

Summary
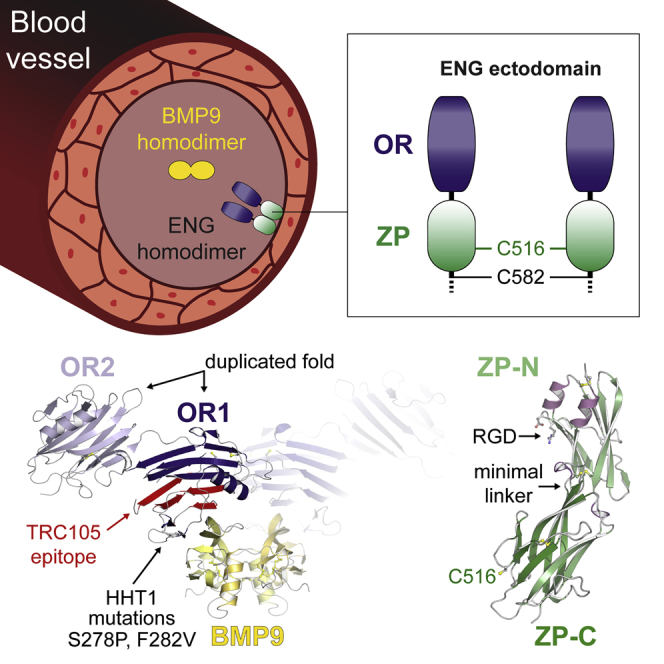
Endoglin (ENG)/CD105 is an essential endothelial cell co-receptor of the transforming growth factor β (TGF-β) superfamily, mutated in hereditary hemorrhagic telangiectasia type 1 (HHT1) and involved in tumor angiogenesis and preeclampsia. Here, we present crystal structures of the ectodomain of human ENG and its complex with the ligand bone morphogenetic protein 9 (BMP9). BMP9 interacts with a hydrophobic surface of the N-terminal orphan domain of ENG, which adopts a new duplicated fold generated by circular permutation. The interface involves residues mutated in HHT1 and overlaps with the epitope of tumor-suppressing anti-ENG monoclonal TRC105. The structure of the C-terminal zona pellucida module suggests how two copies of ENG embrace homodimeric BMP9, whose binding is compatible with ligand recognition by type I but not type II receptors. These findings shed light on the molecular basis of the BMP signaling cascade, with implications for future therapeutic interventions in this fundamental pathway.
Keywords: bone morphogenetic protein receptors, cell surface receptors, endoglin, growth differentiation factor 2, hereditary hemorrhagic telangiectasia, orphan domain, protein interaction domains and motifs, TGF-β superfamily proteins, x-ray crystallography, zona pellucida domain
Graphical Abstract
Highlights
-
•
Crystal structures of human ENG and its complex with BMP9 were determined
-
•
The orphan domain of ENG adopts a fold that explains the effect of HHT1 mutations
-
•
ZP module-mediated dimerization of ENG creates a clamp that secures homodimeric BMP9
-
•
ENG-bound BMP9 can interact with the ALK1 receptor but not the ActRIIB receptor
Endoglin (ENG)/CD105, a key player in angiogenesis and vascular homeostasis, is mutated in the genetic disorder HHT1 and implicated in tumor angiogenesis and preeclampsia. Saito et al. determine structures of human ENG alone and in complex with the physiological ligand BMP9, shedding light onto the molecular basis of BMP signaling.
Introduction
The transforming growth factor β (TGF-β) superfamily consists of type I and II serine/threonine kinase receptors that interact with structurally related ligands, including TGF-βs and bone morphogenetic proteins (BMPs). Ligand-induced receptor complex formation results in phosphorylation of a type I receptor by a type II receptor, which, in turn, leads to phosphorylation of one of several downstream Smad proteins and signal transduction (Horbelt et al., 2012).
ENG is an abundant type I transmembrane glycoprotein of the vascular endothelium that was initially recognized as a co-receptor for TGF-β1 and TGF-β3, associating with TGF-β type I and II receptors (Cheifetz et al., 1992, Gougos and Letarte, 1988a, Gougos and Letarte, 1990, Quackenbush and Letarte, 1985). In most cells, the TGF-β type I receptor is activin receptor-like kinase 5 (ALK5), whereas, in endothelial cells, there is also ALK1. ENG and ALK1 were subsequently shown to bind with much higher affinity to BMP9 (also known as growth differentiation factor 2 [GDF2]) than to TGF-β1 and mediate its proliferation signal (Brown et al., 2005, Nolan-Stevaux et al., 2012, Scharpfenecker et al., 2007). Post-translationally processed homodimeric BMP9 mediates signal transduction via a set of interactions involving ENG, ALK1, and a BMP type II receptor (BMPRII or ActRIIB) (Bidart et al., 2012, Brown et al., 2005, David et al., 2007, Kienast et al., 2016, Scharpfenecker et al., 2007, Upton et al., 2009). Like ALK1, ENG is essential for heart development and vascular homeostasis; Eng−/− mice die at mid-gestation because of defective cardiovascular development and angiogenesis (Bourdeau et al., 1999, Li et al., 1999). ENG is overexpressed in the vascular endothelium of solid tumors (Bernabeu et al., 2009, Miller et al., 1999), and functionally blocking antibodies to ENG can inhibit tumor growth via the ENG/BMP9/ALK1 pathway (Lebrin et al., 2004). Mutations in the ENG gene cause the autosomal dominant disorder hereditary hemorrhagic telangiectasia type 1 (HHT1), a vascular dysplasia with life-threatening consequences that affects 1 in 10,000 individuals and is characterized by arteriovenous malformations, recurrent hemorrhages, and numerous sequelae (Abdalla and Letarte, 2006, Bourdeau et al., 1999, McAllister et al., 1994). Over 500 human ENG gene variants linked to HHT1 are currently reported in the University of Utah Department of Pathology HHT and ENG Database (http://www.arup.utah.edu/database/ENG/ENG_welcome.php; Stenson et al., 2014). Moreover, a circulating form of the ENG ectodomain, suggested to be released upon C-terminal cleavage of ENG by matrix metalloproteinase 14 (MMP-14) (Hawinkels et al., 2010), is increased at early stages of preeclampsia and serves as a predictive marker (Gregory et al., 2014, Venkatesha et al., 2006). Despite the important role of ENG in the vascular system, its association with several diseases and potential use as a target of anti-angiogenic cancer therapy (Bernabeu et al., 2009, Paauwe et al., 2016), no high-resolution structural information is available for the protein and how it interacts with the key ligand BMP9 (Castonguay et al., 2011).
ENG is predominantly expressed as an ∼180 kDa homodimer linked by intermolecular disulfide bonds (Gougos and Letarte, 1988b, Gougos and Letarte, 1990). It is composed of an N-terminal orphan region (OR), a C-terminal bipartite zona pellucida module (ZP), a single transmembrane domain, and a short cytoplasmic peptide that differs in the L and S isoforms of the protein (Figure S1A; Bellón et al., 1993, Bernabeu et al., 2009, Gregory et al., 2014, Litscher and Wassarman, 2015). Extensive attempts to crystallize the full-length ectodomain of ENG (ECTO; residues E26–G586), using different strategies to reduce N-glycan heterogeneity, were not successful. We reasoned that this may be caused by flexibility of the OR/ZP linker (Figure S1A, arrow), which could underlie the different conformations observed by small-angle X-ray scattering (Alt et al., 2012, Van Le et al., 2009) and negative stain electron microscopy (Llorca et al., 2007). To overcome this obstacle, we combined a divide and conquer strategy with a recently developed eukaryotic expression system where proteins of interest are fused to a mammalianized version of bacterial maltose-binding protein (mMBP) (Bokhove et al., 2016a). Using this approach, we first elucidated the crystal structure of ENG OR, which plays a key role in signaling by binding BMP9 (Castonguay et al., 2011) and does not show homology to proteins of known structure (Alt et al., 2012, Llorca et al., 2007); subsequently, we determined the structure of the OR-BMP9 complex as well as the structure of ENG ZP.
