

Background and significance
Tremendous progress has been made identifying the transcription factor networks that drive the specification of cardiac progenitors and direct formation and growth of a developing embryonic heart. The genes are well conserved throughout evolution; key genes were identified through genetic studies in Drosophila cardiogenesis, and these are represented by expanded gene families in vertebrates including humans. For example, transcription factors GATA4, MEF2A, NKX2.5 and TBX5 each have critical roles in vertebrate heart development, maturation and function, working in concert with developmental signaling pathways, environmental influences, and chromatin regulatory machinery. Knockout of any of these genes in mice leads to defects in cardiogenesis and embryonic or early postnatal lethality (1-4). It is challenging to define a precise regulatory hierarchy, as these genes regulate the expression of each other (and themselves), and their encoded proteins interact functionally in complexes that somehow together regulate distinct genesets important for diverse developmental programs, such as chamber growth, septation, valve formation, and outflow tract development.
Given clear developmental roles in animal models, it is gratifying, if not entirely surprising, that mutations in the genes encoding these important transcription factors have been associated with human congenital heart disease (CHD). However, compared to animal models, there are clear differences for the patients. Most notably, the mutations are generally thought to be heterozygous loss of function alleles, so that the CHD phenotypes represent haplo-insufficiency syndromes. This has not been modeled in the mouse, in which both copies of the genes must be inactivated, while heterozygous alleles are developmentally well tolerated, even if the animals may in some cases be physiologically sensitized (5,6). Thus, the cellular and molecular mechanism(s) by which heterozygous point mutations mediate CHD is poorly understood. With the ability to generate personalized induced pluripotent stem cells (iPSCs) carrying heterozygous (or corrected) alleles, and strong protocols for the generation of human cardiac cells in vitro, this is an exciting time for researchers interested in CHD seeking to model patient-specific disease “in a dish”.
In their recently published work, Ang and colleagues exploit the potential of patient-specific iPSCs in their exploration of the molecular origins of a CHD-associated mutation in GATA4 (7). Here the Srivastava group builds on their previous work (8), in which they discovered that the G296S mutation of GATA4 interferes with its interaction with TBX5. That study revealed commonalities in the etiology of CHD arising from mutations in GATA4 or TBX5, suggesting that they act together to regulate a common subset of cardiac genes. A genetic interaction between GATA4 and TBX5 is supported by atrioventricular septal defects in mice that are compound heterozygous for the two genes (9). If common molecular mechanisms of subsets of CHDs are defined, this might provide significant opportunities for targeting pivotal nodes that can be therapeutically manipulated to ameliorate disease severity.
Research findings
In “Disease Model of GATA4 Mutation Reveals Transcription Factor Cooperativity in Human Cardiogenesis”, Ang et al. characterize the in vitro pathological consequences of the GATA4G296S mutation. They derived iPSCs from heterozygous patients, and then also genetically engineered isogenic wildtype lines as the optimal control cells for their studies. They used established protocols to generate cardiomyocytes from the pluripotent cell lines (iPSC-CMs). They then characterized the function of iPSC-CMs through a series of cellular and electrophysiological assays, pinpointing a lack of appropriate response to electrical pacing and deficiencies in force generation and calcium handling in G296S cardiac cells. These findings mirror those observed in patients, demonstrating the utility of the cultured iPSC-CMs to model patient phenotypes. This result is especially important given concern in the field regarding whether the relatively immature fetal-like cardiomyocytes obtained through in vitro differentiation protocols are adequate surrogates for their neonatal or adult counterparts (10).
Following transcriptomic profiling to compare G296S and control iPSC-CMs, the authors identified an overall downregulation in the cardiogenic program with a concomitant upregulation of genes important for endothelial development. They conclude that disturbance of the GATA4-TBX5 (G4T5) interaction ultimately interferes with the ability of a cardiac progenitor to develop or maintain a cardiomyocyte identity. The group then investigated the binding sites of G4T5 in wildtype and mutant cells, and the nearby epigenetic marks. As expected, the transcription factors predominantly co-localized to binding sites in genes that encode for myofibril assembly and cardiac development. They also validated many direct targets previously identified in the mouse, strengthening the value of data obtained from human cells cultured in a dish. They further observed that gene expression was most often lost when binding sites for the two factors were relatively fewer and further apart from each other. This suggests that in wildtype cells, protein-protein interactions provide substantial impact for complex formation above and beyond DNA-binding.
