

Abstract
A high-throughput genotyping system for scoring single nucleotide polymorphisms (SNPs) has been developed. With this system, >1000 SNPs can be analyzed in a single assay, with a sensitivity that allows the use of single haploid cells as starting material. In the multiplex polymorphic sequence amplification step, instead of attaching universal sequences to the amplicons, primers that are unlikely to have nonspecific and productive interactions are used. Genotypes of SNPs are then determined by using the widely accessible microarray technology and the simple single-base extension assay. Three SNP panels, each consisting of >1000 SNPs, were incorporated into this system. The system was used to analyze 24 human genomic DNA samples. With 5 ng of human genomic DNA, the average detection rate was 98.22% when single probes were used, and 96.71% could be detected by dual probes in different directions. When single sperm cells were used, 91.88% of the SNPs were detectable, which is comparable to the level that was reached when very few genetic markers were used. By using a dual-probe assay, the average genotyping accuracy was 99.96% for 5 ng of human genomic DNA and 99.95% for single sperm. This system may be used to significantly facilitate large-scale genetic analysis even if the amount of DNA template is very limited or even highly degraded as that obtained from paraffin-embedded cancer specimens, and to make many unpractical research projects highly realistic and affordable.
With 24,000-35,000 genes in the human genome (Ewing and Green 2000; Roest Crollius et al. 2000; Lander et al. 2001; Venter et al. 2001; Brentani et al. 2003; Pennisi 2003) and a large portion of these genes expressed in each tissue, no biological process in the human body occurs in isolation. Although development of modern molecular tools has allowed researchers to study individual genes in great detail, today's biologists are becoming more and more interested in understanding biological processes in a comprehensive way. Genetic analysis is one of the most powerful tools for this purpose. The recent discovery of millions of single nucleotide polymorphisms (SNPs) has provided a rich resource for such analysis. dbSNP Build 121 of the SNP database maintained by the National Center for Biotechnology Information (NCBI) contains 19,888,389 SNP submissions representing 9,856,125 nonredundant clusters, of which 4,540,241 have been validated as real SNPs. However, how to take advantage of this magnificent resource has been a challenging issue.
To genotype an SNP, two major requirements need to be met: (1) detection of the polymorphic sequence, and (2) discrimination of the allelic variants that differ by a single base. Several available techniques meet the second requirement (for review, see Brennan 2001; Kwok and Chen 2003), many of which are very efficient and cost-effective. The first requirement is usually met after DNA sequence amplification by the polymerase chain reaction (PCR) (Saiki et al. 1985; Mullis and Faloona 1987). PCR is so powerful and convenient that it has become a necessary step in most genotyping systems. However, amplification of a large number of polymorphic sequences separately is very expensive and time-consuming. Several efforts have been made to amplify multiple sequences simultaneously. However, the capacity of multiplex PCR is limited by primer dimerization. Primer dimers from primer dimerization are very deleterious not only because they possess perfect primer-anchoring sequences but also because they are usually much shorter than the amplicons and, therefore, amplify easily. For this reason, the capacity of multiplex PCR was a bottleneck in high-throughput genotyping and nucleic acid detection before success in development of high-throughput multiplex genotyping systems.
Early efforts to enhance the multiplex capacity involved optimizing PCR conditions based on the belief that better PCR conditions may allow more sequences to be amplified simultaneously (for review, see Markoulatos et al. 2002). However, since optimized PCR conditions cannot significantly reduce primer interaction, such efforts have rarely reached the capacity of simultaneous amplification of 10 sequences (for review, see Edwards and Gibbs 1994).
Second-generation methods were initially marked by the protocol developed in our laboratory (Lin et al. 1996), which features using 5′ universal sequences (tails) on specific PCR primers that were attached to the ends of the amplicons during the early stage of PCR amplification. All specific primers could then be replaced by only two primers identical to the 5′ universal tails. This improvement allowed amplification of 26 DNA sequences in a single tube (Lin et al. 1996). It was shown that 41 sequences could be amplified very specifically and the resultant fragments could be resolved by gel electrophoresis (X. Cui and H. Li, unpubl.).
Very recently, the universal tail concept was applied by a few newly developed systems (Yeakley et al. 2002; Hardenbol et al. 2003; Kennedy et al. 2003; Fan et al. 2004; Matsuzaki et al. 2004). With these systems, universal sequences are attached to the amplicons with different approaches before PCR, allowing >1000 SNP-containing sequences to be amplified in a single reaction. This advance has significantly facilitated high-throughput genotyping. The technique initially described by Yeakley et al. (2002) has been commercialized by Illumina and used in ∼65% of the HapMap project (http://www.hapmap.org). The technologies described by Hardenbol et al. (2003) and Kennedy et al. (2003) have also been commercialized and used in largescale genetic analyses (Butcher et al. 2004; Zhou et al. 2004). However, attaching the universal tails to the amplified sequences requires additional experimental steps in comparison with amplification by regular PCR, limiting the detection sensitivity. Some of these procedures also require pooling of individually amplified PCR products from multiple tubes, followed by column purification. Requirements of specialized probes and detection platforms and long oligonucleotides also limit the flexibility and cost-effectiveness of these systems.