The structure of ENG OR reveals a new fold wherein a β helix-like domain with an unprecedented topology has been duplicated, followed by circular permutation. The electron density map of ENG OR in complex with BMP9 shows how the knuckle region of the ligand interacts hydrophobically with a ridge at the edge of OR, including residues previously implicated in BMP9 binding (Mallet et al., 2015). Finally, the crystal structure of ENG ZP reveals a minimal fold with the short ZP-N/ZP-C interdomain linker signature of non-polymerizing ZP modules (Bokhove et al., 2016b). Based on the structures of OR and ZP, a model can be presented of the ENG disulfide-bonded dimer in complex with BMP9 and the type I receptor ALK1.
Results and Discussion
The Crystal Structure of OR Reveals Two Intertwined Domains
The structure of OR (ENG E26–S337) fused to mMBP (MOR; Figures S1B and S2A–S2C) was solved at 2.4 Å resolution by molecular replacement (MR) with MBP (Table S1). The electron density map (Figure S2B) reveals that OR is a monomer and consists of two domains that share a common new fold (OR1/OR2; Figure 1). Each domain consists of 12 β strands, which form a parallel β helix-like structure, and a single α helix. Interestingly, OR2 (β3–β14 and α1) is inserted C-terminally to OR1 β1–β2, after which OR1 (β15–β24 and α2) is completed (Figure 1B). Both domains contain a conserved disulfide bond involving C30–C207 and C53–C182, respectively (Figures 1A–1C; Figure S2D). Two additional Cys in OR1, C242 and C330 (Figure S1A), are likely to form a third disulfide that is not resolved because of structural disorder of residues S329–S337. However, an MOR variant lacking both Cys is secreted as efficiently as wild-type MOR and retains BMP9 binding activity, indicating that C242 and C330 are not crucial for folding and interaction with the ligand (Figure S1C).
Figure 1.

ENG OR Consists of a Duplicated and Circularly Permutated Domain
(A) Cartoon representation of the OR crystal structure, consisting of two domains (OR1/OR2) and rainbow-colored from blue (N terminus) to red (C terminus). Cys residues are shown in ball-and-stick representation.
(B) OR topology with OR1 and OR2 β helices colored dark and light blue, respectively. β Strands (arrows) are numbered.
(C) Superposition of OR1 and OR2, colored as in (B).
(D) Suggested evolution of ENG OR by gene duplication and circular permutation.
(E) Superposition of ENG OR2 and tubulin cofactor C (TBCC).
See also Figures S1–S3 and S6 and Table S1.
OR Is a Result of Gene Duplication Followed by Circular Permutation
OR1 and OR2 are only 18% identical in sequence but can be superimposed with a Cα root-mean-square deviation of 2.2 Å for 102 overlapping residues (Figure 1C; Figure S3A). Although OR2 does not require OR1 for folding and secretion (Figure S3B), an OR1-only construct was not secreted by itself (Figure S3C). Notably, the relative sequence position of the Cys moieties of the conserved disulfide is reversed in OR1 compared with OR2 so that C30 and C207 correspond to C182 and C53, respectively. Moreover, the N-terminal sequence of OR1 (including strands β1 and β2) aligns with the C-terminal part of its OR2 domain (Figure S3A). Taken together, these observations suggest that OR1 was generated by a duplication of an ancestral OR2 gene, followed by circular permutation (Figure 1D). Considering that ENG is only found in amniotes, a possible evolutionary relationship for OR2 may be, in turn, indicated by the fact that its structure distantly resembles tubulin cofactor C (Figure 1E). However, this β-helical molecular chaperone has a markedly different topology with a C-terminal α helix (Nithianantham et al., 2015).
BMP9 Binds to a Conserved Hydrophobic Groove of OR1
To gain insights into how ENG modulates signaling (including cell proliferation) by interacting with ligands, we co-expressed MOR and BMP9 in mammalian cells and crystallized the resulting complex (Figure S4A). Although the crystals only diffracted to ∼4.5 Å, the data could be phased by MR using mMBP, OR1, and an independently determined structure of BMP9 (Figure S4B; Table S1). In the complex crystals, the disulfide-bonded BMP9 homodimer spans two adjacent asymmetric units by lying on a symmetry axis (Figure S4C), and the knuckle of each BMP9 molecule binds to a copy of OR1 (Figure S4D), resulting in a “dimer of heterodimers” (Figure 2A). This arrangement is consistent with co-expression experiments of His- and Myc-tagged MOR with BMP9 followed by His pull-down. Immunoblot analysis shows that Myc-MOR co-elutes with His-MOR only in the presence of BMP9, indicating that the dimeric ligand binds two copies of OR that do not interact with each other (Figure 2B). In agreement with the fact that mMBP and BMP9 are positioned on opposite sides of OR1 in the crystals (Figure S4C), mMBP does not inhibit ENG-BMP9 binding (Figures 2A and 2B), nor does it have the ability to bind BMP9 itself (Figure S4E, lane 11). Moreover, the OR1-BMP9 interaction is consistent with the observation that OR2—which appears to have some degree of flexibility relative to OR1 (Figure S4F)—does not bind BMP9 when expressed by itself (Figure S3B).
Figure 2.

A Hydrophobic Surface of ENG OR1 Binds to the Knuckle Region of BMP9
(A) Crystal structure of the OR-BMP9 complex, showing the dimer generated by crystal symmetry. OR is colored as in Figures 1B and 1C, and BMP9 chains are shown in yellow.
(B) Non-reducing immunoblot and Coomassie (CBB) analysis of His pull-down of MOR/BMP9 co-expression experiments. Two copies of OR bind dimeric BMP9 but do not interact with each other.
(C) Detail of the OR1-BMP9 interface, with the surface of OR1 colored by hydrophobicity (orange, most hydrophobic; white, least hydrophobic).
(D) Alternative view of (C), with both proteins in cartoon representation and interface residues shown in ball-and-stick representation.
(E) BMP9 is pulled down by non-mMBP fused wild-type OR but not by double or triple mutant ORs, indicating that the residues shown in (D) are essential for binding.
See also Figures S4 and S8 and Table S1.
The structure of the complex reveals that the ENG-BMP9 interface is largely hydrophobic (Figure 2C). It comprises OR1 residues that contribute to ENG strand β20 or immediately precede it (I271 and Q270 or N268, respectively; Figure 2D; Figure S3A), which are close to BMP9 residues P412, T413, and Y416 (Figure 2D). Consistent with a nanomolar binding affinity for BMP9 (Table 1; Alt et al., 2012, Castonguay et al., 2011, Gregory et al., 2014), individual mutations of ENG residues N268, Q270, and I271 are not sufficient to disrupt the interaction between unfused OR and BMP9 (Figure S4E, lanes 2, 4, and 5). However, double and triple combinations of these mutations show that OR1 residues Q270 and I271 (Figure 2D; Figure S3A), which are highly conserved in mammals, are essential for BMP9 binding (Figure 2E). Importantly, neither the aforementioned individual mutations nor their combinations affect OR folding because the corresponding proteins are secreted comparably with wild-type OR (Figure S4E, lanes 2, 4, 5, and 10; Figure 2E). Similarly, alanine mutants of D246, Y277, and F290 are secreted normally, although, in this case, the mutant proteins are still able to bind BMP9 (Figure S4E, lanes 1, 6, and 9). In contrast, mutation of the buried hydrophobic residue M269 severely affects OR folding and secretion; however, the fraction of protein that is correctly folded and secreted retains BMP9-binding activity (Figure S4E, lane 3). Interestingly, 9 of 11 residues located in the BMP9 β6–β7 region that contacts OR1 are conserved in BMP10, the other high-affinity ligand of ENG (Castonguay et al., 2011). On the contrary, these residues differ significantly in BMPs that either do not bind ENG or interact only weakly with it (Castonguay et al., 2011, Gregory et al., 2014; Figure S4G). Most importantly, the interface region of ENG also involves conserved S278 and invariant F282, whose mutation in HHT1 patients disrupts BMP9-dependent signaling (Mallet et al., 2015) as well as in vitro binding between OR and BMP9 (Figure 2E; Figure S4E, lanes 7 and 8). Further supporting the cell signaling studies (Mallet et al., 2015) and our pull-down experiments (Figure 2E), quantitative analysis of protein-protein interaction by biolayer interferometry (BLI) shows that the mutations Q270A/I271A, S278P, and F282V completely abolish the binding of unfused OR to BMP9. Moreover, BLI confirms that mMBP fusion does not interfere with the interaction between wild-type OR and BMP9 (Table 1; Figure S8).
