

Abstract
Purpose
Genetic alterations such as activating KRAS and/or inactivating TP53 are thought to be the most common drivers to tumorigenesis. Therefore, we assessed phase I cancer patients with KRAS+/TP53+ mutations.
Results
Approximately 8% of patients referred to phase I clinical trials harbored concurrent KRAS and TP53 mutations. Patients who received a phase I trial therapy (n = 57) had a median OS of 12 months, compared with 4.6 months in those who were not treated (n = 106; p = 0.003). KRAS G13 and TP53 R273 mutations were associated with poor overall survival (OS), while antiangiogenesis and gene aberration-related therapies were associated with prolonged OS. A prognostic model using neutrophilia, thrombocytosis, hypoalbuminemia, body mass index <30 kg/m2, and the absence of lung metastasis was established and validated. Phase I cancer patients in the low-risk group had a median OS of 16.6 months compared with 5.4 months in the high-risk group (p < 0.001). Untreated patients in the low-risk group had a median OS of 6.7 months compared with 3.6 months in the high-risk group (p = 0.033).
Experimental Design
We analyzed 163 consecutive patients with advanced KRAS+/TP53+ mutant cancer who were referred to phase I clinical trials, to identify molecular aberrations, clinical characteristics, survivals, and potentially effective treatment regimens.
Conclusions
This study provided preliminary evidence that besides modulation of the proinflammatory state, antiangiogensis and concomitant gene aberration-related therapies may improve the treatment of KRAS+/TP53+ mutant cancer.
Keywords: KRAS, TP53, chronic inflammation, phase I trial, gene aberration-related therapy
INTRODUCTION
Oncogenic mutations in rat sarcoma viral oncogene homolog (RAS) genes are detected in approximately 30% of human cancers, predominantly in colorectal cancer, pancreatic cancer, and lung adenocarcinomas [1]. These mutations occur most frequently in Kirsten RAS (KRAS), which encodes a small GTPase that mediates downstream signaling from growth factor receptors [2, 3]. KRAS mutations can constructively activate downstream signaling pathways, such as RAS/mitogen-activated protein kinase (MEK)/extracellular signal-regulated kinases (ERK) and phosphoinositide 3-kinase (PI3K)/AKT/mammalian target of rapamycin (mTOR), and this signaling pathway activation triggers nuclear gene transcription and cell differentiation and proliferation [4].
However, KRAS mutation alone, which occurs in the early process of tumorigenesis, is not sufficient to induce malignant transformation of normal epithelial cells [5, 6]. Additional loss of tumor suppressor genes, such as TP53 [7, 8], is required for cancer development, which arises through sequential accumulation of oncogenic mutations and loss of tumor suppressor genes. Somatic TP53 mutation is the most common genetic aberration in tumor suppressor genes, occurring in 10% to 96% of human cancers [9]. Functional TP53 mutations lead to ablation of cell cycle arrest and DNA damage repair, as well as overexpression of nuclear target genes, resulting in genomic instability and tumor development [10]. Dual mutations in TP53 and KRAS (KRAS+/TP53+, + indicates positive hotspot test) occur in up to 20% of advanced solid tumors [11–14]. In genetically engineered mouse models, mice harboring both the TP53 R172H and KRAS G12D mutations had a significantly shortened latency, and thus more tumors than mice with the KRAS G12D mutation alone [7, 15].
Because concurrent KRAS and TP53 mutations manifest potentially synergistic biologic effects, cancers carrying both KRAS and TP53 mutations (KRAS+/TP53+) might represent a unique cancer subtype with distinct and aggressive biologic behaviors [16]. Blockade of downstream signaling pathways such as RAF/MEK or PI3K/AKT/mTOR in KRAS-mutant cancer [17] and antiangiogenic-based therapy in TP53-mutant cancer would be appropriate therapeutic strategies [18, 19]. Unfortunately, effective therapies directly targeting TP53 or KRAS mutations are not available and these mutations are currently considered undruggable [20, 21].
Many phase I clinical trials include patients with malignancies arising from undruggable genetic mutations, but it is unclear which types of therapies are most promising for the treatment of these malignancies. Therefore, it is of great scientific interest and clinical urgency to explore potential therapeutic options for malignancies with undruggable genetic mutations. In the current study, we reviewed demographics and clinical outcomes of patients with advanced KRAS+/TP53+ mutant cancers who were referred to phase I clinical trials at The University of Texas MD Anderson Cancer Center. Our aims were to investigate specific genetic aberrations associated with clinical outcomes and to identify potential therapeutic regimens for the treatment of advanced KRAS+/TP53+ mutant cancers.
RESULTS
Patient characteristics
From March 2102 to October 2014, 2, 144 consecutive patients with advanced cancers were referred to phase I clinical trials at MD Anderson and underwent molecular tests for tumor genetic aberrations. Among these patients, 167 (7.8%) harbored concurrent KRAS and TP53 hotspot mutations (KRAS+/TP53+ mutant cancer), 182 (8.5%) harbored KRAS+/TP53– hotspot mutations, and 839 (39.1%) harbored KRAS–/TP53+ hotspot mutations (- indicates negative hotspot test). Four patients with KRAS+/TP53+ mutant cancer had insufficient clinical data and were not included in our analysis. The baseline characteristics of the remaining 163 patients are summarized in Table 1.