In this communication, we describe a simple genotyping system that requires a single round of multiplex PCR followed by a single step to generate single-stranded DNA (ssDNA) before genotype determination. We have shown that 20 μL of PCR product amplified from 5 ng of genomic DNA is sufficient for routine detection of >1000 SNPs. Highly reliable results have also been obtained from analyzing single haploid sperm cells.
Results
High-throughput multiplex amplification and genotype determination
SNP selection
SNPs were selected from the dbSNP database (ftp://ftp.ncbi.nih.gov/snp/human/chr_rpts/) maintained by NCBI. To ensure that the selected SNPs were real and suitable for the multiplex system, a series of filters was used for selection. These filters excluded SNPs that were flanked by a significant number of short tandem repeats and closely located SNPs (i.e., SNPs separated by <130 bases), which may significantly affect the specificity of amplification. All selected SNP sequences were passed through the BLAST (http://www.ncbi.nlm.nih.gov/BLAST) and BLAT (http://www.genome.ucsc.edu/cgi-bin/hgBlat?db=hg8) searches at the Web sites maintained by NCBI and the University of California, Santa Cruz (UCSC), respectively. SNPs with flanking sequences having more than one hit in the human genome were also excluded in the study. When information was available, SNPs with their heterozygosities >0.18 were chosen. In order to use a twocolor fluorescent labeling system (which uses the cyanine dyes Cy3 and Cy5) for genotype determination, only transition SNPs (A/T to G/C changes or vice versa) were selected (note that transversion SNPs may be included either by using a four-color system or by grouping the suitable types together). Sequences for the SNPs used in the present study are given online at http://www2.umdnj.edu/lilabweb/Publications/Multiplex3G.
Primer design
So far, all approaches toward enhancing multiplex amplification capacity have been based on minimizing primer-primer interaction or by mitigating such interactions by making additional experimental efforts. If primers with no predictable productive interaction (i.e., lacking significant complementarity that might cause in primer dimerization) are selected, all experimental effort toward minimizing and avoiding primer-primer interaction may become unnecessary. We have shown that this can be accomplished by selecting primer sequences with minimum complementarity between their 3′-ends and globally as specified in the Methods section. Primer sequences used in the present study are given online at http://www2.umdnj.edu/lilabweb/Publications/Multiplex3G.
To determine the generality of our program for primer design, a simulation study was performed. Three input groups with 1200 or more SNPs in each were selected with the filtering criteria described in Methods. Another three groups of comparable size were selected randomly from the dbSNP database. Primers could be designed for ∼90% of both filtered and randomly selected groups, indicating that our program can be used for the vast majority of randomly selected SNPs (Table 1).
Table 1.
Assay conversion rates for filtered and randomly selected SNPs
| Selection | Starting SNPs | Converted | Conversion rate |
|---|---|---|---|
| Filtered | 1200 | 1093 | 91.08 |
| Filtered | 1200 | 1057 | 88.08 |
| Filtered | 1203 | 1093 | 90.86 |
| Average | 1201 | 1081 | 90.01 |
| Random | 1206 | 1059 | 87.81 |
| Random | 1207 | 1085 | 89.89 |
| Random | 1200 | 1081 | 90.08 |
| Average | 1204 | 1075 | 89.26 |
Multiplex amplification and polymorphic sequence detection
Polymorphic sequences of each group were amplified by a single multiplex reaction (Fig. 1A). A 1-2-μL aliquot of the amplified product was used as template for ssDNA synthesis under the same conditions used in PCR but with only one primer added for each SNP. These primers were designed in such a way that their 3′- ends would anneal next to the polymorphic sites (Fig. 1B), and, therefore, they could also be used as probes (primer-probes) on the microarray for genotyping (see below). For each SNP, two such primer-probes were designed in opposite directions so that they could be used to generate ssDNA in different directions. Resulting ssDNA was hybridized to the probes arrayed onto a glass slide. During hybridization, different ssDNA species from the multiplex amplification bind to their respective probes on the glass slide allowing the ssDNA species to be resolved (Fig. 1E).
Figure 1.