Table 1.
Kinetics Analysis of ENG-BMP9 Interaction by BLI
| KD (M) | kon (M−1s−1) | koff (s−1) | |
|---|---|---|---|
| ECTO | 9.81 × 10−9 | 2.0 × 105 ± 1.69 × 103 | 1.98 × 10−3 ± 1.15 × 10−5 |
| OR | 2.95 × 10−7 | 2.95 × 105 ± 8.96 × 103 | 8.71 × 10−2 ± 9.08 × 10−4 |
| MOR | 1.79 × 10−7 | 3.49 × 105 ± 1.3 × 104 | 6.22 × 10−2 ± 7.76 × 10−4 |
| OR F282V | NBD | ||
| OR S278P | NBD | ||
| OR Q270A/I271A | NBD | ||
| mMBP | NBD |
NBD, no binding detected under the tested experimental conditions. See also Figure S8.
Interestingly, the epitope of TRC105/SN6j, a monoclonal antibody to ENG used in clinical trials and acting as a tumor suppressor (She and Seon, 2001, Bernabeu et al., 2009, Nolan-Stevaux et al., 2012, Paauwe et al., 2016), overlaps with the BMP9-binding region of OR1 (Figure 2A, red). This explains why TRC105, but not SN6h, whose target epitope is elsewhere on ENG (Figure 2A, cyan), inhibits BMP9-dependent signaling (Nolan-Stevaux et al., 2012). Similarly, overlap of the BMP9 binding site with the prodomain region of pro-BMP9 (Mi et al., 2015) (Figure S4H) explains why the prodomain is displaced upon interaction with ENG (Kienast et al., 2016).
The ZP Module of ENG Adopts a Minimal Fold with a Short ZP-N/ZP-C Linker
Whereas the BMP9-binding activity of ENG depends on OR, the ZP moiety mediates its dimerization and spatial organization within the complex. Whereas an unfused ZP construct was barely secreted (Bokhove et al., 2016a), mMBP-fused ENG P338–G581 (MZP; Figure S1B) could be efficiently expressed as a mixture of monomer and intermolecularly disulfide-bonded homodimer (Figure S5A, left). Although the relative ratio of the latter species significantly increased upon sample concentration (Figure S5A, right), crystallization selected the monomer (Figure S5B). The 2.7 Å structure of MZP displays the hallmarks of a typical ZP module composed of ZP-N and ZP-C domains with C1-C4, C2-C3 and C5-C7 disulfide connectivity, respectively (Bokhove et al., 2016b, Han et al., 2010, Litscher and Wassarman, 2015; Figures 3A and 4A; Figures S5C, S5D, and S6); however, it also contains several specific features. Consistent with a role of ENG in integrin-mediated cell adhesion (Gougos and Letarte, 1990, Rossi et al., 2015), the RGD motif of the protein constitutes a short exposed loop of ZP-N (Figure 3A, left). Moreover, following the disordered OR/ZP linker residues P338–D348, C350 before αA forms a disulfide with C382 located after αBC (Figure 3A; Figure S5D). This is reminiscent of a comparable disulfide in uromodulin (UMOD), another ZP module-containing protein (Bokhove et al., 2016b). In agreement with an important structural role of this disulfide, loss of C382 is associated with HHT1 (Stenson et al., 2014; University of Utah Department of Pathology HHT and ENG Database, http://www.arup.utah.edu/database/ENG/ENG_welcome.php), and mutation of C350 and/or C382 disrupts ENG folding and secretion (Figures 4B and 4C).
Figure 3.

ENG Contains a Minimal ZP Module with Closely Interacting ZP-N and ZP-C Domains
(A) Crystal structure of ENG ZP, colored according to secondary structure. Canonical disulfide bonds and the RGD motif are shown in ball-and-stick representation. The extra ZP-N disulfide (C350–C382) and the free ZP-C Cys involved in dimerization (C516) are also indicated.
(B) Side-by-side comparison of the ENG and UMOD ZP modules. The secondary structure elements of ENG ZP-N and ZP-C are only spaced by five residues; thus, these domains are much closer to each other than the corresponding domains of UMOD, which are separated by a long linker (red). As a result, whereas the ZP-N homodimerization surface of UMOD is available to pair with another molecule (black/gray), the equivalent region of ENG interacts with ZP-C and maintains the protein in a monomeric state.
See also Figures S1, S5, and S6 and Table S1.
Figure 4.

Intermolecular Disulfide Bonding of ENG ZP C516 Generates a Homodimeric Clamp for the BMP9 Ligand
(A) The disulfide bonds observed in the structures of OR (Figures 1A and 1B; Figure S2D) and ZP (Figure 3A; Figure S5D) are indicated in red. Because neither ENG E26-S337 (Castonguay et al., 2011) nor MOR (Figure 2B; Figure S1C) make intermolecular disulfide bonds, the pattern suggests that homodimerization of ENG is mediated by a C516-C516 disulfide in addition to C582-C582 (Guerrero-Esteo et al., 2002).
(B) A non-reducing anti-His immunoblot of conditioned medium (M) shows secretion of monomeric and dimeric ENG ZP constructs (white and black arrowheads, respectively). Immunoblot of cell lysates (L) indicates that all constructs are expressed in equal amounts. Consistent with the ENG architecture proposed in (A), mutation of C516 abolishes homodimerization of MZP. Moreover, the additional C350–C382 disulfide found in ENG ZP-N (Figure 3A; Figure S5D) is essential for MZP secretion.
(C) Disulfide connectivity and secretion state of the MZP constructs analyzed in (B). Lack of secretion is symbolized by a red cross.
(D) Immunoblot and Coomassie analyses of co-expression experiments under non-reducing conditions show that two ENG molecules, preferentially a disulfide-bonded dimer, bind homodimeric BMP9.
(E) SEC-MALS analysis of purified His-ECTO-BMP9 complex produced in HEK293S cells reveals two conformations whose common molecular mass (red line) is consistent with a 2:2 stoichiometry.
(F) Theoretical model of the complete extracellular region of homodimeric ENG in complex with BMP9. The relative orientation of the ZP-C domains cross-linked by the C516-C516 intermolecular disulfide (dashed red ellipse) is compatible with the C582-C582 disulfide (Guerrero-Esteo et al., 2002) at the C terminus of the ectodomain (dashed black ellipse).
See also Figures S1 and S6.
Although the structure of ZP does not support a role for C350 in ENG dimerization (Alt et al., 2012), Cys mutagenesis reveals that, in addition to a C582-C582 disulfide (Guerrero-Esteo et al., 2002), dimerization involves C516 (Figures 4A–4C). This agrees with the observation that, in the monomeric ZP structure, C516 is exposed on the surface of ZP-C (Figure 3A). By consisting of only 133 residues and containing a single intramolecular disulfide bond, the latter represents a simplified version of the ZP-C domain found in other ZP modules proteins (Bokhove et al., 2016b, Han et al., 2010) (Figure 3B), including closely related betaglycan (BG)/TGF-β receptor III (Bernabeu et al., 2009, Diestel et al., 2013, Lin et al., 2011) (Figure S6).
In the ZP module of ENG, a minimal interdomain linker forces a 310 helix within the FG loop of ZP-C to pack against the βA-βG surface of ZP-N; remarkably, the corresponding surface of UMOD mediates ZP-N/ZP-N homodimerization and is essential for protein polymerization (Bokhove et al., 2016b) (Figures 3A and 3B). Taken together, these observations support the idea that, in addition to the lack of a consensus cleavage site within the FG loop of ZP-C (Lin et al., 2011), close spacing of the ZP-N and ZP-C domains of ENG and BG (which also contains a helix within its ZP-C FG loop; Figure S6; Lin et al., 2011) underlies their inability to polymerize like other ZP module proteins (Bokhove et al., 2016b) (Figure 3B).