As the capstone of the paper, Ang et al. examined MED1 occupancy to identify super-enhancer elements (SEs) in the wildtype and G296S human cardiomyocytes. They discovered loss of some cardiac SEs in G296S cells, with concomitant gain of SEs associated with the endothelial gene program, underscoring their transcriptome data (Figure 1). Knockdown of GATA4 led to a downregulation of this SE network, confirming its biological relevance. Interestingly, TBX5 occupancy was better correlated with SE location than GATA4, suggesting that although GATA4 is thought to initiate the protein-DNA interaction, TBX5 location is more correlated with key stabilized nodes.
Figure 1.
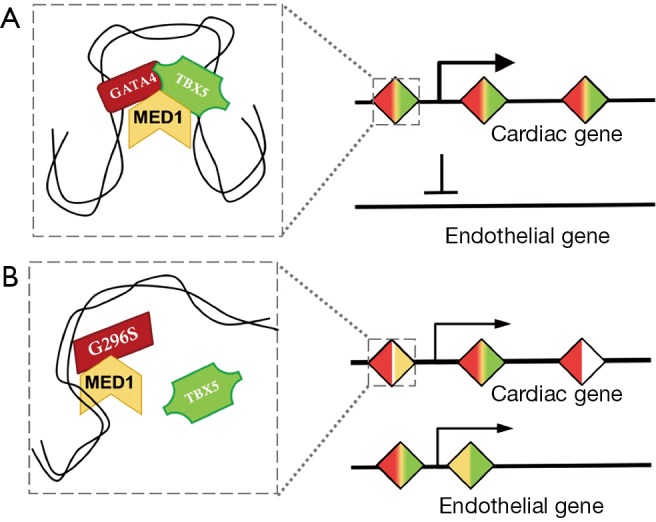
GATA4G296S mutant protein leads to altered localization of transcription factor complexes and changes in gene expression. (A) In wildtype cardiac cells, cardiac gene expression is activated by cooperative binding of GATA4 and TBX5, which may also recruit super-enhancer marker MED1. The expression of endothelial genes is suppressed, presumably by co-repressors in this complex (not shown); (B) in the presence of GATA4G296S mutant protein, which does not bind TBX5, the transcription factor complex is disturbed, decreasing the expression of cardiac genes. Untethered TBX5 mislocalizes to endothelial genes, apparently driving their aberrant expression.
Finally, the group used a systems biology strategy to predict a G4T5-regulated network of genes with either physical or functional interactions. A key finding from this analysis was the prevalence of genes in the PI3 kinase (PI3K) signaling pathway at central nodes. Although GATA4 activity has previously been linked to PI3K signaling (11,12), these studies confirmed the importance of this relationship for cardiac identity and function. Strikingly, the response to small molecule-mediated activation of the PI3K pathway was magnified in G296S cardiomyocytes, inviting the hypothesis that hypersensitivity to PI3K signaling could be targeted to treat disease arising from disturbances to the G4T5 network.
Conclusions and perspectives
In summary, Ang et al. maximized many of the most cutting-edge molecular and cellular technologies to rigorously and comprehensively dissect the mechanisms by which a mutation in GATA4 compromises cardiomyocyte function. Several key observations can be highlighted. First, the study shows that iPSC-CMs can be used, even in the context of a relatively simple two-dimensional cell culture platform, to model at least some aspects of human CHD. This is extremely good news, as it might have been predicted that CHD, thought to be driven largely by defects in embryonic morphogenesis, would lose phenotypic disease identity when taken out of the context of a whole developing organ system. This is significant because the simple 2D platform lends itself to high-throughput drug screening strategies. This highlights an exciting strategy for potentially repurposing FDA-approved drugs for patient-specific diseases [see for example, (13)]. Second, the systems biology approach identified PI3K as a relevant interaction network, suggesting that G4T5 interactions normally function to limit PI3K, an actionable therapeutic target. Finally, the dysregulation of a cardiac genetic program in the context of the GATA4 G296S mutant allele was perhaps not unexpected. However, the finding that genetic programs associated with endocardium and endothelial cells fail to be properly down-regulated is not intuitive and could not have been predicted. This is particularly interesting in the context of CHD in which endocardial dysfunction could lead indirectly to septal and valve defects, and can now be considered a potential driver of pathology. Furthermore, the identification of SE sites in loci not previously associated with the heart suggests that the group has identified some new critical cardiac genes that invite future studies.
