

Abstract
Olfactory receptor (OR) genes constitute the basis of the sense of smell and are encoded by the largest mammalian gene superfamily, with >1000 members. In humans, but not in mice or dogs, the majority of OR genes have become pseudogenes, suggesting that OR genes in humans evolve under different selection pressures than in other mammals. To explore this further, we compare the OR gene repertoire of human with its closest living evolutionary relative, by taking advantage of the recently sequenced genome of the chimpanzee. In agreement with previous reports based on a small number of ORs, we find that humans have a significantly higher proportion of OR pseudogenes than chimpanzees. Moreover, we can reject the possibility that humans have been accumulating OR pseudogenes at a constant neutral rate since the divergence of human and chimpanzee. The comparison of the two repertoires reveals two chimpanzee-specific OR subfamily expansions and three expansions specific to humans. It also suggests that a subset of OR genes are under positive selection in either the human or the chimpanzee lineage. Thus, although overall there is relaxed constraint on human olfaction relative to chimpanzee, species-specific sensory requirements appear to have shaped the evolution of the functional OR gene repertoires in both species.
Olfactory receptor (OR) genes provide the basis for the sense of smell, and with >1000 genes, are the largest gene superfamily in mammalian genomes (Buck and Axel 1991; Ben-Arie et al. 1994; Glusman et al. 2001; Zhang and Firestein 2002). OR proteins are members of the seven-transmembrane domain G-protein coupled receptor (GPCR) gene hyperfamily, but they also share several sequence motifs not found in other GPCRs (Buck and Axel 1991). In mammals, OR genes are typically organized in gene clusters and are found on multiple chromosomes (Trask et al. 1998).
Based on protein sequence similarity, mammalian OR genes are divided into two classes, 17 families and ∼250 subfamilies (Glusman et al. 2000). Class I OR genes are closely related to OR genes that are found in fish, and hence, are referred to as “fish-like,” while class II OR genes are specific to tetrapods. OR genes from the same subfamily are defined as sharing 60% or more of their amino acid sequence. Previous studies suggest that OR genes from the same subfamily may bind the same type of odorants, while differing in their specificity to close structural variants (Malnic et al. 1999; Godfrey et al. 2004).
The completion of the human genome enabled the identification of the entire human OR gene repertoire (Glusman et al. 2001). The most recent version of the Human Olfactory Receptor Data Exploratorium (HORDE: http://bip.weizmann.ac.il/HORDE/) lists 862 OR genes. Of these, 56% carry one or more coding-region disruptions, and hence, are annotated as nonfunctional pseudogenes. Now that the entire OR gene repertoires of mouse (Young et al. 2002; Zhang and Firestein 2002) and dog (Quignon et al. 2003; Olender et al. 2004) are available, it has become possible to compare the human OR gene repertoire with that of other species. These comparisons revealed that the OR repertoires of mouse and dog are of roughly the same size and are ∼20% larger than that of human. Moreover, using the above definition to identify nonfunctional OR genes, the proportion of pseudogenes in mouse and dog is only ∼20% (Young et al. 2002; Quignon et al. 2003). Thus, the number of putatively functional OR genes is three times larger in mouse and dog relative to human.
When only the intact (putatively functional) OR gene repertoires of the three species are contrasted, it appears that although humans have a sharply reduced functional OR repertoire, >150 of the different OR gene subfamilies are shared by all three species (Quignon et al. 2003; Godfrey et al. 2004; Olender et al. 2004). These observations led to the suggestion that, although humans may be less sensitive to certain odors compared with dog and mouse, the repertoire of odors that can be sensed by the three species may be similar (Godfrey et al. 2004; Malnic et al. 2004).
Recently, Gilad et al. (2003b) analyzed the coding sequence of 50 OR genes in five different primates, and found that the human lineage has accumulated OR pseudogenes almost four times more rapidly than any nonhuman primate lineage. As a result, apes and Old World monkeys have many fewer OR pseudogenes than humans. Nonetheless, the proportion of OR pseudogenes in these nonhuman primates is still significantly higher than those of dog or mouse (Rouquier et al. 2000; Gilad et al. 2003b). Taken together, the data suggest that a deterioration of the olfactory repertoire occurred during primate evolution, with a particularly steep decline in the human lineage.
Concurrently, however, an analysis of polymorphism and divergence data at 20 OR genes suggested that a subset of intact human OR genes evolve under positive selection (Gilad et al. 2000, 2003a). In contrast to humans, intact OR genes in chimpanzees appeared to evolve under strong evolutionary constraint (Gilad et al. 2003a), consistent with the observation of fewer OR pseudogenes in chimpanzees.