Table 1. Patient baseline characteristics (n=163).
| Characteristics | Patient number | Percentage (%) |
|---|---|---|
| Age (median, range) | 55 (17-83) | |
| Gender | ||
| Male | 97 | 60 |
| Female | 66 | 40 |
| Race | ||
| White | 103 | 63 |
| African American | 25 | 15 |
| Hispanic | 23 | 14 |
| Asian | 4 | 3 |
| Others | 8 | 5 |
| Type of cancer | ||
| Colorectal | 104 | 64 |
| Pancreatic | 28 | 17 |
| Lung* | 8 | 5 |
| Others** | 23 | 14 |
| With second primary cancer | ||
| Yes | 20 | 12 |
| No | 143 | 88 |
| Sites of metastasis | ||
| Lung | 116 | 71 |
| Liver | 113 | 69 |
| Lymph node | 61 | 37 |
| Peritoneal | 37 | 23 |
| Bone | 23 | 14 |
| Retroperitoneal | 20 | 12 |
| Adrenal | 17 | 10 |
| Soft tissue | 12 | 7 |
| Brain | 8 | 5 |
| Cutaneous | 7 | 4 |
| Renal | 6 | 4 |
| Spleen | 6 | 4 |
| Ovarian | 4 | 2 |
| Vaginal | 4 | 2 |
| Initial diagnosis with metastasis | ||
| Yes | 89 | 55 |
| No | 74 | 45 |
*Lung cancers included adenocarcinoma (n=5), adenosquamous (n=2) and neuroendocrine (n=1). **Other cancers included cholangiocarcinoma (n=3), esophageal (n=1), gastric (n=1), duodenal (n=1), uterine (n=4), ovarian (2), vaginal (n=1), bladder (n=1), sinonasal (n=1), thyroid (n=1), appendiceal (n=2), skin squamous (n=1) and cancer of unknown primary (n=4).
Molecular aberrations
In the 163 patients with KRAS+/TP53+ mutant cancer, G12 (n = 107; 66%) and G13 (n = 25; 15%) mutations constituted the majority of KRAS hotspot mutations. In patients with pancreatic cancers, G12 mutations occurred more frequently (p = 0.003), but G13 mutations were not found. In the total cohort of patients (n = 163), 83 types of TP53 mutations were found, of which 44% were common hotspot mutations: R273 (n = 26; 16%), R175 (n = 19; 12%), R248 (n = 12; 7%), G245 (n = 9; 6%), and R282 (n = 5; 3%). Association of a TP53 hotspot mutation with a specific cancer was not observed. Other concurrent genetic aberrations were found in most patients (n = 125; 77%), and more than one concomitant genetic aberration was found in 87 patients (53%): APC (n = 65; 40%), PIK3CA (n = 37; 23%), KIT (n = 34; 21%), SMAD4 (n = 18; 11%), FBXW7 (n = 11; 7%), MET (n = 10; 6%), JAK3 (n = 9; 6%), CDKN2A (n = 9; 6%), PTEN (n = 6; 4%), and STK11 (n = 5; 3%).
Antitumor activity and PFS
Approximately one-third of patients (n = 57) received a total of 78 phase I trial therapies under 50 different phase I clinical trials. These therapies yielded 2 PRs and 17 SDs (24% of disease control), associated with a median PFS of 2.1 months (95% confidence interval [CI] 1.8-2.4). Among patients treated with an antiangiogenic agent (n = 15), 11 (73%) had PR or SD and the median PFS was 3.7 months (95% CI 2.8-4.6), which was significantly better than among patients who were not treated with an antiangiogenic agent (8/39 [21%] PR or SD, p < 0.001; PFS 1.8 months [95% CI 1.6-2.0], p = 0.043). In patients who received therapies with one agent targeting a concomitant genetic aberration or its downstream proteins (gene aberration-related therapy), the disease control rate was 65% (17/26) and the median PFS was 3.7 months (95% CI 2.6-4.8), which was significantly better than among patients who did not receive this type of treatment (2/28 [7%] PR or SD, p < 0.001; PFS 1.6 months [95% CI 1.2-2.0], p < 0.001). In patients receiving gene aberration-related phase I clinical trial therapy, PFS was similar to that observed with previous standard of care therapy before phase I clinical trial referral (2.5 months [95% CI 1.4-3.6], p = 0.866).
Overall survival
A median OS of 6.7 months (95% CI 4.9-8.5) was observed in the 163 patients with KRAS+/TP53+ mutant cancer who were referred to phase I clinical trials at MD Anderson. Patients who received therapy in a phase I clinical trial had a median OS of 12 months (95% CI 5.6-18.4), which was significantly better than the median OS in those who did not (4.6 months [95% CI 3.6-5.6], p = 0.003). Patients receiving phase I clinical trial therapies with an antiangiogenic agent had a median OS of 13.4 months (95% CI 5.5-20.2), and those receiving gene aberration-related phase I clinical trial therapies had a median OS of 13.5 months (95% CI 5.3-20.6). These OS times compared favorably with those of patients who did not receive these treatments (no antiangiogenic therapy: median OS 8.8 months [95% CI 3.0-14.6], p = 0.6; and no gene aberration-related phase I clinical trial therapy: median OS 7.6 months [95% CI 7.1-8.1], p = 0.2) respectively.
Association of OS with genetic aberrations
Further analysis in 163 patients with KRAS+/TP53+ mutant cancer revealed that patients harboring G13 mutations (n = 25) had a median OS of 4.8 months (95% CI 2.5-7.1), which was significantly worse than among those without the G13 mutation (n = 138, median OS 7.3 months [95% CI 4.8-9.8], p = 0.016). No survival difference was observed between patients with G12 mutations and those without. In patients with colorectal cancers, G13 mutations remained associated with reduced OS (n = 22, median OS 4.8 months [95% CI 2.5-7.1]) compared with patients without G13 mutations (n = 82, median OS 8.4 months [95% CI 5.3-11.5]; p = 0.012), as shown in Figure 1. Patients with a TP53 R273 mutation (n = 14) had a median OS of 5.7 months [95% CI 3.0-8.4], which was worse than in patients without the R273 mutation (n = 90, median OS 8.5 months [95% CI 5.8-11.2]; p = 0.048), as shown in Figure 2.