ZP-Mediated Dimerization of ENG Clamps BMP9 within the Signaling Complex
To investigate the solution stoichiometry of the ENG-BMP9 complex, we co-expressed His-MECTO (∼120 kDa), ECTO-Myc (∼80 kDa), and BMP9 (2 × 12.1 kDa) (Figure 4D). This generates five distinct species, shown in colored boxes: His-MECTO homodimer (∼240 kDa, blue), His-MECTO/ECTO-Myc heterodimer (∼200 kDa, green), ECTO-Myc homodimer (∼160 kDa, red), His-MECTO monomer (cyan), and ECTO-Myc monomer (magenta). His pull-down followed by anti-Myc immunoblot detects disulfide-bonded His-MECTO/ECTO-Myc heterodimers bound to BMP9 (Figure 4D, lane 1). Because no monomeric ECTO-Myc is present in lane 1, BMP9 is preferentially bound by disulfide-bonded dimeric ENG. Furthermore, the same lane shows lack of homodimeric ECTO-Myc among the material pulled down by immobilized metal affinity chromatography (IMAC), excluding that BMP9 binds more than one ENG dimer or higher oligomeric forms of the protein. Analysis of a purified ECTO-BMP9 complex by size exclusion chromatography with multi-angle light scattering (SEC-MALS) confirmed these results by detecting a molecular mass of ∼150 kDa (Figure 4E).
Based on the combination of our crystallographic and biochemical data (Figures 1, 2, 3, and 4A–4E), we have modeled the BMP9-bound state of homodimeric ENG (Figure 4F). As a result of the intermolecular cross-linking provided by ZP disulfide C516-C516 (Figures 4B and 4C) as well as membrane-proximal C582-C582 (Guerrero-Esteo et al., 2002), the two ENG molecules generate a clamp-like structure whose OR arms protrude into the extracellular space to secure the knuckles of the BMP9 homodimer. Consistent with this proposed architecture, which is compatible with MMP-14 cleavage of ENG at G586|L587 (Hawinkels et al., 2010), pull-down experiments suggest that ENG ZP does not interact directly with BMP9 (Figure S5E). However, the ZP must indirectly lower the dissociation of BMP9 from OR, because, as previously observed by surface plasmon resonance (SPR) using an OR construct truncated at P338 (Alt et al., 2012), the dissociation rate constant (koff) of the OR-BMP9 complex is significantly higher than that of ECTO-BMP9 (Table 1). Together with the structure and SEC-MALS-derived stoichiometry (Figure 4E), this observation suggests that, by covalently cross-linking two protein chains (Figure 4B), ZP confers an antibody-like architecture to ENG. This has the effect of increasing the avidity of the ENG-BMP9 interaction by presenting two closely spaced OR1 binding sites that both match the homodimeric structure of the ligand and counteract its dissociation from a single OR1 site (Figure 4F). Consistent with this idea, longer OR constructs that are non-physiologically cross-linked into dimers by pairs of C350 (which normally makes an intramolecular disulfide with C382; Figure 3A; Figure S5D) dissociate much slower from BMP9 than monomeric OR (Alt et al., 2012, Castonguay et al., 2011).
Taken together, these considerations suggest that ZP not only provides a structural scaffold that spaces OR from the plasma membrane, positioning BMP9 for interaction with type I and II receptors such as ALK1 and ActRIIB, but also affects signaling by indirectly increasing ligand retention.
The ENG-BMP9 Interface Is Compatible with Binding of ALK1, but not ActRIIB, to BMP9
BMP9-dependent signaling in endothelial cells is thought to be regulated by the association of the ligand with ENG and the type I receptor ALK1 (Scharpfenecker et al., 2007). By merging the model of ENG-BMP9 (Figure 4F) with the structure of ALK1-BMP9 in crystals of ALK1-BMP9-ActRIIB (Townson et al., 2012), the extracellular part of an ENG-BMP9-ALK1 ternary complex can be straightforwardly modeled (Figures 5A and 5B). The resulting assembly suggests that, whereas ALK1 binds to the wrist of BMP9 (Townson et al., 2012), the knuckle region of the ligand is recognized non-competitively by ENG OR1 (Figures 2A–2D). Also considering that ENG and ALK1 are physically spaced from each other by the finger/knuckle region of the ligand (Figure 5A), this agrees with SPR binding studies suggesting that ENG and ALK1 bind independently and non-cooperatively to BMP9 (Alt et al., 2012, Castonguay et al., 2011). Accordingly, a soluble triple complex could be detected upon co-expression of BMP9, ECTO, and the extracellular part of ALK1 (Figure 5C). On the other hand, comparison of the ternary complex model (Figure 5B) with the structure of ALK1-BMP9-ActRIIB (Townson et al., 2012; Figure 5D) indicates that OR1 and the type II receptor ActRIIB compete for the same region of BMP9. This is consistent with SPR-based evidence that ENG and ActRIIB bind to BMP9 in a mutually exclusive way (Castonguay et al., 2011) and likely also applies to BMPRII because previous structural studies showed that type II receptors always interact with the knuckle region of growth factors (Hinck et al., 2016).
Figure 5.

Theoretical Model of the ENG-BMP9-ALK1 Ternary Complex
(A) Combination of the homodimeric ENG-BMP9 model shown in Figure 4F with structural information on how the type I receptor ALK1 binds BMP9 (Townson et al., 2012) suggests a possible architecture for the co-receptor-BMP ligand-type I receptor ternary complex on the plasma membrane.
(B) Top view of the ENG-BMP9-ALK1 model.
(C) SDS-PAGE analysis of ENG ECTO-FLAG, ALK1-His, and BMP9 co-expression followed by His pull-down confirms that the proteins form a triple complex in solution. Consistent with the model depicted in (A) and (B), ALK1 is only able to pull down ECTO when BMP9 is present. All samples were analyzed under non-reducing conditions, except for that marked with R (reducing conditions).
(D) Top view of the crystal structure of the ALK1-BMP9-ActRIIB complex (Townson et al., 2012). Comparison with the ENG-BMP9-ALK1 model (B) suggests that ENG and ActRIIB compete for the same binding region on BMP9.
Together with the observation that ENG binds BMP9 with comparable affinity to ALK1 (Alt et al., 2012) but is significantly more abundant than the type I receptor in endothelial cells (Abdalla et al., 2000, Cheifetz et al., 1992, Ohta et al., 1987, Quackenbush and Letarte, 1985), these findings support a model where homodimeric BMP9 is first efficiently captured and retained on the surface of endothelial cells by the bivalent architecture of ENG, and a ternary complex is then formed upon non-competitive recruitment of ALK1 (Figures 5A–5C). Presentation of the ligand to a type II receptor, a step requiring BMP9 release from its binding site on ENG, would finally initiate signal transduction. Mirroring the case of antagonistic TGF-β family ligands (Aykul and Martinez-Hackert, 2016), this model would corroborate the idea that different receptors can compete for the same ligand to modulate signaling (Mahlawat et al., 2012). Although it remains to be established how captured BMP9 becomes available to ActRIIB/BMPRII, the fact that the ligand has a relatively high koff for a single OR1 site (Table 1) suggests that this interaction may be triggered by transient dissociation of homodimeric BMP9 from one of its two binding sites on ENG. This would temporarily free one of the knuckle regions of BMP9 for recognition by a type II receptor located close to the ternary ENG-BMP9-ALK1 complex on the plasma membrane. Considering that ActRIIB has a lower koff than OR (Table 1; Alt et al., 2012, Aykul and Martinez-Hackert, 2016, Townson et al., 2012), subsequent dissociation of the second knuckle of BMP9 from ENG would effectively result in transfer of the ALK1-bound ligand to the type II receptor. Elucidation of the details of the signal transduction mechanism will require further structural studies of the intact ternary complex. This may adopt a range of conformations not recapitulated by our crystal structure-based model as a result of flexibility at the ENG OR/ZP junction—possibly underlying the two ECTO-BMP9 states observed by SEC-MALS (Figure 4E)—as well as membrane anchoring.
Structural Basis of HHT1
Mapping of almost 100 gene variants linked to HHT1 (Mallet et al., 2015, Paquet et al., 2001, Stenson et al., 2014; University of Utah Department of Pathology HHT and ENG database, http://www.arup.utah.edu/database/ENG/ENG_welcome.php) onto the structure of ENG indicates that, with the exception of previously discussed OR1 residues S278 and F282 (Figures 2D and 2E), the majority of HHT1 missense mutations affect buried hydrophobic residues (Figure S7).