The inability to compare the human OR repertoire with a complete OR repertoire from another primate limited the scope of previous studies to estimating the fraction of pseudogenes and inferring the nature of selection acting on a sample of human OR gene clusters. In particular, it was not possible to systematically investigate which human OR genes evolved under positive selection, to ask when humans started the more rapid accumulation of OR pseudogenes (relative to other apes), or to identify specific OR subfamilies that show an increased or decreased proportion of pseudogenes in humans. To address these questions, we identified the entire chimpanzee OR gene repertoire from the publicly available sequence of the chimpanzee genome and compared it with the human one. We also used the mouse OR gene repertoire to obtain trios of putative OR orthologs in mouse, chimpanzee, and human. The analysis of these trios allowed us to identify individual OR genes evolving under different selection pressures in human and chimpanzee lineages.
Results
The chimpanzee OR gene repertoire
We identified 1091 putative OR genes in the recently completed draft of the chimpanzee genome. Of these, 192 sequences were shorter than 300 bp (corresponding to 1/3 of the entire OR protein length) and were excluded from subsequent analyses. Of 899 chimpanzee OR genes, 353 (39%) have an uninterrupted (intact) open reading frame, and hence, may be considered functional. This fraction of intact chimpanzee OR genes may be an underestimate due to sequencing errors that have been incorporated into the chimpanzee genome assembly and that appear to disrupt coding regions. In order to test this possibility, we used the previously published sequences of 30 chimpanzee intact OR genes (Gilad et al. 2003b) and compared them with the corresponding sequences in the chimpanzee genome. We found an average of 0.71% sequence differences between the sequences obtained by Gilad et al. (2003b) and those of the chimpanzee assembly. Seven (23.3%) of the OR genes annotated as “intact” by Gilad et al. (2003b) contain either nonsense mutations or single base-pair insertions/deletions in the chimpanzee assembly that lead to one or more in-frame premature stop codons. If these disruptions are in fact sequencing errors, then, extrapolating to the whole repertoire, the corrected fraction of intact genes in the chimpanzee OR gene repertoire is ∼50%.
Next, we used the full-length (>800 bp) OR sequences from human and chimpanzee in order to build a distance-based phylogenetic tree of both OR gene repertoires (Fig. 1). Following the family-subfamily classification of OR genes (Glusman et al. 2000, 2001), the overlap of the represented OR subfamilies in the repertoires of human and chimpanzee is nearly complete (Fig. 1) and, in particular, in most OR subfamilies, there is a human ortholog for almost every chimpanzee OR gene. However, there are also some species-specific expansions. A chimpanzee expansion within OR subfamily 4C (Fig. 2A), and three human expansions in subfamilies 2A, 4F (also noted by Linardopoulou et al. 2001), and 6C (Fig. 2B,C,D). The high-sequence similarity between lineage-specific OR genes in subfamilies 4F, 2A, and 4C, (98.6%-99.3%) suggests that recent duplications underlie these expansions. In contrast, the average sequence similarity between the human-specific OR genes in subfamily 6C is only 70%. This suggests that these genes existed in the common ancestor of human and chimpanzee, and that their orthologs were either deleted from the chimpanzee genome, or were not found by us (possibly due to properties of the assembly). In addition, the chimpanzee has roughly ∼60% more loci from the 7E subfamily compared with human (84 and 132 7E OR genes in human and chimpanzee, respectively). The 7E OR subfamily in human consists almost entirely of pseudogenes (Newman and Trask 2003); similarly, there is only one intact OR gene among the chimpanzee 7E OR subfamily sequences.
Figure 1.

A neighbor-joining tree of the olfactory receptor repertoires of human and chimpanzee. The sequence of the bovine rhodopsin protein was used as outgroup (indicated). Numbers indicate the different OR gene families. Human external branches are red, chimpanzee ones are blue.
Figure 2.

Distance matrix trees for specific OR subfamilies in human and chimpanzee. The first letter of the OR name indicate the species name (H and C for human and chimpanzee, respectively). Human OR sequence H5U512 was used as an outgroup in all cases. (A) Subfamily 4C: 20 human sequences (10 intact) and 27 chimpanzee sequences (12 intact) (B) Subfamily 2A: 14 human sequences (nine intact) and nine chimpanzee sequences (seven intact) (C) Subfamily 4F: 16 human sequences (nine intact) and 11 chimpanzee sequences (four intact) (D) Subfamily 6C: 17 human sequences (10 intact) and nine chimpanzee sequences (seven intact).
Estimating the age of human pseudogenes
We identified 761 clear cases of human-chimpanzee OR gene orthologous pairs (see Methods). Of these, the number of apparent pseudogenes in human and chimpanzee is 403 and 440, respectively. We compared the conceptual protein sequences in order to identify all coding-region disruptions in each pseudogene. We then defined two groups of human OR pseudogenes as follows: (1) shared pseudogenes, i.e., those that share at least one coding-region disruption with their chimpanzee ortholog, and hence, were most likely pseudogenes in the human-chimpanzee common ancestor, and (2) human-specific pseudogenes, i.e., those that do not share any disruption with their chimpanzee orthologs, and most likely were intact in the common ancestor of human and chimpanzee.