Figure 1. Kaplan-Meier overall survival (OS) curves in patients with KRAS+/TP53+ mutant colorectal cancer who received therapy in a phase I clinical trial, stratified by KRAS G13 mutation status (due to sample size, all p values are unadjusted).
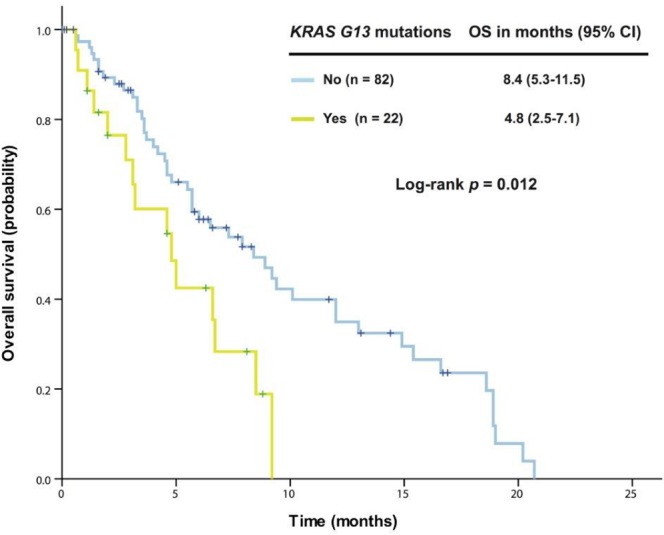
.
Figure 2. Kaplan-Meier overall survival (OS) curves in patients with KRAS+/TP53+ mutant colorectal cancer who received therapy in a phase I clinical trial, stratified by TP53 R273 mutation status (due to sample size, all p values are unadjusted).
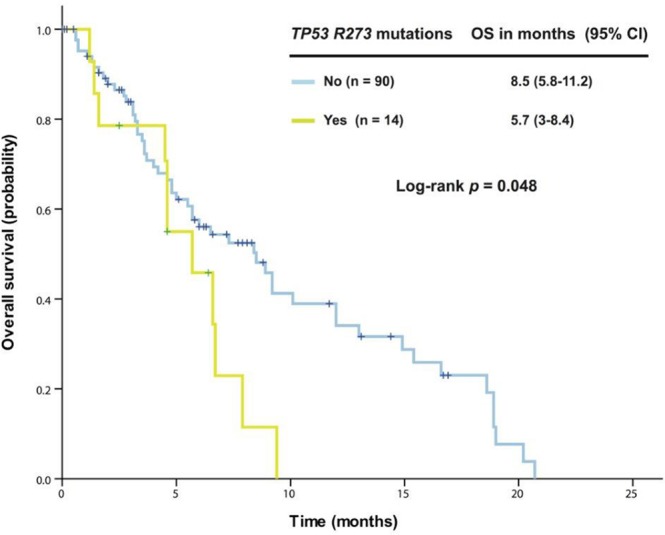
.
Exploratory study of a prognostic model
We were unable to apply the Royal Marsden Hospital score [22] or the MD Anderson prognostic score [23] to the 57 patients who received therapy in a phase I clinical trial. Therefore, we decided to explore a prognostic model specific to patients with KRAS+/TP53+ mutant cancer. First, we analyzed the association of OS with potential risk factors using univariate and multivariate analyses in these 57 patients (Table 2). Five independent poor risk factors were identified for predicting individual survival outcome: neutrophilia, thrombocytosis, hypoalbuminemia, body mass index <30 kg/m2, and the absence of lung metastasis. These parameters were then extracted using binary subgroups (no = 0, yes = 1) to explore a risk prognostic model predictive of OS after the initial phase I clinical trial visit. This model classified the patients into one of two risk cohorts (p < 0.001; Figure 3): a low-risk group (score ≤ 1, n = 40) associated with a median OS of 16.6 months (95% CI 12.9-20.4) or a high-risk group (score > 1, n = 17) associated with a median OS of 5.4 months (95% CI 3.7-7.1).
Table 2. Univariate and multivariate analyses of OS in 57 patients who received a phase I trial therapy.