Fifteen representative mutation sites, ten in OR and five in ZP, are highlighted in Figures 6A and 6B, respectively. The structure of ENG reveals that mutations L32R/H, G52D/V, L82H, V125D, L162R, L170P, L221P, I252T, L262P, I384T, and S407Q/N interfere with hydrophobic core packing, explaining the impaired expression and/or secretion of the corresponding mutants (Ali et al., 2011, Mallet et al., 2015, Paquet et al., 2001, Pece-Barbara et al., 1999). The same phenotype is also caused by mutations C53R, C207R, C363Y, C382W/G, C412Y/S, C493Y, and R529C, which—as observed in the case of the mutant C382S (Figure 4B)—disrupt folding by interfering with correct disulfide bond formation (Figures S2D and S5D; Ali et al., 2011, Mallet et al., 2015, Paquet et al., 2001, Pece-Barbara et al., 1999). The two effects are combined in the case of the mutation W149C, which severely impairs trafficking of ENG to the cell surface (Ali et al., 2011, Pece-Barbara et al., 1999) by replacing a largely buried hydrophobic side chain (Figure 6A) with a reactive residue that could introduce a spurious disulfide bond. Finally, ENG secretion is also affected by mutations such as D391Y and R437W (Ali et al., 2011, Mallet et al., 2015), which substitute solvent-exposed residues (Figure 6B) with hydrophobic amino acids.
Figure 6.

HHT1-Associated Mutations Disrupt the Folding of ENG
(A and B) Overview and close-ups of ENG OR (A) and ZP (B), with patient mutation sites highlighted in magenta. Disulfide bonds and C516 are shown in ball-and-stick representation; coloring is according to Figures 2 and 3. Relative percentages of solvent accessibilities of residue side chains are indicated in parenthesis. See also Figure S7.
In summary, analysis of the ENG structure suggests that most HHT1 mutations impair its folding rather than directly hinder its ability to engage in protein-protein interactions. This leads to reduced amount of functional ENG at the cell surface, thereby causing HHT1 by haploinsufficiency (Paquet et al., 2001).
Conclusions
By suggesting how the bivalent architecture of ENG secures a pool of BMP9 molecules on the plasma membrane, where they can bind to type I receptor ALK1 and become transiently available for interaction with the type II receptors ActRIIB and BMPRII, our studies shed light on some of the mechanisms underlying BMP pathway regulation. At the same time, structural knowledge of human ENG and its interaction with BMP9 constitutes a valuable framework for understanding the molecular basis of HHT. This disease is caused primarily (>85%) by mutations in ENG (HHT1; Johnson et al., 1996, McAllister et al., 1994) and ALK1 (HHT2; Johnson et al., 1996, McAllister et al., 1994), but also by mutations in the common downstream signaling SMAD4 (Gallione et al., 2004) as well as in BMP9 itself in a very similar vascular anomaly syndrome (Wooderchak-Donahue et al., 2013). Our findings also contribute to clarifying the tumor-suppressing activity of monoclonal TRC105, which targets the ENG-BMP9 interface (Nolan-Stevaux et al., 2012). Future studies are needed to understand how ENG forms complexes with other ligands of the TGF-β superfamily, for which it has much lower affinities than for BMP9, and to elucidate how ENG contributes to type II receptor-mediated signaling.
Experimental Procedures
DNA Constructs
For secreted expression of unfused proteins, cDNA fragments including tag-encoding sequences were cloned into mammalian expression vector pHLsec (Aricescu et al., 2006). Except in the case of ECTO and OR constructs, which were expressed using the native signal peptide of ENG (residues M1–A25), ENG inserts were cloned in-frame with the Crypα signal peptide-encoding sequence of pHLsec. Constructs encoding non-mMBP-fused, C-terminally 8His-tagged OR1 with its native signal peptide (ENG M1-T46-(GGGS)2-L203-S329-C242S) and extracellular C-terminally 6His-tagged ALK1 (residues D22–G114) with its native signal peptide (residues M1–G21) were synthesized (DNA2.0/ATUM) and cloned into pHLsec. mMBP fusion proteins were expressed using pHLmMBP vectors (Bokhove et al., 2016a), which are available at Addgene (https://www.addgene.org/Luca_Jovine/). C-terminally FLAG-tagged ECTO was expressed using a previously described pCMV5 vector (Gregory, 2011). The cDNA encoding residues M1–R429 of human BMP9 was cloned into pHLsec without any tags so that purification of mature BMP9 depended on its ability to form a complex with His-tagged ENG partners.
Mutations were generated by overlap extension PCR, and all constructs were verified by DNA sequencing (Eurofins Genomics).
Protein Expression
Large-scale production of MOR and the MOR-BMP9 complex for crystallographic studies was performed in HEK293S cells expressing Endoglycosidase H (Endo H)-sensitive, high mannose-type glycoproteins (Chang et al., 2007, Reeves et al., 2002). HEK293S cells were grown in Dulbecco's modified Eagle's medium (DMEM) with 4 mM L-glutamine (Life Technologies/Thermo Fisher Scientific) and 10% (v/v) fetal bovine serum (Saveen & Werner) at 37°C, 5% CO2 using T flasks (150 cm2, BD Biosciences) or cell factories (2,528 cm2, Thermo Fisher Scientific). Transient transfections were essentially performed as described previously (Aricescu et al., 2006, Bokhove et al., 2016a). MOR-BMP9 and ECTO-BMP9 co-transfections were performed using a 3:7 DNA mass ratio of MOR versus BMP9. Because MZP does not contain N-glycosylation sites, it was expressed in HEK293T cells.
For functional assays, complexes were obtained by co-expression of His-MOR, Myc-MOR, and BMP9 in HEK293T cells using a 2.5:2.5:5 DNA ratio. His-MECTO, ECTO-Myc, and BMP9 complexes were prepared using a 2:3:5 DNA ratio.
Protein Purification
Three days post-transfection, conditioned medium containing secreted proteins was harvested, 0.22 μm-filtered (Sarstedt), and adjusted to 20 mM sodium 2-[4-(2-hydroxyethyl)piperazin-1-yl]ethanesulfonate (Na-HEPES) (pH 7.8), 150 mM NaCl, 5 mM imidazole (IMAC binding buffer). 10 mL nickel-nitrilotriacetic acid (Ni-NTA) agarose slurry (QIAGEN) was added per liter of medium and incubated overnight at 4°C on an overhead shaker (Heidolph). IMAC beads were then collected in a column, washed with binding buffer, and batch-eluted with a binding buffer containing 500 mM imidazole. The eluted material was concentrated using a spin filter (Millipore) with an appropriate molecular weight cutoff. Concentrated protein was subjected to SEC directly after IMAC (HEK293T-produced MZP) or after deglycosylation (HEK293S-produced MOR or MOR-BMP9 complex). Endo H deglycosylation of MOR and MOR-BMP9 was performed for 1 hr at 37°C in 120 mM sodium-potassium phosphate (pH 6.0) using a 1:10 mass ratio of enzyme to substrate. Concentrated proteins were applied to a Superdex 200 26/60 SEC column connected to an ÄKTAFPLC system (GE Healthcare). The composition of the SEC running buffer was 20 mM Na-HEPES (pH 7.8), 100 mM NaCl, and 1.5 mM D-maltose for MOR; 500 mM and 200 mM NaCl were used for the MOR-BMP9 complex and MZP, respectively. Protein-containing fractions were pooled, concentrated, and used for crystallization trials.
For functional assays, media were harvested from HEK293T cells transfected in 150 cm2 T flasks (Sarstedt). Small-scale IMAC or IMAC/SEC pull-downs were performed using the same protocol as above, linearly scaled down. Following pull-down, IMAC beads with bound ENG or MZP constructs were incubated with medium of HEK293T cells expressing BMP9 (consecutive pull-downs). Media obtained from His-MOR, Myc-MOR, and BMP9 co-expression were purified using IMAC and SEC, whereas media obtained from His-MECTO, ECTO-Myc, MZP, ZP, and BMP9 co-expression or ALK1-His, ECTO-FLAG, and BMP9 co-expression were purified by IMAC. In parallel, we established that mMBP does not pull down BMP9 (Figure S4E, lane 11).
