation of antiviral major histocompatibility complex class I-restricted T cells in the absence of CD8 coreceptors. J Virol. 2008;82:4697–4705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Weck KE, Barkon ML, Yoo LI, et al. Mature B cells are required for acute splenic infection, but not for establishment of latency, by murine gammaherpesvirus 68. J Virol. 1996;70:6775–6780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hogquist K, Jameson SC, Heath WR, et al. T cell receptor antagonist peptides induce positive selection. Cell. 1994;76:17–27. [DOI] [PubMed] [Google Scholar]
- 38.Bickerstaff A, Pelletier R, Wang JJ, et al. An experimental model of acute humoral rejection of renal allografts associated with concomitant cellular rejection. Am J Pathol. 2008;173:347–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kemball CC, Lee ED, Vezys V, et al. Late priming and variability of epitope-specific CD8+ T cell responses during a persistent virus infection. J Immunol. 2005;174:7950–7960. [DOI] [PubMed] [Google Scholar]
- 40.Chalasani G, Dai Z, Konieczny B, et al. Recall and propagation of allospecific memory T cells independent of secondary lymphoid organs. Proc Natl Acad Sci U S A. 2002;99:6175–6180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Floyd TL, Orr SB, Coley SM, et al. High-frequency alloreactive T cells augment effector function of low-frequency CD8+ T cell responses under CD28/CD154 blockade. Transplantation. 2010;89:1208–1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Swimm AI, Bornmann W, Jiang M, et al. Abl family tyrosine kinase regulate sialylated ganglioside receptors for polyomavirus. J Virol. 2010;84:4243–4251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wang T, Ahmed EB, Chen L, et al. Infection with the intracellular bacterium, Listeria monocytogenes, overrides established tolerance in a mouse cardiac allograft model. Am J Transplant. 2010;10:1524–1533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sagedal S, Nordal KP, Hartmann A, et al. The impact of cytomegalovirus infection and disease on rejection episodes in renal allograft recipients. Am J Transplant. 2002;2:850–856. [DOI] [PubMed] [Google Scholar]
- 45.Vilchez RA, Dauber J, McCurry K, et al. Parainfluenza virus infection in adult lung transplant recipients: an emergent clinical syndrome with implications on allograft function. Am J Transplant. 2003;3:116–120. [DOI] [PubMed] [Google Scholar]
- 46.Doherty PC, Christensen JP, Belz GT, et al. Dissecting the host response to a gamma-herpesvirus. Philos Trans R Soc Lond B Biol Sci. 2001;356:581–593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Pambrun E, Mengelle C, Fillola G, et al. An association between BK Virus replication in bone marrow and cytopenia in kidney-transplant recipients. J Transplant. 2014;2014:252914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Costa C, Cavallo R. Polyomavirus-associated nephropathy. World J Transplant. 2012;2:84–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Beus JM, Hashmi SS, Selvaraj SA, et al. Heterologous immunity triggered by a single, latent virus in mus musculus: combined costimulation- and adhesion-blockade decrease rejection. PLoS One. 2013;8:e71221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Stapler D, Lee E, Selvaraj S, et al. Expansion of effector memory TCR Vbeta4+ CD8+ T cells is associated with latent infection-mediated resistance to transplantation tolerance. J Immunol. 2008;180:3190–3200. [DOI] [PubMed] [Google Scholar]
- 51.Gray KS, Collins CM, Speck SH. Characterization of omental immune aggregates during establishment of a latent gammaherpesvirus infection. PLoS One. 2012;7:e43196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Mannon RB, Hoffmann SC, Kampen RL, et al. Molecular evaluation of BK polyomavirus nephropathy. Am J Transplant. 2005;5:2883–2893. [DOI] [PubMed] [Google Scholar]
- 53.Gago M, Cornell L, Kremers W, et al. Kidney allograft inflammation and fibrosis, causes and consequences. Am J Transplant. 2012;12:1199–1207. [DOI] [PubMed] [Google Scholar]
- 54.Zeng G, Huang Y, Huang Y, et al. Antigen-specificity of T cell infiltrates in biopsies with T cell mediated rejection and BK polyomavirus viremia: analysis by next generation sequencing. Am J Transplant. 2016;16:3131–3138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Vu D, Sakharkar P, Shah T, et al. Association of interferon gamma gene polymorphisms with BK virus infection among Hispanic renal allograft recipients. Transplantation. 2014;97:660–667. [DOI] [PubMed] [Google Scholar]
- 56.Russell PS, Chase CM, Colvin RB, et al. Kidney transplants in mice. An analysis of the immune status of mice bearing long-term, H-2 incompatible transplants. J Exp Med. 1978;147:1449–1468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Barbosa D, Kahwaji J, Puliyanda D, et al. Polyomavirus BK viremia in kidney transplant recipients after desensitization with IVIG and rituximab. Transplantation. 2014;97:755–761. [DOI] [PubMed] [Google Scholar]
- 58.Wiseman AC. Polyomavirus nephropathy: a current perspective and clinical considerations. Am J Kidney Dis. 2009;54:131–142. [DOI] [PubMed] [Google Scholar]
- 59.Sigdel T, Bestard O, Salomonis N, et al. Intragraft antiviral-specific gene expression as a distinctive transcriptional signature for studies in polyomavirus-associated nephropathy. Transplantation. 2016;100:2062–2070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Rowe W. The epidemiology of mouse polyoma virus infection. Bacteriol Rev. 1961;25:18–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Carroll J, Dey D, Kreisman L, et al. Receptor-binding and oncogenic properties of polyoma viruses isolated from feral mice. PLoS Pathog. 2007;3:e179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Blattman J, Antia R, Sourdive D, et al. Estimating the precursor frequency of naive antigen-specific CD8 T cells. J Exp Med. 2002;195:657–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Drachenberg CB, Hirsch HH, Papadimitriou JC, et al. Polyomavirus BK versus JC replication and nephropathy in renal transplant recipients: a prospective evaluation. Transplantation. 2007;84:323–330. [DOI] [PubMed] [Google Scholar]
- 64.Randhawa P, Baksh F, Aoki N, et al. JC virus infection in allograft kidneys: analysis by polymerase chain reaction and immunohistochemistry. Transplantation. 2001;71:1300–1303. [DOI] [PubMed] [Google Scholar]