| Potential Risk Factors | Patient Number | Median OS (months, 95% CI) | p value | |
|---|---|---|---|---|
| Univariate | Multivariate | |||
| Age < 65 years | Yes (n=46) | 10.1 (5.2-15) | 0.121 | 0.185 |
| No (n=11) | 17.9 (6.7-29.1) | |||
| Male | Yes (n=37) | 15.2 (6.3-24.1) | 0.334 | 0.228 |
| No (n=20) | 12 (4.1-19.9) | |||
| Colorectal cancer | Yes (n=39) | 12 (6.6-17.4) | 0.773 | 0.582 |
| No (n=18) | 7.3 (0-16.5 | |||
| Presence of a second primary cancer | Yes (n=5) | 10.1 (0, infinity) | 0.086 | 0.106 |
| No (n=52) | 13 (4.6-21.4) | |||
| Metastasis at initial diagnosis | Yes (n=28) | 7.3 (4.2-10.4) | 0.176 | 0.978 |
| No (n=29) | 13 (8-18.1) | |||
| Number of metastatic sites≤ 2 | Yes (n=18) | 13 (2.1-23.9) | 0.572 | 0.402 |
| No (n=39) | 10.1 (4.2-16) | |||
| Lung metastasis | Yes (n=43) | 15.4 (8.3-22.5) | 0.015 | 0.01 |
| No (n=14) | 7.3 (4.5-10.1) | |||
| Liver metastasis | Yes (n=41) | 8.5 (4.6-12.4) | 0.571 | 0.593 |
| No (n=16) | 15.2 (7-23.4) | |||
| Eastern Cooperative Oncology Group (ECOG) performance status of 0 | Yes (n=8) | 16.6 (3.1-30.1) | 0.077 | 0.66 |
| No (n=49) | 10.1 (3.1-17.1) | |||
| Neutrophilia | Yes (n=6) | 3.4 (2-4.8) | <0.001 | <0.001 |
| No (n=51) | 13 (6.6-19.4) | |||
| Lymphopenia | Yes (n=16) | 7.3 (1.9-12.7) | 0.103 | 0.386 |
| No (n=41) | 13 (7.4-18.6) | |||
| Anemia | Yes (n=41) | 13 (4.8-21.2) | 0.778 | 0.097 |
| No (n=16) | 10.1 (3.9-16.3) | |||
| Thrombocytosis | Yes (n=1) | 2.6 (0, infinity) | <0.001 | 0.022 |
| No (n=56) | 12 (5.5-18.5) | |||
| Normal lactate dehydrogenase | Yes (n=33) | 15.2 (11.3-19.1) | 0.11 | 0.119 |
| No (n=24) | 6.5 (5-8) | |||
| Hypoalbuminemia | Yes (n=2) | 2.5 (0, infinity) | 0.016 | 0.029 |
| No (n=55) | 13 (6.2-19.8) | |||
| Normal creatinine | Yes (n=56) | 12 (5.6-18.4) | 0.811 | 0.984 |
| No (n=1) | 2.7 (0, infinity) | |||
| Hyperbilirubinemia | Yes (n=11) | 10.1 (4.5-15.7) | 0.86 | 0.039 |
| No (n=46) | 12 (4.2-20) | |||
| Venous thromboembolism | Yes (n=12) | 12 (3.6-20.4) | 0.593 | 0.281 |
| No (n=45) | 13 (6.8-19.2) | |||
| Body mass index (BMI) ≥30 kg/m2 | Yes (n=11) | 12 (0-26.9) | 0.05 | 0.023 |
| No (n=46) | 10.1 (4.2-16) | |||
Figure 3. A prognostic model was established from 57 patients with advanced KRAS+/TP53+ mutant cancer who received therapy in a phase I clinical trial.
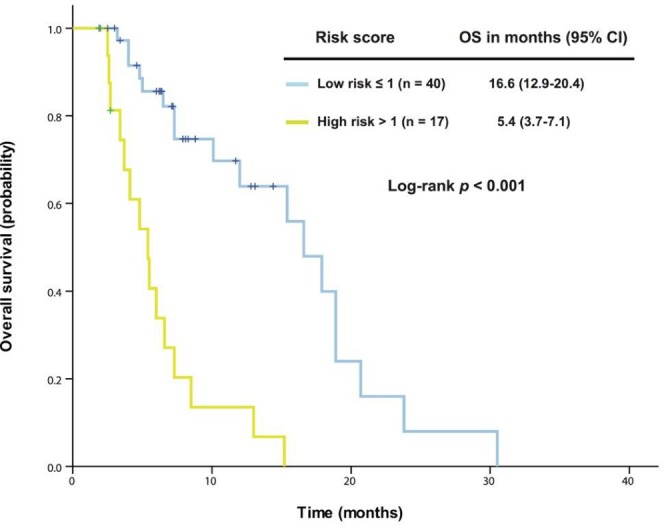
Kaplan-Meier overall survival (OS) curves are shown, stratified by risk score (low-risk group: score ≤1, high-risk group: score >1) (due to sample size, all p values are unadjusted).
To support this model, we used another cohort of patients who were referred to a phase I clinical trial but did not receive the therapy. In this cohort, patients in the low-risk group (n = 56) had a median OS of 6.7 months (95% CI 3.4-10.0), which was significantly better than that of those in the high-risk group (n = 48, median OS 3.6 months [95% CI 2.4-4.8], p = 0.033), as shown in Figure 4.
Figure 4. The established prognostic model was validated in 104 patients with advanced KRAS+/TP53+ mutant cancer who did not receive therapy in a phase I clinical trial.
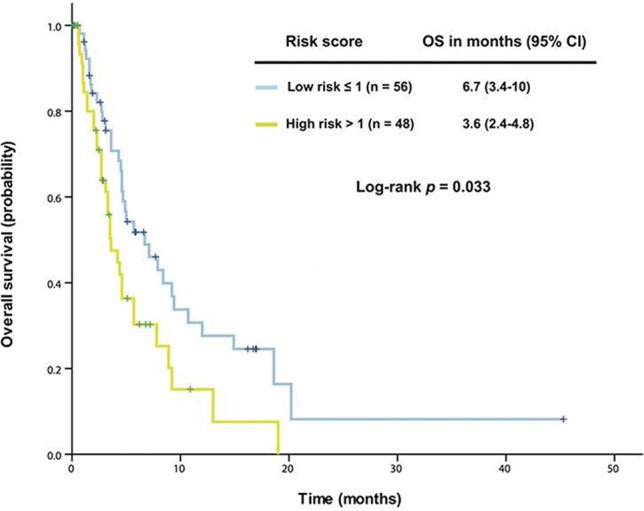
Kaplan-Meier overall survival (OS) curves are shown, stratified by risk score (low-risk group: score ≤1, high-risk group: score >1) (due to sample size, all p values are unadjusted).