Protein Analysis
Cell lysates were prepared by resuspending cells from a 10 cm2 culture well in lysis buffer (50 mM Tris-HCl/50 mM 3-(N-Morpholino)propanesulfonic acid [MOPS] [pH 7.7], 0.1% SDS, and 1 mM EDTA) supplied with protease inhibitors (Roche) and benzonase (New England Biolabs), followed by 30 min incubation at 4°C and filtration with a 0.22 μm spin filter (Millipore). Conditioned medium was centrifuged for 5 min at 500 × g, and the cell pellet was discarded. The supernatant was filtered using a 0.22 μm syringe filter or centrifuged at 4°C at 18,000 × g for 15 min.
Proteins separated by SDS-PAGE were detected with SimplyBlue SafeStain (Invitrogen/Thermo Fisher Scientific) or transferred to nitrocellulose membranes (GE Healthcare) for immunoblotting with Penta·His mouse monoclonal antibody (1:1,000, QIAGEN) or 9E10 anti-Myc mouse monoclonal antibody (1:2,000, Sigma-Aldrich). Chemiluminescence detection was performed with Western Lightning ECL Plus (PerkinElmer).
Protein-Protein Interactions
Experimental details for BLI measurements and SEC-MALS analysis of the ENG-BMP9 interaction are reported in the Supplemental Experimental Procedures.
Protein Crystallization
Crystallization experiments were carried out by hanging drop vapor diffusion at room temperature using a mosquito crystallization robot (TTP Labtech). Droplet-shaped crystals of C-terminally 6His-tagged MOR (6 mg/mL) were obtained in 0.1 M Tris-HCl (pH 8.5) and 30% (w/v) PEG1000 (Figure S2A) and directly flash-cooled in liquid nitrogen for data collection. Because of the pH sensitivity of the ENG-BMP9 interaction, BMP9 crystals fortuitously appeared in drops set up with the MOR-BMP9 complex (11 mg/mL) and a mother liquor consisting of 1.0 M LiCl, 4% (w/v) PEG6000, and 0.1 M Na-citrate (pH 3.5). For cryocooling, BMP9 crystals were adjusted to 1.0 M LiCl, 6% (w/v) PEG6000, 0.1 M Na-citrate (pH 4.0), and 30% (v/v) glycerol. Hexagonal bipyramidal crystals of N-terminally 6His-tagged MOR in complex with BMP9 (MOR-BMP9, 6 mg/mL) grew in 1.1 M ammonium tartrate (pH 7.0) and 25% saturated sucrose (Figure S4A). For data collection, crystals were directly flash-cooled in liquid nitrogen. Rod-shaped crystals of MZP (9 mg/mL) were obtained using 11.5% (v/v) 2-Methyl-2,4-pentanediol (MPD), 11.5% (w/v) PEG1000 and 11.5% (w/v) PEG3350, and 100 mM 2-(N-Morpholino)ethanesulfonic acid (MES)/imidazole (pH 6.5) (Figure S5B). Before flash-cooling in liquid nitrogen, crystals were stabilized using a 50% (v/v) precipitant mix containing 10% (v/v) glycerol.
X-Ray Diffraction Data Collection and Structure Determination
Experimental details for data collection, structure determination, as well as sequence and structure analysis are described in the Supplemental Experimental Procedures. Data collection, refinement, and validation statistics are summarized in Table S1.
Author Contributions
T.S., M.B., R.C., S.Z.C., and L.H. expressed proteins and performed biological assays. T.S. and M.B. crystallized proteins. T.S., M.B., L.J., and D.d.S. collected crystallographic data. M.B. and L.J. determined and analyzed structures. T.S., M.B., R.C., S.Z.C., M.L., and L.J. wrote the manuscript.
Acknowledgments
We thank A. Gregory (University of Toronto) for initial work on this project, K. Nishimura (Karolinska Institutet) and M. Selmer (Uppsala University) for help with BMP9 expression and data collection, A. Stsiapanava (Karolinska Institutet) for comments, and the European Synchrotron Radiation Facility (ESRF, Grenoble) and Diamond Light Source (DLS, Oxford) for beamtime (ESRF, mx1639/mx1749; DLS, mx8492-34). We are also extremely grateful to Edwin de Grijs and Tim Heiseler (PALL FortéBio) for help with BLI measurements and Christoph Albermann and Christian Ackerschott (Wyatt Technology Europe) for SEC-MALS analysis. This work was supported by the Karolinska Institutet, the Center for Innovative Medicine, Swedish Research Council grant 2012-5093, the Göran Gustafsson Foundation for Research in Natural Sciences and Medicine, the Sven and Ebba-Christina Hagberg Foundation; an EMBO Young Investigator award, and the European Research Council (ERC) under the European Union’s Seventh Framework Programme (FP7/2007-2013)/ERC grant agreement 260759 (to L.J.). Crystallographic data collection was also supported by FP7/2007-2013 under BioStruct-X (grant agreement 283570).
Published: May 30, 2017
Footnotes
Supplemental Information includes Supplemental Experimental Procedures, eight figures, and one table and can be found with this article online at http://dx.doi.org/10.1016/j.celrep.2017.05.011.
Accession Numbers
The accession numbers for the atomic coordinates and structure factors for human ENG OR, BMP9, the ENG OR-BMP9 complex, and ENG ZP reported in this paper are PDB: 5I04, 5I05, 5HZW, and 5HZV, respectively.
Supplemental Information
References
- Abdalla S.A., Letarte M. Hereditary haemorrhagic telangiectasia: current views on genetics and mechanisms of disease. J. Med. Genet. 2006;43:97–110. doi: 10.1136/jmg.2005.030833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abdalla S.A., Pece-Barbara N., Vera S., Tapia E., Paez E., Bernabeu C., Letarte M. Analysis of ALK-1 and endoglin in newborns from families with hereditary hemorrhagic telangiectasia type 2. Hum. Mol. Genet. 2000;9:1227–1237. doi: 10.1093/hmg/9.8.1227. [DOI] [PubMed] [Google Scholar]
- Ali B.R., Ben-Rebeh I., John A., Akawi N.A., Milhem R.M., Al-Shehhi N.A., Al-Ameri M.M., Al-Shamisi S.A., Al-Gazali L. Endoplasmic reticulum quality control is involved in the mechanism of endoglin-mediated hereditary haemorrhagic telangiectasia. PLoS ONE. 2011;6:e26206. doi: 10.1371/journal.pone.0026206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alt A., Miguel-Romero L., Donderis J., Aristorena M., Blanco F.J., Round A., Rubio V., Bernabeu C., Marina A. Structural and functional insights into endoglin ligand recognition and binding. PLoS ONE. 2012;7:e29948. doi: 10.1371/journal.pone.0029948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aricescu A.R., Lu W., Jones E.Y. A time- and cost-efficient system for high-level protein production in mammalian cells. Acta Crystallogr. D Biol. Crystallogr. 2006;62:1243–1250. doi: 10.1107/S0907444906029799. [DOI] [PubMed] [Google Scholar]
- Aykul S., Martinez-Hackert E. Transforming Growth Factor-β family ligands can function as antagonists by competing for type II receptor binding. J. Biol. Chem. 2016;291:10792–10804. doi: 10.1074/jbc.M115.713487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellón T., Corbí A., Lastres P., Calés C., Cebrián M., Vera S., Cheifetz S., Massague J., Letarte M., Bernabéu C. Identification and expression of two forms of the human transforming growth factor-β-binding protein endoglin with distinct cytoplasmic regions. Eur. J. Immunol. 1993;23:2340–2345. doi: 10.1002/eji.1830230943. [DOI] [PubMed] [Google Scholar]
- Bernabeu C., Lopez-Novoa J.M., Quintanilla M. The emerging role of TGF-β superfamily coreceptors in cancer. Biochim. Biophys. Acta. 2009;1792:954–973. doi: 10.1016/j.bbadis.2009.07.003. [DOI] [PubMed] [Google Scholar]