Species-specific and shared coding-region disruptions in OR genes have been described in the past (Rouquier et al. 1998). Here, we concentrated on human-specific disruptions in the shared pseudogenes (by definition, at least one disruption in these loci is shared with chimpanzee, but any additional ones may be human specific). We assume that these disruptions are neutral mutations, as they occurred in pseudogenes. As expected under the hypothesis of a neutral molecular clock, the number of disruptions per shared pseudogene appears to be approximately Poisson distributed (Fig. 3). If we fit a Poisson distribution to the data, the estimate of the mean is 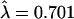 . Assuming six million years (Myr) since the common ancestor of a human and a chimpanzee sequence, this corresponds to a neutral OR gene disruption rate of 1.17·10-6 per gene (∼1 kb) per year. This calculation provides a general estimate of the rate at which neutral gene disruptions accumulate in OR genes, and possibly in other human genes with similar GC content.
. Assuming six million years (Myr) since the common ancestor of a human and a chimpanzee sequence, this corresponds to a neutral OR gene disruption rate of 1.17·10-6 per gene (∼1 kb) per year. This calculation provides a general estimate of the rate at which neutral gene disruptions accumulate in OR genes, and possibly in other human genes with similar GC content.
Figure 3.

The distribution of human-specific OR gene disruptions. The broken line is the Poisson fit for the data (λ = 0.701).
Next, we tabulated the number of coding-region disruptions in the human-specific pseudogenes. Interestingly, we could not reject a Poisson distribution for these disruptions either (by χ2, excluding the zero count class, P = 0.58). We then proceeded by assuming that at a certain point in human evolution, a subset of OR genes became unnecessary and were free to accumulate coding-region disruptions. In order to estimate this time point, we used the mean of the Poisson distribution, which we estimated to be 0.480. Under our model, assuming 6 Myr since the common ancestor of a human and a chimpanzee sequence, we estimate that the relaxation of selective constraint started (0.451/0.701)·6 Mya = 3.86 Mya, with 3.28-4.56 Myr as rough 95% confidence intervals (obtained by parametrical bootstrapping 10,000 times). Thus, we can reject the hypothesis that OR genes have been accumulating disruptions at a neutral rate over the past 6 Myr. This hypothesis can also be rejected by testing whether the Poisson distribution obtained for neutral disruptions over 6 Myr fits the data for human-specific disruptions; it does not (P < 0.01).
OR genes under selection
Previous studies of human and chimpanzee OR genes indicated that this gene superfamily evolves under different selection pressures in each species (Gilad et al. 2003a). Here, we asked whether we can identify specific OR genes that may have been a target of natural selection in one of the species. Specifically, we use an analysis that is sensitive to differences between the species in the type of selection pressures acting on a given locus.
As a starting point for our analysis, we used the previously published set of human-mouse OR gene orthologs (Man et al. 2004). We then used the “reciprocal best hit” humanchimpanzee OR gene list in order to identify the corresponding chimpanzee orthologs. Thus, we obtained clear humanchimpanzee-mouse ortholog trios for 201 OR genes. By using the mouse ortholog as outgroup, we were able to estimate the OR gene sequences of the human-chimpanzee common ancestor, and thereby infer lineage-specific substitutions for each OR gene.
In order to test for differences in selection pressures among the species, we compared the rate of synonymous and nonsynonymous divergence on each lineage. Under the null model, there is a single ratio of nonsynonymous to synonymous divergence (Dn/Ds) for the trio of species. Under the alternative, each lineage is allowed a separate Dn/Ds ratio. For each OR gene, we maximized the likelihood of the parameters given the data. We then used a likelihood ratio test (LRT) to test the null model. In this way, we could reject the null model for 52 OR genes (P < 0.05; five genes are expected to be significant by chance, after excluding genes with zero counts in any class of substitutions).
Since our main goal in this section was to identify specific genes that are most likely to evolve under positive selection, we concentrated on 18 OR genes that were significant at a false discovery rate (Benjamini and Hochberg 1995) of 1% (Table 1). We inspected the Dn/Ds values for each of these OR genes on individual lineages to help interpret the rejection of the null model. In six cases, the Dn/Ds value for substitutions on the chimpanzee lineage was below one, while the Dn/Ds value for the human lineage was higher than 1.2 (Table 1). This suggests that the rejection of the null model in these cases is due to positive selection driving the evolution of the human, but not the chimpanzee OR gene. Similarly, we find three cases for which it seems that the chimpanzee, but not the human gene, has evolved under positive selection. In nine cases, the Dn/Ds value for both lineages is lower than 1.2. In these cases, the rejection of the null model may be due to strong purifying selection on one lineage (possibly the mouse) and relaxed constraint on at least one of the others.