DISCUSSION
Our findings suggest that the KRAS G13 and TP53 R273 mutations are associated with poor outcome in patients with KRAS+/TP53+ mutant cancer, and antiangiogenic therapy combined with therapy targeting specific genetic aberrations may be an effective treatment strategy. To the best of our knowledge, the current study is the first to analyze clinical outcomes of patients with advanced hotspot KRAS+/TP53+ mutant cancers who were referred to a phase I clinical trial program at MD Anderson.
KRAS and TP53 are frequently mutated in many types of cancer. Although they are highly attractive therapeutic targets, they remain outside of the reach of direct pharmacologic intervention [20]. Until a breakthrough is achieved with a direct pharmacologic approach, alternative strategies for addressing these undruggable targets remain under investigation [24]. Unfortunately, we found that only approximately one-third of patients with advanced hotspot KRAS+/TP53+ mutant cancers received treatment in a phase I clinical trial, much less than the overall rate of 55% of all patients who were referred to phase I clinical trials at the same institution [25].
A median OS of 12 months was observed in patients with KRAS+/TP53+ mutant cancer who had received treatment in a phase I clinical trial, consistent with a previous study showing a median OS of 10 months in 1,181 consecutive cancer patients treated in phase I clinical trials [23]. Other studies have reported a median OS of 8 months in 365 patients harboring hotspot KRAS mutations [13] and 14.6 months in 188 patients harboring hotspot TP53 mutations at the same institution [14]. These findings indicate that outcomes for patients with hotspot KRAS+/TP53+ mutant cancer who enroll in phase I clinical trials are better than in those with hotspot KRAS mutations [13] but worse than in those with hotspot TP53 mutations [14]. The differential outcomes for patients with specific cancer genetics [26] may reflect the reality that there are many phase I clinical trials of antiangiogenic-based therapeutic regimens but few studies appropriate for those with hotspot KRAS mutations [25]. These findings also suggest that patients harboring hotspot TP53 mutations may benefit from antiangiogenic-based therapeutic regimens [19]. The evidence that the Royal Marsden Hospital score or the MD Anderson prognostic score could not be used to predict outcomes of the patients with KRAS+/TP53+ mutant cancer who received a phase I clinical trial therapy may indicate that the outcome was related to their unique biological characteristics, and availability of effective phase I trial therapy.
In our cohort of patients with KRAS+/TP53+ mutant cancer, approximately two-thirds of patients had KRAS G12 mutations and one-sixth had G13 mutations. Although the absence of a G13 mutation is usually associated with poor prognosis in pancreatic cancer, the presence of a G13 mutation was associated with significantly shorter OS than other KRAS mutations in our full cohort of patients with KRAS+/TP53+ mutations and in those with colorectal cancer. This is consistent with previous findings showing that the KRAS G13 mutation was an independent prognostic factor for poor metastasis-free survival in colon cancer compared with either wild-type KRAS or G12 mutation [27, 28].
We observed a total of 83 types of TP53 mutations in our cohort, and most were located in the DNA binding domain. In contrast with a previous study showing that patients with hotspot TP53 R273 mutant ovarian cancer had significantly longer median OS than those with other hotspot TP53 mutations [29], our study revealed that a hotspot TP53 R273 mutation was associated with poor survival in patients with metastatic colorectal cancer. These inconsistent data imply that different cell contexts may lead to different outcomes, which warrants further investigation clinically and preclinically.
Although genetics likely play an important role in tumorigenesis, the inflammatory process is initiated by the movement of innate immune system cells to the microenvironment, followed by the secretion of proinflammatory cytokines, growth factors, and reactive oxygen species, causing DNA damage and promoting neoplastic development, as has been found in many tumor types [6]. Our multivariable analysis revealed that five independent baseline factors (neutrophilia, thrombocytosis, hypoalbuminemia, body mass index <30 kg/m2, and the absence of lung metastasis) were able to predict individual outcome not only in patients with KRAS+/TP53+ mutant cancer who had received therapy in a phase I clinical trial, but also in those who had not received therapy. Four of these prognostic factors are related to the proinflammatory state, which works alongside KRAS and TP53 mutations to enhance tumor progression and develop resistance to cancer therapy, resulting in poor clinical outcomes [30–32]. Therefore, a thorough understanding of the mechanisms of the proinflammatory state in conjunction with cancer-related gene aberrations may provide a scientific rationale to develop effective therapeutic strategies for advanced KRAS+/TP53+ mutant cancer. Though we cannot completely explain association of the absence of lung metastasis with poor outcome, we did observe that phase I metastatic colorectal cancer patients with pulmonary metastasis had a relatively slow process for tumor progression, which might reflect different biologic properties of these tumors, and requires further investigation.
Our study has limitations. First, the retrospective setup and limited sample size might yield statistical bias. Due to multiplicity of statistical testing in such small sample size, all p values are exploratory and unadjusted. Second, data from patients with hotspot KRAS+/TP53-, KRAS-/TP53+, and KRAS-/TP53- cancer were not available for our analysis of patients with metastatic KRAS+/TP53+ mutant cancer, which limited our ability to reach conclusions from data comparison among these four groups of patients.
PATIENTS AND METHODS
Patients
We retrospectively reviewed 2,144 consecutive patients with advanced cancers who were referred to phase I clinical trials at MD Anderson from March 2102 to October 2014 and who had sufficient tumor tissue specimens available for next generation sequencing. Among these patients, 167 harbored concurrent hotspot mutations in the KRAS and TP53 genes. Patient baseline demographics, laboratory results, gene aberrations, status of phase I clinical trial therapy, and clinical outcomes were obtained from electronic medical records. All patients were followed until death or censored on March 10, 2016. Trial conduct, data collection, and subsequent data analysis were performed in accordance with the guidelines of the MD Anderson Institutional Review Board (IRB) after the IRB approval for the research and a waiver of informed consent were obtained.