- Bidart M., Ricard N., Levet S., Samson M., Mallet C., David L., Subileau M., Tillet E., Feige J.-J., Bailly S. BMP9 is produced by hepatocytes and circulates mainly in an active mature form complexed to its prodomain. Cell. Mol. Life Sci. 2012;69:313–324. doi: 10.1007/s00018-011-0751-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bokhove M., Sadat Al Hosseini H., Saito T., Dioguardi E., Gegenschatz-Schmid K., Nishimura K., Raj I., de Sanctis D., Han L., Jovine L. Easy mammalian expression and crystallography of maltose-binding protein-fused human proteins. J. Struct. Biol. 2016;194:1–7. doi: 10.1016/j.jsb.2016.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bokhove M., Nishimura K., Brunati M., Han L., de Sanctis D., Rampoldi L., Jovine L. A structured interdomain linker directs self-polymerization of human uromodulin. Proc. Natl. Acad. Sci. USA. 2016;113:1552–1557. doi: 10.1073/pnas.1519803113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourdeau A., Dumont D.J., Letarte M. A murine model of hereditary hemorrhagic telangiectasia. J. Clin. Invest. 1999;104:1343–1351. doi: 10.1172/JCI8088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown M.A., Zhao Q., Baker K.A., Naik C., Chen C., Pukac L., Singh M., Tsareva T., Parice Y., Mahoney A. Crystal structure of BMP-9 and functional interactions with pro-region and receptors. J. Biol. Chem. 2005;280:25111–25118. doi: 10.1074/jbc.M503328200. [DOI] [PubMed] [Google Scholar]
- Castonguay R., Werner E.D., Matthews R.G., Presman E., Mulivor A.W., Solban N., Sako D., Pearsall R.S., Underwood K.W., Seehra J. Soluble endoglin specifically binds bone morphogenetic proteins 9 and 10 via its orphan domain, inhibits blood vessel formation, and suppresses tumor growth. J. Biol. Chem. 2011;286:30034–30046. doi: 10.1074/jbc.M111.260133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang V.T., Crispin M., Aricescu A.R., Harvey D.J., Nettleship J.E., Fennelly J.A., Yu C., Boles K.S., Evans E.J., Stuart D.I. Glycoprotein structural genomics: solving the glycosylation problem. Structure. 2007;15:267–273. doi: 10.1016/j.str.2007.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheifetz S., Bellón T., Calés C., Vera S., Bernabeu C., Massagué J., Letarte M. Endoglin is a component of the transforming growth factor-β receptor system in human endothelial cells. J. Biol. Chem. 1992;267:19027–19030. [PubMed] [Google Scholar]
- David L., Mallet C., Mazerbourg S., Feige J.-J., Bailly S. Identification of BMP9 and BMP10 as functional activators of the orphan activin receptor-like kinase 1 (ALK1) in endothelial cells. Blood. 2007;109:1953–1961. doi: 10.1182/blood-2006-07-034124. [DOI] [PubMed] [Google Scholar]
- Diestel U., Resch M., Meinhardt K., Weiler S., Hellmann T.V., Mueller T.D., Nickel J., Eichler J., Muller Y.A. Identification of a Novel TGF-β-Binding Site in the Zona Pellucida C-terminal (ZP-C) Domain of TGF-β-Receptor-3 (TGFR-3) PLoS ONE. 2013;8:e67214. doi: 10.1371/journal.pone.0067214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallione C.J., Repetto G.M., Legius E., Rustgi A.K., Schelley S.L., Tejpar S., Mitchell G., Drouin E., Westermann C.J.J., Marchuk D.A. A combined syndrome of juvenile polyposis and hereditary haemorrhagic telangiectasia associated with mutations in MADH4 (SMAD4) Lancet. 2004;363:852–859. doi: 10.1016/S0140-6736(04)15732-2. [DOI] [PubMed] [Google Scholar]
- Gougos A., Letarte M. Biochemical characterization of the 44G4 antigen from the HOON pre-B leukemic cell line. J. Immunol. 1988;141:1934–1940. [PubMed] [Google Scholar]
- Gougos A., Letarte M. Identification of a human endothelial cell antigen with monoclonal antibody 44G4 produced against a pre-B leukemic cell line. J. Immunol. 1988;141:1925–1933. [PubMed] [Google Scholar]
- Gougos A., Letarte M. Primary structure of endoglin, an RGD-containing glycoprotein of human endothelial cells. J. Biol. Chem. 1990;265:8361–8364. [PubMed] [Google Scholar]
- Gregory A.L. University of Toronto; 2011. Structural and Functional Characteristics of a Soluble Form of Endoglin in the Context of Preeclampsia. MSc. [Google Scholar]
- Gregory A.L., Xu G., Sotov V., Letarte M. Review: the enigmatic role of endoglin in the placenta. Placenta. 2014;35(Suppl):S93–S99. doi: 10.1016/j.placenta.2013.10.020. [DOI] [PubMed] [Google Scholar]
- Guerrero-Esteo M., Sanchez-Elsner T., Letamendia A., Bernabeu C. Extracellular and cytoplasmic domains of endoglin interact with the transforming growth factor-β receptors I and II. J. Biol. Chem. 2002;277:29197–29209. doi: 10.1074/jbc.M111991200. [DOI] [PubMed] [Google Scholar]
- Han L., Monné M., Okumura H., Schwend T., Cherry A.L., Flot D., Matsuda T., Jovine L. Insights into egg coat assembly and egg-sperm interaction from the X-ray structure of full-length ZP3. Cell. 2010;143:404–415. doi: 10.1016/j.cell.2010.09.041. [DOI] [PubMed] [Google Scholar]
- Hawinkels L.J., Kuiper P., Wiercinska E., Verspaget H.W., Liu Z., Pardali E., Sier C.F., ten Dijke P. Matrix metalloproteinase-14 (MT1-MMP)-mediated endoglin shedding inhibits tumor angiogenesis. Cancer Res. 2010;70:4141–4150. doi: 10.1158/0008-5472.CAN-09-4466. [DOI] [PubMed] [Google Scholar]
- Hinck A.P., Mueller T.D., Springer T.A. Structural biology and evolution of the TGF-β family. Cold Spring Harb. Perspect. Biol. 2016;8:a022103. doi: 10.1101/cshperspect.a022103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horbelt D., Denkis A., Knaus P. A portrait of Transforming Growth Factor β superfamily signalling: Background matters. Int. J. Biochem. Cell Biol. 2012;44:469–474. doi: 10.1016/j.biocel.2011.12.013. [DOI] [PubMed] [Google Scholar]
- Johnson D.W., Berg J.N., Baldwin M.A., Gallione C.J., Marondel I., Yoon S.J., Stenzel T.T., Speer M., Pericak-Vance M.A., Diamond A. Mutations in the activin receptor-like kinase 1 gene in hereditary haemorrhagic telangiectasia type 2. Nat. Genet. 1996;13:189–195. doi: 10.1038/ng0696-189. [DOI] [PubMed] [Google Scholar]
- Kienast Y., Jucknischke U., Scheiblich S., Thier M., de Wouters M., Haas A., Lehmann C., Brand V., Bernicke D., Honold K., Lorenz S. Rapid activation of bone morphogenic protein 9 by receptor-mediated displacement of pro-domains. J. Biol. Chem. 2016;291:3395–3410. doi: 10.1074/jbc.M115.680009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lebrin F., Goumans M.-J., Jonker L., Carvalho R.L.C., Valdimarsdottir G., Thorikay M., Mummery C., Arthur H.M., ten Dijke P. Endoglin promotes endothelial cell proliferation and TGF-β/ALK1 signal transduction. EMBO J. 2004;23:4018–4028. doi: 10.1038/sj.emboj.7600386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li D.Y., Sorensen L.K., Brooke B.S., Urness L.D., Davis E.C., Taylor D.G., Boak B.B., Wendel D.P. Defective angiogenesis in mice lacking endoglin. Science. 1999;284:1534–1537. doi: 10.1126/science.284.5419.1534. [DOI] [PubMed] [Google Scholar]
- Lin S.J., Hu Y., Zhu J., Woodruff T.K., Jardetzky T.S. Structure of betaglycan zona pellucida (ZP)-C domain provides insights into ZP-mediated protein polymerization and TGF-β binding. Proc. Natl. Acad. Sci. USA. 2011;108:5232–5236. doi: 10.1073/pnas.1010689108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Litscher E.S., Wassarman P.M. John Wiley & Sons, Inc.; Hoboken, New Jersey: 2015. A guide to zona pellucida domain proteins. [Google Scholar]