Table 1.
LRT results and Ka/Ks value for human and chimpanzee OR genes
| Locus (human name) | Humana Dn/Ds | Chimpanzeea Dn/Ds | LRT | P valueb |
|---|---|---|---|---|
| OR52H1 | 2.80 | 0.97 | 34.24 | 0.000001 |
| OR5M3 | 0.78 | 0.48 | 25.98 | 0.000002 |
| OR5M8 | 0.47 | 1.13 | 21.34 | 0.000023 |
| OR11L1 | 1.00 | 0.78 | 20.23 | 0.000040 |
| OR1L8 | 0.66 | 1.20 | 19.01 | 0.000074 |
| OR52B2 | 0.79 | 0.33 | 18.26 | 0.000108 |
| OR4F29 | 0.48 | 3.20 | 17.30 | 0.000175 |
| OR6K2 | 2.03 | 0.71 | 17.13 | 0.000190 |
| OR51G1 | 1.35 | 0.69 | 15.76 | 0.000378 |
| OR4D11 | 1.73 | 0.68 | 15.76 | 0.000379 |
| OR10G7 | 0.60 | 1.14 | 15.34 | 0.000468 |
| OR4C11 | 0.67 | 0.84 | 15.11 | 0.000525 |
| OR5AP2 | 1.10 | 0.92 | 14.19 | 0.000829 |
| OR4D10 | 0.65 | 0.94 | 14.16 | 0.000841 |
| OR51Q1 | 0.78 | 0.94 | 13.70 | 0.001059 |
| OR56B2P | 0.62 | 2.49 | 13.29 | 0.001297 |
| OR1P1P | 1.60 | 0.33 | 13.08 | 0.001444 |
| OR4F13P | 2.20 | 0.43 | 12.91 | 0.001576 |
Dn/Ds values that seem indicative of positive selection are in bold.
P values of the Likelihood Ratio test (LRT) of the null vs. alternative models.
Discussion
We analyzed the complete chimpanzee OR gene repertoire and compared it with the repertoire of OR genes in human. On a first pass, the number of chimpanzee genomic segments that our algorithm identified as OR gene candidates is 26% higher than the number of human OR genes. Moreover, we could only find clear human orthologs for 761 (69%) of the chimpanzee candidate OR genes. However, when we only considered the 899 chimpanzee OR sequences that are longer than 300 bp, the size of the chimpanzee OR repertoire becomes similar to that of human, and the proportion of chimpanzee loci with a human ortholog is 85%. This suggests that many of the short sequences identified as OR genes result from imperfections of the chimpanzee genome assembly. It is not improbable that these sequences should have been collapsed in the assembly, rather than be represented as unique (short) genomic segments.
The discrepancy in ortholog matches does not completely disappear when the chimpanzee short sequences are excluded. In most remaining cases, we can explain the lack of an ortholog for ∼15% of OR genes by lineage-specific relative expansions.
These expansions include both intact OR genes as well as pseudogenes and are probably the product of a neutral process of duplication and deletion (Nei et al. 2000). Alternatively, these expansions could be the result of species-specific sensory needs, as the number of functional genes within any given OR subfamily may be proportional to the breadth of binding sites within a subfamily (Malnic et al. 1999).
We also noted a difference between human and chimpanzee in the size of the 7E OR subfamily. This OR subfamily consists almost exclusively of pseudogenes and was shown to have expanded in the human lineage (Newman and Trask 2003). Our findings suggest a similar, even more pronounced, expansion of family 7E in chimpanzee. The selective advantage of this expansion, if any, remains unclear.
Relaxation of constraint on the human lineage
We used the previously published sequence of 30 chimpanzee intact OR genes (Gilad et al. 2003b) in order to estimate the number of sequencing errors that lead to an apparent coding-region disruption in the chimpanzee genome draft. Our corrected estimate of the proportion of pseudogenes in the chimpanzee OR repertoire (∼50%) is still significantly higher than the estimate from Gilad et al. (2003a,b). This is probably due to the high number of subfamily 7E OR pseudogenes in chimpanzee. OR genes from this subfamily were excluded from the analysis of Gilad et al. (2003a,b), since a recent expansion has been observed for this subfamily (Newman and Trask 2003), and, except for one sequence, all of the 7E ORs are pseudogenes. If we exclude the 7E subfamily from our analysis, the proportion of pseudogenes in human and chimpanzee are 51% and 41%, respectively (P <10-3). These values are within the 95% CI of the observations of Gilad et al. (2003a,b). We note that if we underestimated the number of sequence errors that result in an apparent disrupted coding region, the correct proportion of OR pseudogenes in chimpanzee may be lower. Thus, the use of the entire repertoire confirms that a greater proportion of OR genes evolve under no or little constraint in humans relative to chimpanzees.