Molecular analysis
For somatic hotspot mutation analysis, DNA was extracted, purified, and quantified from microdissected, paraffin-embedded tumor specimens. Next generation sequencing for hotspot mutations was performed using the Ion Ampliseq Cancer Panel (Life Technologies, Grand Island, NY) in a Clinical Laboratory Improvement Amendments-certified Molecular Diagnostics Laboratory at MD Anderson [19, 33]. A panel of 46 genes was initially tested and then expanded to 50 genes, as described previously [34].
Treatment and evaluation
The decision to enroll an eligible patient in a phase I clinical trial depended on the protocol availability and the discretion of the treating physicians. Tumor responses (CR = complete remission, PR = partial response, SD = stable disease, and PD = progressive disease) were evaluated according to Response Evaluation Criteria in Solid Tumors (RECIST) version 1.0 or 1.1 [35, 36], depending on individual protocols. Progression-free survival (PFS) was calculated from the date of initiation of a phase I clinical trial therapy to the date of first objective documentation of PD, death, or censor date. PFS for patients alive and progression-free at last evaluation should be censored at date of last clinical evaluation. Overall survival (OS) was calculated from the date of the initial phase I clinical trial visit to the date of death or censor date. Time to death for patients alive at last contact should be censored at date of last contact.
Statistical analysis
Continuous interval-scaled data were summarized as median (range). Categorical data were summarized as frequencies and relative frequencies. Associations between categorical variables were tested using the chi-squared and Fisher exact tests. PFS and OS curves were estimated using the Kaplan-Meier method and compared using log rank tests. Cox proportional hazards regression analysis was used for multivariable analysis. All tests were two-sided and considered significant when p < 0.05. Analyses were performed using SPSS version 23.0 (SPSS, Chicago, IL).
CONCLUSIONS
We found that hotspot KRAS+/TP53+ mutations occurred in approximately 8% of cancer patients referred to our institution for phase I clinical trials, and that the KRAS G13 mutation, as well as the TP53 R273 mutation, were associated with poor OS. Antiangiogenesis and gene aberration-related therapies may improve overall survival in patients with concurrent KRAS+/TP53+ hotspot mutant cancer. Also our data also suggest that the proinflammatory state is a key event in cancer development, facilitated through evolving gene aberrations. The current study has provided further support that the combination of modulating the proinflammatory state via immunotherapeutic agents [37] with expanding pharmacologic manipulation to address undruggable molecular cancer targets may lead to novel and effective approaches to the treatment of KRAS+/TP53+ mutant advanced cancer.
Acknowledgments
The authors thank patients, faculty, and staff in the Department of Investigational Cancer Therapeutics and the Institute of Personalized Cancer Therapeutics at The University of Texas MD Anderson Cancer Center for their participation in phase I clinical trials and patient care; Le Hung, PhD in the Department of Investigational Cancer Therapeutics for assistance with the patient database search; and Erica A Goodoff in the Department of Scientific Publications at MD Anderson for assistance with editing the manuscript. ZW is supported in part by the National Natural Sciences Foundation, China (81101778 and 81472206) and Beijing Natural Science Foundation, China (7172045).
Footnotes
CONFLICTS OF INTEREST
The authors declare that there are no conflicts of interest regarding the publication of this paper.
REFERENCES
- 1.Cox AD, Fesik SW, Kimmelman AC, Luo J, Der CJ. Drugging the undruggable RAS: Mission possible? Nature reviews Drug discovery. 2014;13:828–851. doi: 10.1038/nrd4389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kandoth C, McLellan MD, Vandin F, Ye K, Niu B, Lu C, Xie M, Zhang Q, McMichael JF, Wyczalkowski MA, Leiserson MD, Miller CA, Welch JS, et al. Mutational landscape and significance across 12 major cancer types. Nature. 2013;502:333–339. doi: 10.1038/nature12634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Singh H, Longo DL, Chabner BA. Improving Prospects for Targeting RAS. Journal of clinical oncology. 2015;33:3650–3659. doi: 10.1200/JCO.2015.62.1052. [DOI] [PubMed] [Google Scholar]
- 4.Okumura S, Janne PA. Molecular pathways: the basis for rational combination using MEK inhibitors in KRAS-mutant cancers. Clinical cancer research. 2014;20:4193–4199. doi: 10.1158/1078-0432.CCR-13-2365. [DOI] [PubMed] [Google Scholar]
- 5.Tsuchida N, Murugan AK, Grieco M. Kirsten Ras* oncogene: Significance of its discovery in human cancer research. Oncotarget. 2016;7:46717–46733. doi: 10.18632/oncotarget.8773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Szylberg L, Janiczek M, Popiel A, Marszalek A. Large Bowel Genetic Background and Inflammatory Processes in Carcinogenesis—Systematic Review. Advances in clinical and experimental medicine. 2015;24:555–563. doi: 10.17219/acem/31239. [DOI] [PubMed] [Google Scholar]