- Llorca O., Trujillo A., Blanco F.J., Bernabeu C. Structural model of human endoglin, a transmembrane receptor responsible for hereditary hemorrhagic telangiectasia. J. Mol. Biol. 2007;365:694–705. doi: 10.1016/j.jmb.2006.10.015. [DOI] [PubMed] [Google Scholar]
- Mahlawat P., Ilangovan U., Biswas T., Sun L.Z., Hinck A.P. Structure of the Alk1 extracellular domain and characterization of its bone morphogenetic protein (BMP) binding properties. Biochemistry. 2012;51:6328–6341. doi: 10.1021/bi300942x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mallet C., Lamribet K., Giraud S., Dupuis-Girod S., Feige J.J., Bailly S., Tillet E. Functional analysis of endoglin mutations from hereditary hemorrhagic telangiectasia type 1 patients reveals different mechanisms for endoglin loss of function. Hum. Mol. Genet. 2015;24:1142–1154. doi: 10.1093/hmg/ddu531. [DOI] [PubMed] [Google Scholar]
- McAllister K.A., Grogg K.M., Johnson D.W., Gallione C.J., Baldwin M.A., Jackson C.E., Helmbold E.A., Markel D.S., McKinnon W.C., Murrell J. Endoglin, a TGF-β binding protein of endothelial cells, is the gene for hereditary haemorrhagic telangiectasia type 1. Nat. Genet. 1994;8:345–351. doi: 10.1038/ng1294-345. [DOI] [PubMed] [Google Scholar]
- Mi L.Z., Brown C.T., Gao Y., Tian Y., Le V.Q., Walz T., Springer T.A. Structure of bone morphogenetic protein 9 procomplex. Proc. Natl. Acad. Sci. USA. 2015;112:3710–3715. doi: 10.1073/pnas.1501303112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller D.W., Graulich W., Karges B., Stahl S., Ernst M., Ramaswamy A., Sedlacek H.H., Müller R., Adamkiewicz J. Elevated expression of endoglin, a component of the TGF-β-receptor complex, correlates with proliferation of tumor endothelial cells. Int. J. Cancer. 1999;81:568–572. doi: 10.1002/(sici)1097-0215(19990517)81:4<568::aid-ijc11>3.0.co;2-x. [DOI] [PubMed] [Google Scholar]
- Nithianantham S., Le S., Seto E., Jia W., Leary J., Corbett K.D., Moore J.K., Al-Bassam J. Tubulin cofactors and Arl2 are cage-like chaperones that regulate the soluble αβ-tubulin pool for microtubule dynamics. eLife. 2015;4:e08811. doi: 10.7554/eLife.08811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nolan-Stevaux O., Zhong W., Culp S., Shaffer K., Hoover J., Wickramasinghe D., Ruefli-Brasse A. Endoglin requirement for BMP9 signaling in endothelial cells reveals new mechanism of action for selective anti-endoglin antibodies. PLoS ONE. 2012;7:e50920. doi: 10.1371/journal.pone.0050920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohta M., Greenberger J.S., Anklesaria P., Bassols A., Massagué J. Two forms of transforming growth factor-β distinguished by multipotential haematopoietic progenitor cells. Nature. 1987;329:539–541. doi: 10.1038/329539a0. [DOI] [PubMed] [Google Scholar]
- Paauwe M., Heijkants R.C., Oudt C.H., van Pelt G.W., Cui C., Theuer C.P., Hardwick J.C.H., Sier C.F.M., Hawinkels L.J.A.C. Endoglin targeting inhibits tumor angiogenesis and metastatic spread in breast cancer. Oncogene. 2016;35:4069–4079. doi: 10.1038/onc.2015.509. [DOI] [PubMed] [Google Scholar]
- Paquet M.E., Pece-Barbara N., Vera S., Cymerman U., Karabegovic A., Shovlin C., Letarte M. Analysis of several endoglin mutants reveals no endogenous mature or secreted protein capable of interfering with normal endoglin function. Hum. Mol. Genet. 2001;10:1347–1357. doi: 10.1093/hmg/10.13.1347. [DOI] [PubMed] [Google Scholar]
- Pece-Barbara N., Cymerman U., Vera S., Marchuk D.A., Letarte M. Expression analysis of four endoglin missense mutations suggests that haploinsufficiency is the predominant mechanism for hereditary hemorrhagic telangiectasia type 1. Hum. Mol. Genet. 1999;8:2171–2181. doi: 10.1093/hmg/8.12.2171. [DOI] [PubMed] [Google Scholar]
- Quackenbush E.J., Letarte M. Identification of several cell surface proteins of non-T, non-B acute lymphoblastic leukemia by using monoclonal antibodies. J. Immunol. 1985;134:1276–1285. [PubMed] [Google Scholar]
- Reeves P.J., Callewaert N., Contreras R., Khorana H.G. Structure and function in rhodopsin: high-level expression of rhodopsin with restricted and homogeneous N-glycosylation by a tetracycline-inducible N-acetylglucosaminyltransferase I-negative HEK293S stable mammalian cell line. Proc. Natl. Acad. Sci. USA. 2002;99:13419–13424. doi: 10.1073/pnas.212519299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossi E., Lopez-Novoa J.M., Bernabeu C. Endoglin involvement in integrin-mediated cell adhesion as a putative pathogenic mechanism in hereditary hemorrhagic telangiectasia type 1 (HHT1) Front. Genet. 2015;5:457. doi: 10.3389/fgene.2014.00457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scharpfenecker M., van Dinther M., Liu Z., van Bezooijen R.L., Zhao Q., Pukac L., Löwik C.W.G.M., ten Dijke P. BMP-9 signals via ALK1 and inhibits bFGF-induced endothelial cell proliferation and VEGF-stimulated angiogenesis. J. Cell Sci. 2007;120:964–972. doi: 10.1242/jcs.002949. [DOI] [PubMed] [Google Scholar]
- She X.W., Seon B.K. Epitope mapping of endoglin, a TGF-β receptor, using recombinant fragments and twelve monoclonal antibodies. Proc. Am. Assoc. Cancer Res. 2001;42:825. [Google Scholar]
- Stenson P.D., Mort M., Ball E.V., Shaw K., Phillips A., Cooper D.N. The Human Gene Mutation Database: building a comprehensive mutation repository for clinical and molecular genetics, diagnostic testing and personalized genomic medicine. Hum. Genet. 2014;133:1–9. doi: 10.1007/s00439-013-1358-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Townson S.A., Martinez-Hackert E., Greppi C., Lowden P., Sako D., Liu J., Ucran J.A., Liharska K., Underwood K.W., Seehra J. Specificity and structure of a high affinity activin receptor-like kinase 1 (ALK1) signaling complex. J. Biol. Chem. 2012;287:27313–27325. doi: 10.1074/jbc.M112.377960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Upton P.D., Davies R.J., Trembath R.C., Morrell N.W. Bone morphogenetic protein (BMP) and activin type II receptors balance BMP9 signals mediated by activin receptor-like kinase-1 in human pulmonary artery endothelial cells. J. Biol. Chem. 2009;284:15794–15804. doi: 10.1074/jbc.M109.002881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Le B., Franke D., Svergun D.I., Han T., Hwang H.Y., Kim K.K. Structural and functional characterization of soluble endoglin receptor. Biochem. Biophys. Res. Commun. 2009;383:386–391. doi: 10.1016/j.bbrc.2009.02.162. [DOI] [PubMed] [Google Scholar]
- Venkatesha S., Toporsian M., Lam C., Hanai J., Mammoto T., Kim Y.M., Bdolah Y., Lim K.H., Yuan H.T., Libermann T.A. Soluble endoglin contributes to the pathogenesis of preeclampsia. Nat. Med. 2006;12:642–649. doi: 10.1038/nm1429. [DOI] [PubMed] [Google Scholar]
- Wooderchak-Donahue W.L., McDonald J., O’Fallon B., Upton P.D., Li W., Roman B.L., Young S., Plant P., Fülöp G.T., Langa C. BMP9 mutations cause a vascular-anomaly syndrome with phenotypic overlap with hereditary hemorrhagic telangiectasia. Am. J. Hum. Genet. 2013;93:530–537. doi: 10.1016/j.ajhg.2013.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.