In our attempt to date the time since humans have started to rapidly accumulate OR pseudogenes, we made the simplistic assumption that all human intact OR genes were under evolutionary constraint until some point in human evolution. Then, a subset of OR genes became unnecessary, and hence, neutrally evolving. We know, however, that not all ORs are under constraint in nonhuman primates (Gilad et al. 2003b). Thus, a more realistic model might include a background rate of OR disruptions for all primates, with additional sets of OR genes becoming unnecessary at various time points during human evolution. Unfortunately, without additional information, it is difficult to make inferences about such a model. However, by assuming no background rate of OR gene disruptions in our calculation, our estimate is an upper bound on the time since humans experienced relaxed evolutionary constraint relative to other primates. Hence, we are able to exclude the possibility that humans have been accumulating OR pseudogenes at a neutral rate since human and chimpanzee last had a common ancestor.
Positive selection on OR genes
Previously, Gilad et al. (2003a) suggested that OR genes in human evolve under positive selection, but found no evidence for such adaptation in chimpanzee. The authors found that most chimpanzee intact OR genes evolve under strong evolutionary constraint and suggested that this may reduce the power to detect positive selection. Here, we take advantage of the identification of 201 human-chimpanzee-mouse ortholog trios. Our approach is similar to that used by Clark et al. (2003) to detect rapidly evolving proteins in human and chimpanzee.
We find 52 OR genes whose phylogenetic trees are significantly more likely under a model where Dn/Ds varies among evolutionary lineages. A significant LRT result could reflect differences in selective constraint between orthologs, or might result from positive selection acting on an OR gene in only one of the lineages. By inspecting the data further, we highlighted several OR genes, both in human and in chimpanzee, as probable candidates for adaptations. These OR genes experienced, on average, seven amino acid substitutions per gene. Interestingly, in some cases (OR4D11 and OR1P1P in human, and OR1L8 in chimpanzee), we find that amino acid substitutions occurred in the putative-binding site of the OR protein (Man et al. 2004). These changes may have functional significance. However, in the other OR proteins that are inferred to have evolved under selection, amino acid substitutions are scattered with no clear pattern. In addition, we find several substitutions in positions that are otherwise extremely conserved across OR proteins (such as the DRY motif) (Buck and Axel 1991). Substitutions in these positions may result in a dysfunctional receptor (Young et al. 2002). We are unable to provide a satisfactory explanation for our observation of Dn/Ds ratios well above one for these genes.
The signature of selection on OR genes can be corroborated by the analysis of polymorphism data (e.g., Hamblin and Di Rienzo 2000; Hamblin et al. 2002). Targets of selection identified from the analysis of polymorphism and divergence are promising candidates for human- and chimpanzee-specific chemosensory traits. A natural next step is to collect data from additional primates to establish whether selective pressures are truly exclusive to one species. Finally, studies to associate OR genes to their primary odorants will determine whether the genes identified in this study truly underlie species-specific sensitivity.
Methods
Identification of chimpanzee OR genes
We used Gene-IT's BioFacet software (Gene-IT) to compare the chimpanzee genome draft (PCAP1026, NCBI Build 1.1, November 2003, http://www.ncbi.nlm.nih.gov/; R. Waterston, pers. comm.) to all nucleotide sequences in HORDE v.40 (http://bip.weizmann.ac.il/HORDE/), with an expectation value cutoff of 0.00001. We selected all resulting alignments with a Smith-Waterman score >50 and over 70% identity and coalesced overlapping results, thus obtaining 1091 genomic segments. The most frequent length of these genomic segments was ∼930 bp, corresponding to a complete OR gene (Pilpel and Lancet 1999). We generated a library of potential chimp OR genes by extracting these genomic ranges, padding them with 200 bp in each direction (where possible) and masked repeats using RepeatMasker (http://repeatmasker.systemsbiology.net/). Using FASTX (Pearson et al. 1997), we compared each potential chimp OR gene to the intact protein sequences in HORDE v.40, with an expectation value cutoff of 0.01, and kept up to 10 results. We then used the protein match with highest identity to the query to reconstruct a conceptual translation for each chimp OR gene.