- 7.O’Dell MR, Huang JL, Whitney-Miller CL, Deshpande V, Rothberg P, Grose V, Rossi RM, Zhu AX, Land H, Bardeesy N, Hezel AF. Kras(G12D) and p53 mutation cause primary intrahepatic cholangiocarcinoma. Cancer research. 2012;72:1557–1567. doi: 10.1158/0008-5472.CAN-11-3596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chow OS, Kuk D, Keskin M, Smith JJ, Camacho N, Pelossof R, Chen CT, Chen Z, Avila K, Weiser MR, Berger MF, Patil S, Bergsland E, et al. KRAS and combined KRAS/TP53 mutations in locally advanced rectal cancer are independently associated with decreased response to neoadjuvant therapy. Annals of surgical oncology. 2016;23:2548–55. doi: 10.1245/s10434-016-5205-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rivlin N, Brosh R, Oren M, Rotter V. Mutations in the p53 tumor suppressor gene: important milestones at the various steps of tumorigenesis. Genes Cancer. 2011;2:466–474. doi: 10.1177/1947601911408889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bigi A, Beltrami E, Trinei M, Stendardo M, Pelicci PG, Giorgio M. Cyclophilin D counteracts P53-mediated growth arrest and promotes Ras tumorigenesis. Oncogene. 2016;35:5132–43. doi: 10.1038/onc.2016.42. [DOI] [PubMed] [Google Scholar]
- 11.Cancer Genome Atlas Research N Comprehensive molecular profiling of lung adenocarcinoma. Nature. 2014;511:543–550. doi: 10.1038/nature13385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bailey P, Chang DK, Nones K, Johns AL, Patch AM, Gingras MC, Miller DK, Christ AN, Bruxner TJ, Quinn MC, Nourse C, Murtaugh LC, Harliwong I, et al. Genomic analyses identify molecular subtypes of pancreatic cancer. Nature. 2016;531:47–52. doi: 10.1038/nature16965. [DOI] [PubMed] [Google Scholar]
- 13.Said R, Ye Y, Falchook GS, Janku F, Naing A, Zinner R, Blumenschein GR, Jr, Fu S, Hong DS, Piha-Paul SA, Wheler JJ, Kurzrock R, Palmer GA, et al. Outcomes of patients with advanced cancer and KRAS mutations in phase I clinical trials. Oncotarget. 2014;5:8937–8946. doi: 10.18632/oncotarget.2339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Said R, Ye Y, Hong DS, Janku F, Fu S, Naing A, Wheler JJ, Kurzrock R, Thomas C, Palmer GA, Hess KR, Aldape K, Tsimberidou AM. Characteristics and survival of patients with advanced cancer and p53 mutations. Oncotarget. 2014;5:3871–3879. doi: 10.18632/oncotarget.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Acin S, Li Z, Mejia O, Roop DR, El-Naggar AK, Caulin C. Gain-of-function mutant p53 but not p53 deletion promotes head and neck cancer progression in response to oncogenic K-ras. The Journal of pathology. 2011;225:479–489. doi: 10.1002/path.2971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ji H, Ramsey MR, Hayes DN, Fan C, McNamara K, Kozlowski P, Torrice C, Wu MC, Shimamura T, Perera SA, Liang MC, Cai D, Naumov GN, et al. LKB1 modulates lung cancer differentiation and metastasis. Nature. 2007;448:807–810. doi: 10.1038/nature06030. [DOI] [PubMed] [Google Scholar]
- 17.Hunter SM, Anglesio MS, Ryland GL, Sharma R, Chiew YE, Rowley SM, Doyle MA, Li J, Gilks CB, Moss P, Allan PE, Stephens AN, Huntsman DG, et al. Molecular profiling of low grade serous ovarian tumours identifies novel candidate driver genes. Oncotarget. 2015;6:37663–37677. doi: 10.18632/oncotarget.5438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hou MM, Wang Z, Janku F, Piha-Paul S, Naing A, Hong D, Westin S, Coleman RL, Sood AK, Tsimberidou AM, Subbiah V, Wheler J, Zinner R, et al. Continuous anti-angiogenic therapy after tumor progression in patients with recurrent high-grade epithelial ovarian cancer: phase I trial experience. Oncotarget. 2016;7:35132–43. doi: 10.18632/oncotarget.9048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fu S, Hou MM, Naing A, Janku F, Hess K, Zinner R, Subbiah V, Hong D, Wheler J, Piha-Paul S, Tsimberidou A, Karp D, Araujo D, et al. Phase I study of pazopanib and vorinostat: a therapeutic approach for inhibiting mutant p53-mediated angiogenesis and facilitating mutant p53 degradation. Annals of oncology. 2015;26:1012–1018. doi: 10.1093/annonc/mdv066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lazo JS, Sharlow ER. Drugging Undruggable Molecular Cancer Targets. Annual review of pharmacology and toxicology. 2016;56:23–40. doi: 10.1146/annurev-pharmtox-010715-103440. [DOI] [PubMed] [Google Scholar]
- 21.Hantschel O, Grebien F, Superti-Furga G. Targeting allosteric regulatory modules in oncoproteins: “drugging the undruggable”. Oncotarget. 2011;2:828–829. doi: 10.18632/oncotarget.354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Arkenau HT, Barriuso J, Olmos D, Ang JE, de Bono J, Judson I, Kaye S. Prospective validation of a prognostic score to improve patient selection for oncology phase I trials. Journal of clinical oncology. 2009;27:2692–2696. doi: 10.1200/JCO.2008.19.5081. [DOI] [PubMed] [Google Scholar]