Perhaps one of the more intriguing questions that remains unanswered from this work relates to the specificity of the G296S mutant allele. When mapping GATA4 binding sites in the mutant cells, the assays would not have distinguished contributions in complexes from wildtype or mutant proteins. Many mutations in GATA4 (or the sister genes GATA5 and GATA6) have been associated with human CHD. Many of these mutant alleles are thought to be loss-of-function through defects that eliminate the ability of the protein to bind DNA. The G296S allele is primarily defective in protein complex interaction with TBX5, and while this may impact DNA-binding, it is unclear if other mutations will have similar or distinct alterations in the GATA4-directed regulatory networks, such as endothelial program repression. This question, so central to a better understanding of haplo-insufficiency CHD syndromes, can now be further addressed by the strategies and platforms presented by Ang et al. The work nicely illustrates how iPSC-CMs can provide a unique window into cardiac disease pathology and a blueprint for future studies toward the lofty goal of personalized medicine.
Acknowledgements
Funding: Todd Evans is supported by an Outstanding Investigator Award from the NHLBI (R35-HL135778).
Footnotes
Conflicts of Interest: The authors have no conflicts of interest to declare.
References
- 1.Watt AJ, Battle MA, Li J, et al. GATA4 is essential for formation of the proepicardium and regulates cardiogenesis. Proc Natl Acad Sci U S A 2004;101:12573-8. 10.1073/pnas.0400752101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Naya FJ, Black BL, Wu H, et al. Mitochondrial deficiency and cardiac sudden death in mice lacking the MEF2A transcription factor. Nat Med 2002;8:1303-9. 10.1038/nm789 [DOI] [PubMed] [Google Scholar]
- 3.Tanaka M, Chen Z, Bartunkova S, et al. The cardiac homeobox gene Csx/Nkx2.5 lies genetically upstream of multiple genes essential for heart development. Development 1999;126:1269-80. [DOI] [PubMed] [Google Scholar]
- 4.Bruneau BG, Nemer G, Schmitt JP, et al. A murine model of Holt-Oram syndrome defines roles of the T-box transcription factor Tbx5 in cardiogenesis and disease. Cell 2001;106:709-21. 10.1016/S0092-8674(01)00493-7 [DOI] [PubMed] [Google Scholar]
- 5.Bisping E, Ikeda S, Kong SW, et al. Gata4 is required for maintenance of postnatal cardiac function and protection from pressure overload-induced heart failure. Proc Natl Acad Sci U S A 2006;103:14471-6. 10.1073/pnas.0602543103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Oka T, Maillet M, Watt AJ, et al. Cardiac-specific deletion of Gata4 reveals its requirement for hypertrophy, compensation, and myocyte viability. Circ Res 2006;98:837-45. 10.1161/01.RES.0000215985.18538.c4 [DOI] [PubMed] [Google Scholar]
- 7.Ang YS, Rivas RN, Ribeiro AJ, et al. Disease Model of GATA4 Mutation Reveals Transcription Factor Cooperativity in Human Cardiogenesis. Cell 2016;167:1734-49.e22. 10.1016/j.cell.2016.11.033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Garg V, Kathiriya IS, Barnes R, et al. GATA4 mutations cause human congenital heart defects and reveal an interaction with TBX5. Nature 2003;424:443-7. 10.1038/nature01827 [DOI] [PubMed] [Google Scholar]
- 9.Maitra M, Schluterman MK, Nichols HA, et al. Interaction of Gata4 and Gata6 with Tbx5 is critical for normal cardiac development. Dev Biol 2009;326:368-77. 10.1016/j.ydbio.2008.11.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yang X, Pabon L, Murry CE. Engineering adolescence: maturation of human pluripotent stem cell-derived cardiomyocytes. Circ Res 2014;114:511-23. 10.1161/CIRCRESAHA.114.300558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bisping E, Ikeda S, Sedej M, et al. Transcription factor GATA4 is activated but not required for insulin-like growth factor 1 (IGF1)-induced cardiac hypertrophy. J Biol Chem 2012;287:9827-34. 10.1074/jbc.M111.338749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.McMullen JR, Shioi T, Huang WY, et al. The insulin-like growth factor 1 receptor induces physiological heart growth via the phosphoinositide 3-kinase(p110alpha) pathway. J Biol Chem 2004;279:4782-93. 10.1074/jbc.M310405200 [DOI] [PubMed] [Google Scholar]
- 13.Zeng H, Guo M, Zhou T, et al. An Isogenic Human ESC Platform for Functional Evaluation of Genome-wide-Association-Study-Identified Diabetes Genes and Drug Discovery. Cell Stem Cell 2016;19:326-40. 10.1016/j.stem.2016.07.002 [DOI] [PMC free article] [PubMed] [Google Scholar]