Phylogenetic analysis
We selected those human OR genes that had a nucleotide sequence of at least 800 bp. Since the chimpanzee collection of ORs was more likely to contain fragments, we used an alternative criterion to select chimpanzee OR genes; if the conceptually translated nucleotide sequence was flanked on both sides by untranslated sequence, then the conceptually translated region had to span at least 800 bp. Since the protein sequences of genes are better conserved than the nucleotide sequences, we chose a protein multiple-sequence alignment as a starting point for the phylogenetic analysis. We used ClustalX v1.83 (Chenna et al. 2003) in “Profile Alignment” mode to align the conceptual translations of the selected OR genes against a template alignment—a previously published, manually curated, OR multiple sequence alignment that contained representatives from all OR families (Man et al. 2004). An overlap of at least 70 amino acids in the alignment was selected as a criterion to determine whether two genes could be compared. We scanned the resultant alignment for pairs of sequences that did not meet this criterion. We then excluded a minimal number of sequences from our set of human and chimp genes, so that all pairs of sequences had an overlap that is longer than the cutoff. The remaining sequences, 694 from chimpanzee and 762 from human, were aligned again against the template alignment. We used seaview (Galtier et al. 1996) to correct any obvious errors in the alignment. We manually added the protein sequence of bovine rhodopsin to the alignment, according to a previously published alignment (Man et al. 2004). We then back-translated the resultant protein sequence alignment into a nucleotide sequence alignment, from which we computed a distance matrix using only overlap regions for each pair of sequences. We constructed a phylogenetic tree with the the neighbor program from the PHYLIP package, using bovine rhodopsin as an outgroup. Trees were drawn using TreeExplorer (K. Tamura; http://evolgen.biol.metro-u.ac.jp/TE/TE_man.html).
Identification of human-chimpanzee orthologs
We generated the human-chimpanzee OR gene ortholog list by using a novel statistical approach for comparing and ranking sequence alignments (G. Glusman and A. Siegel, in prep.). This method (1) generates all pairwise alignments between human and chimpanzee OR protein sequences, (2) compares every pair of alignments to determine whether they could be randomly generated using an equivalent background model, (3) sorts the alignments by ranking higher those with statistically significantly higher identity levels, or in the case of statistical equivalence, preferring longer alignments, and (4) generates the list of potential orthologs by scanning the ranked alignments, accepting best-matching human-chimpanzee pairs, and discarding pairs involving previously assigned sequences.
Identification of shared and human-specific pseudogenes
Using conceptual translation, we identified all of the coding-region disruptions in human OR pseudogenes that are present in the human-chimpanzee OR ortholog list. If an uninterrupted ORF was found, the gene was annotated as intact. If no ORF was identified, the gene was annotated as a pseudogene. This approach probably results in an underestimate of the proportion of pseudogenes, as not all OR genes with an intact coding region are functional. Mutations in promoter or control regions of OR genes may lead to reduced or no expression. Similarly, radical missense mutations in highly conserved positions of the OR protein may result in dysfunction (Young et al. 2002; Menashe et al. 2003). Although it is known that there are several highly conserved positions among OR genes, it is not always straightforward to ascertain which, if any, of these positions is necessary to retain function. Some changes will alter, rather than completely abolish the function of the receptor (Gaillard et al. 2004). We therefore chose the most straightforward definition of a pseudogene, a gene without a full open reading frame.
We then preformed a pairwise alignment of the conceptual protein sequence of each human pseudogene with its conceptually translated chimpanzee ortholog. A coding-region disruption was considered to be “shared” between the two species if the same codon carried the mutation (a stop codon, or a single basepair insertion/deletion within a codon). In all cases, we noted how many coding-region disruptions are shared versus human specific. If no shared disruptions were found, the locus was inferred to be a human-specific pseudogene.
Estimation of the time since human rapid accumulation of OR pseudogenes
We assumed that the number of coding-region disruptions per locus is Poisson distributed (i.e., that disruption mutations occur at a constant rate, are independent and infrequent). Let n be the number of genes with disruptions and T be the total number of observed disruptions in human-specific pseudogenes. We cannot directly observe the number of human OR genes that could have been disrupted (i.e., are under no constraint) but by chance were not. Instead, we observe all intact genes, a subset of which were not disrupted by chance and a subset of which are probably intact due to evolutionary constraint. Thus, we are missing information about the number of loci with zero disruptions, X. Conditional on X loci with 0 disruptions, λ = T/(X+n). In order to estimate X and λ jointly, we solved for the λ that minimized the sum of χ2 deviations (across classes, for zero to infinity observations), setting X =e[-λ]. To assess the error associated with our estimate of the mean, we performed the following bootstrapping procedure: We drew repeatedly from a Poisson distribution with mean 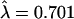 until there were T (or more) total observations, then estimated the sample mean. As our ∼95% confidence interval, we took the central 95 percentile of the distribution of sample means across 10,000 replicates.
until there were T (or more) total observations, then estimated the sample mean. As our ∼95% confidence interval, we took the central 95 percentile of the distribution of sample means across 10,000 replicates.
PAML analysis
We used the PAML package (Yang 1997), with substitution model (4; HKY85), in order to infer the sequence ancestral to human and chimpanzee for each OR gene in the ortholog trio list. We also used PAML in order to assess the likelihood of two models of protein evolution given our data. The null model (H0), allows one Dn/Ds parameter for the entire tree, while the alternative model (H1) permits a separate Dn/Ds ratio for each lineage. We use a likelihood ratio test (LRT) to test the null model and a χ2 distribution with two degrees of freedom to obtain p-values. Since sequence divergence between human and chimpanzee is only ∼1.2% (Chen et al. 2001; Ebersberger et al. 2002), in some cases, there were no sequence differences in one or more substitution categories (synonymous or nonsynonymous substitutions) in one or more lineages. In these cases, we could not estimate meaningful Dn/Ds ratios for all the lineages, and we therefore excluded these loci from the analysis.