- 23.Wheler J, Tsimberidou AM, Hong D, Naing A, Falchook G, Piha-Paul S, Fu S, Moulder S, Stephen B, Wen S, Kurzrock R. Survival of 1,181 patients in a phase I clinic: the MD Anderson Clinical Center for targeted therapy experience. Clinical cancer research. 2012;18:2922–2929. doi: 10.1158/1078-0432.CCR-11-2217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chen Z, Cheng K, Walton Z, Wang Y, Ebi H, Shimamura T, Liu Y, Tupper T, Ouyang J, Li J, Gao P, Woo MS, Xu C, et al. A murine lung cancer co-clinical trial identifies genetic modifiers of therapeutic response. Nature. 2012;483:613–617. doi: 10.1038/nature10937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fu S, McQuinn L, Naing A, Wheler JJ, Janku F, Falchook GS, Piha-Paul SA, Tu D, Howard A, Tsimberidou A, Zinner R, Hong DS, Kurzrock R. Barriers to study enrollment in patients with advanced cancer referred to a phase I clinical trials unit. The oncologist. 2013;18:1315–1320. doi: 10.1634/theoncologist.2013-0202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Janku F, Hong DS, Fu S, Piha-Paul SA, Naing A, Falchook GS, Tsimberidou AM, Stepanek VM, Moulder SL, Lee JJ, Luthra R, Zinner RG, Broaddus RR, et al. Assessing PIK3CA and PTEN in early-phase trials with PI3K/AKT/mTOR inhibitors. Cell reports. 2014;6:377–387. doi: 10.1016/j.celrep.2013.12.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Peeters M, Douillard JY, Van Cutsem E, Siena S, Zhang K, Williams R, Wiezorek J. Mutant KRAS codon 12 and 13 alleles in patients with metastatic colorectal cancer: assessment as prognostic and predictive biomarkers of response to panitumumab. Journal of clinical oncology. 2013;31:759–765. doi: 10.1200/JCO.2012.45.1492. [DOI] [PubMed] [Google Scholar]
- 28.Ilm K, Kemmner W, Osterland M, Burock S, Koch G, Herrmann P, Schlag PM, Stein U. High MACC1 expression in combination with mutated KRAS G13 indicates poor survival of colorectal cancer patients. Molecular cancer. 2015;14:38. doi: 10.1186/s12943-015-0316-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Seagle BL, Yang CP, Eng KH, Dandapani M, Odunsi-Akanji O, Goldberg GL, Odunsi K, Horwitz SB, Shahabi S. TP53 hot spot mutations in ovarian cancer: selective resistance to microtubule stabilizers in vitro and differential survival outcomes from The Cancer Genome Atlas. Gynecologic oncology. 2015;138:159–164. doi: 10.1016/j.ygyno.2015.04.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Serresi M, Gargiulo G, Proost N, Siteur B, Cesaroni M, Koppens M, Xie H, Sutherland KD, Hulsman D, Citterio E, Orkin S, Berns A, van Lohuizen M. Polycomb Repressive Complex 2 Is a Barrier to KRAS-Driven Inflammation and Epithelial-Mesenchymal Transition in Non-Small-Cell Lung Cancer. Cancer cell. 2016;29:17–31. doi: 10.1016/j.ccell.2015.12.006. [DOI] [PubMed] [Google Scholar]
- 31.Aguilera-Aguirre L, Bacsi A, Radak Z, Hazra TK, Mitra S, Sur S, Brasier AR, Ba X, Boldogh I. Innate inflammation induced by the 8-oxoguanine DNA glycosylase-1-KRAS-NF-kappaB pathway. Journal of immunology. 2014;193:4643–4653. doi: 10.4049/jimmunol.1401625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pal S, Bhattacharjee A, Ali A, Mandal NC, Mandal SC, Pal M. Chronic inflammation and cancer: potential chemoprevention through nuclear factor kappa B and p53 mutual antagonism. Journal of inflammation. 2014;11:23. doi: 10.1186/1476-9255-11-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hou MM, Liu X, Wheler J, Naing A, Hong D, Coleman RL, Tsimberidou A, Janku F, Zinner R, Lu K, Kurzrock R, Fu S. Targeted PI3K/AKT/mTOR therapy for metastatic carcinomas of the cervix: A phase I clinical experience. Oncotarget. 2014;5:11168–11179. doi: 10.18632/oncotarget.2584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Meric-Bernstam F, Brusco L, Shaw K, Horombe C, Kopetz S, Davies MA, Routbort M, Piha-Paul SA, Janku F, Ueno N, Hong D, De Groot J, Ravi V, et al. Feasibility of large-scale genomic testing to facilitate enrollment onto genomically matched clinical trials. Journal of clinical oncology. 2015;33:2753–2762. doi: 10.1200/JCO.2014.60.4165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Therasse P, Arbuck SG, Eisenhauer EA, Wanders J, Kaplan RS, Rubinstein L, Verweij J, Van Glabbeke M, van Oosterom AT, Christian MC, Gwyther SG. New guidelines to evaluate the response to treatment in solid tumors. European Organization for Research and Treatment of Cancer, National Cancer Institute of the United States, National Cancer Institute of Canada. J Natl Cancer Inst. 2000;92:205–216. doi: 10.1093/jnci/92.3.205. [DOI] [PubMed] [Google Scholar]
- 36.Eisenhauer EA, Therasse P, Bogaerts J, Schwartz LH, Sargent D, Ford R, Dancey J, Arbuck S, Gwyther S, Mooney M, Rubinstein L, Shankar L, Dodd L, et al. New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1) Eur J Cancer. 2009;45:228–247. doi: 10.1016/j.ejca.2008.10.026. [DOI] [PubMed] [Google Scholar]
- 37.Baniyash M, Sade-Feldman M, Kanterman J. Chronic inflammation and cancer: suppressing the suppressors. Cancer immunology, immunotherapy. 2014;63:11–20. doi: 10.1007/s00262-013-1468-9. [DOI] [PMC free article] [PubMed] [Google Scholar]