Electronic database information
All chimpanzee OR sequences were submitted to the HORDE (http://bip.weizmann.ac.il/HORDE/) database.
Acknowledgments
We thank Gene-IT for providing the Biofacet software, as well as M. Przeworski and S. Pääbo for stimulating discussions and helpful comments and discussions. Y.G. is supported by an EMBO postdoctoral fellowship.
Footnotes
Article and publication are at http://www.genome.org/cgi/doi/10.1101/gr.2846405.
References
- Ben-Arie, N., Lancet, D., Taylor, C., Khen, M., Walker, N., Ledbetter, D.H., Carrozzo, R., Patel, K., Sheer, D., Lehrach, H., et al. 1994. Olfactory receptor gene cluster on human chromosome 17: Possible duplication of an ancestral receptor repertoire. Hum. Mol. Genet. 3: 229-235. [DOI] [PubMed] [Google Scholar]
- Benjamini, Y. and Hochberg, Y. 1995. Controlling the false discovery rate—A practical and powerful approach to multiple testing. J. R. Stat. Soc. B Met. 57: 289-300. [Google Scholar]
- Buck, L. and Axel, R. 1991. A novel multigene family may encode odorant receptors: A molecular basis for odor recognition. Cell 65: 175-187. [DOI] [PubMed] [Google Scholar]
- Chen, F.C., Vallender, E.J., Wang, H., Tzeng, C.S., and Li, W.H. 2001. Genomic divergence between human and chimpanzee estimated from large-scale alignments of genomic sequences. J. Hered. 92: 481-489. [DOI] [PubMed] [Google Scholar]
- Chenna, R., Sugawara, H., Koike, T., Lopez, R., Gibson, T.J., Higgins, D.G., and Thompson, J.D. 2003. Multiple sequence alignment with the Clustal series of programs. Nucleic Acids Res. 31: 3497-3500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark, A.G., Glanowski, S., Nielsen, R., Thomas, P.D., Kejariwal, A., Todd, M.A., Tanenbaum, D.M., Civello, D., Lu, F., Murphy, B., et al. 2003. Inferring nonneutral evolution from human-chimp-mouse orthologous gene trios. Science 302: 1960-1963. [DOI] [PubMed] [Google Scholar]
- Ebersberger, I., Metzler, D., Schwarz, C., and Paabo, S. 2002. Genomewide comparison of DNA sequences between humans and chimpanzees. Am. J. Hum. Genet. 70: 1490-1497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaillard, I., Rouquier, S., Chavanieu, A., Mollard, P., and Giorgi, D. 2004. Amino-acid changes acquired during evolution by olfactory receptor 912-93 modify the specificity of odorant recognition. Hum. Mol. Genet. 13: 771-780. [DOI] [PubMed] [Google Scholar]
- Galtier, N., Gouy, M., and Gautier, C. 1996. SEAVIEW and PHYLO_WIN: Two graphic tools for sequence alignment and molecular phylogeny. Comput. Appl. Biosci. 12: 543-548. [DOI] [PubMed] [Google Scholar]
- Gilad, Y., Segre, D., Skorecki, K., Nachman, M.W., Lancet, D., and Sharon, D. 2000. Dichotomy of single-nucleotide polymorphism haplotypes in olfactory receptor genes and pseudogenes. Nat. Genet. 26: 221-224. [DOI] [PubMed] [Google Scholar]
- Gilad, Y., Bustamante, C.D., Lancet, D., and Paabo, S. 2003a. Natural selection on the olfactory receptor gene family in humans and chimpanzees. Am. J. Hum. Genet. 73: 489-501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilad, Y., Man, O., Paabo, S., and Lancet, D. 2003b. Human specific loss of olfactory receptor genes. Proc. Natl. Acad. Sci. 100: 3324-3327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glusman, G., Bahar, A., Sharon, D., Pilpel, Y., White, J., and Lancet, D. 2000. The olfactory receptor gene superfamily: Data mining, classification, and nomenclature. Mamm. Genome 11: 1016-1023. [DOI] [PubMed] [Google Scholar]
- Glusman, G., Yanai, I., Rubin, I., and Lancet, D. 2001. The complete human olfactory subgenome. Genome Res. 11: 685-702. [DOI] [PubMed] [Google Scholar]
- Godfrey, P.A., Malnic, B., and Buck, L.B. 2004. The mouse olfactory receptor gene family. Proc. Natl. Acad. Sci. 101: 2156-2161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamblin, M.T. and Di Rienzo, A. 2000. Detection of the signature of natural selection in humans: Evidence from the Duffy blood group locus. Am. J. Hum. Genet. 66: 1669-1679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamblin, M.T., Thompson, E.E., and Di Rienzo, A. 2002. Complex signatures of natural selection at the Duffy blood group locus. Am. J. Hum. Genet. 70: 369-383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linardopoulou, E., Mefford, H.C., Nguyen, O., Friedman, C., van den Engh, G., Farwell, D.G., Coltrera, M., and Trask, B.J. 2001. Transcriptional activity of multiple copies of a subtelomerically located olfactory receptor gene that is polymorphic in number and location. Hum. Mol. Genet. 10: 2373-2383. [DOI] [PubMed] [Google Scholar]
- Malnic, B., Hirono, J., Sato, T., and Buck, L.B. 1999. Combinatorial receptor codes for odors. Cell 96: 713-723. [DOI] [PubMed] [Google Scholar]
- Malnic, B., Godfrey, P.A., and Buck, L.B. 2004. The human olfactory receptor gene family. Proc. Natl. Acad. Sci. 101: 7205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Man, O., Gilad, Y., and Lancet, D. 2004. Prediction of the odorant binding site of olfactory receptor proteins by human-mouse comparisons. Protein Sci. 13: 240-254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menashe, I., Man, O., Lancet, D., and Gilad, Y. 2003. Different noses for different people. Nat. Genet. 34: 143-144. [DOI] [PubMed] [Google Scholar]
- Nei, M., Rogozin, I.B., and Piontkivska, H. 2000. Purifying selection and birth-and-death evolution in the ubiquitin gene family. Proc. Natl. Acad. Sci. 97: 10866-10871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newman, T. and Trask, B.J. 2003. Complex evolution of 7E olfactory receptor genes in segmental duplications. Genome Res. 13: 781-793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olender, T., Fuchs, T., Linhart, C., Shamir, R., Adams, M., Kalush, F., Khen, M., and Lancet, D. 2004. The canine olfactory subgenome. Genomics 83: 361-372. [DOI] [PubMed] [Google Scholar]
- Pearson, W.R., Wood, T., Zhang, Z., and Miller, W. 1997. Comparison of DNA sequences with protein sequences. Genomics 46: 24-36. [DOI] [PubMed] [Google Scholar]
- Pilpel, Y. and Lancet, D. 1999. The variable and conserved interfaces of modeled olfactory receptor proteins. Protein Sci. 8: 969-977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quignon, P., Kirkness, E., Cadieu, E., Touleimat, N., Guyon, R., Renier, C., Hitte, C., Andre, C., Fraser, C., and Galibert, F. 2003. Comparison of the canine and human olfactory receptor gene repertoires. Genome Biol. 4: R80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rouquier, S., Taviaux, S., Trask, B.J., Brand-Arpon, V., van den Engh, G., Demaille, J., and Giorgi, D. 1998. Distribution of olfactory receptor genes in the human genome. Nat. Genet. 18: 243-250. [DOI] [PubMed] [Google Scholar]
- Rouquier, S., Blancher, A., and Giorgi, D. 2000. The olfactory receptor gene repertoire in primates and mouse: Evidence for reduction of the functional fraction in primates. Proc. Natl. Acad. Sci. 97: 2870-2874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trask, B.J., Massa, H., Brand-Arpon, V., Chan, K., Friedman, C., Nguyen, O.T., Eichler, E., van den Engh, G., Rouquier, S., Shizuya, H., et al. 1998. Large multi-chromosomal duplications encompass many members of the olfactory receptor gene family in the human genome. Hum. Mol. Genet. 7: 2007-2020. [DOI] [PubMed] [Google Scholar]
- Yang, Z. 1997. PAML: A program package for phylogenetic analysis by maximum likelihood. Comput. Appl. Biosci. 13: 555-556. [DOI] [PubMed] [Google Scholar]
- Young, J.M., Friedman, C., Williams, E.M., Ross, J.A., Tonnes-Priddy, L., and Trask, B.J. 2002. Different evolutionary processes shaped the mouse and human olfactory receptor gene families. Hum. Mol. Genet. 11: 535-546. [DOI] [PubMed] [Google Scholar]
- Zhang, X. and Firestein, S. 2002. The olfactory receptor gene superfamily of the mouse. Nat. Neurosci. 5: 124-133. [DOI] [PubMed] [Google Scholar]
WEB SITE REFERENCES
- http://bip.weizmann.ac.il/HORDE/; Human Olfactory Receptor Data Exploratorium.
- http://evolgen.biol.metro-u.ac.jp/TE/TE_man.html; TreeExplorer.
- http://repeatmasker.systemsbiology.net/; RepeatMasker.