Schematic illustration of the multiplex genotyping procedure. Only one SNP is shown. Primers and probes are shown as arrowed lines. Microarray spots are indicated as ellipsoids. (A) Amplification of the polymorphic sequence. Two allelic sequences use the same set of primers, P1 and P2. (B) Generation of ssDNA by using the primer-probes in both directions in separate tubes. Only the two allelic template strands in each reaction are shown. (C) ssDNA generated from B. (D) Addition of the ssDNA to the respective microarrays containing probes in different directions. (E) ssDNA templates hybridized to their probes on the microarrays. (F) Labeling probes by incorporating fluorescently labeled ddNTPs. (G) Labeled probes after washing off all other reagents.
The next step was to determine the allelic state of the sequences hybridized to their probes on the microarray. This was accomplished by the single-base-extension assay (Shumaker et al. 1996; Pastinen et al. 1997, 2000; Syvanen 1999; Lindblad-Toh et al. 2000). As mentioned above, the 3′-ends of the oligonucleotide probes hybridizing to the ssDNA were immediately next to the polymorphic sites (Fig. 1E). Using ssDNA as a template, each probe was extended by a single dideoxynucleoside triphosphate (ddNTP) conjugated to a fluorescent chromophore (Cy3 or Cy5). In this way, probes hybridizing to different allelic sequences were labeled with different fluorescent colors (Fig. 1F). After labeling, everything but the labeled probes was washed off, and the microarray was ready for scanning.
Because DNA sequences are double stranded, ssDNA can be generated in two directions and used for independent genotyping with the corresponding probes. Results generated using such a dual-probe method can be compared. Inconsistent genotypes can then be discarded to ensure a very high level of genotyping accuracy. To simplify the study, all probes incorporating ddA and/or ddG were always used together and were designated as AG probes. Similarly, all CT probes were also used together for genotyping in the opposite direction. The relative correspondence between these primer-probes and the regular primers is shown in Figure 1. Primer-probe sequences used in the present study are given online at http://www2.umdnj.edu/lilabweb/Publications/Multiplex3G. A typical microarray image and a scatter plot from this image are shown in Figure 2, A and B.
Figure 2.

(A) A microarray image from genotyping one individual with Group II SNPs. Each probe was printed twice and shown as neighboring spots. Spots in red and green, homozygous; yellow, heterozygous; white, pink, and light green, spots with strong signal that have exceeded the linear range; and dark, low signal but not necessarily mean no signal or too low for genotype calls. (B) Scatter plot based on the color intensities from the microarray image shown in A. Two horizontal lines are the cutoffs (natural logarithms of the ratios [Cy3/Cy5] at 2 and -2) to divide the spots into three genotype groups. (C) A plot simply based on the two color intensities for the 24 samples (two spots for each sample) of an SNP. Values of the signal intensities indicated on the axes should be multiplied by 1000. Note that since different parameters are used, the color orientations are different in B and C.
Genotype determination
Theoretically, when hybridizing ssDNA contains a homozygous SNP, a probe should predominantly incorporate one color (signal color that is specific) over the other (background color), while a probe hybridizing to ssDNA containing a heterozygous SNP should incorporate both colors equally. Experimentally, the color intensity is affected by various factors such as nonspecific hybridization, the bandwidth of the light filters, and the ratio between photomultiplier gains selected for each wavelength during scanning. Because of the impact of these experimental factors, three major issues need to be addressed when determining genotypes from the digitized data produced from a microarray image: (1) normalization of the two color intensities, (2) background subtraction of each color, and (3) genotype determination. A computer program, AccuTyping, was developed to address these issues using the algorithms below.
We take advantage of our ability to analyze a large number of SNPs in a single assay and use the color intensities from homozygous SNPs as internal controls. Our computer program first sorted the SNPs based on the ratio between their two color intensities. For each individual, the maximal fraction of heterozygous SNPs is expected to be 50%, and the other 50% would be homozygous with 25% for each of the two alleles. To be conservative, we treat 20% of SNPs with the highest ratio and 20% with the lowest ratio as homozygous.
A given homozygous SNP has two color intensities, that is, the background color intensity and the signal color intensity. The background color intensity can be used for background subtraction. However, the intensities of the two signal colors of heterozygous SNPs may differ globally and often deviate from a 1:1 ratio because of experimental variables. Such a difference can be calibrated based on the signal color intensities of these two groups of homozygous SNPs, that is, those with the highest and lowest ratios between the two color intensities.
After normalization and background subtraction, the genotypes were determined based on the log ratios between the two normalized color intensities by using empirical linear values as cutoffs, which divided SNPs into three groups, two homozygous and one heterozygous. The cutoff values were validated by comparing the microarray results with those obtained by using independent genotyping methods.
Three multiplex groups with >1000 SNPs in each have been established (Table 2). Consideration was taken to select SNPs evenly distributed along the corresponding chromosomes. The chromosomes that these SNPs belong to and the average distances between adjacent SNPs are listed in Table 2.
Table 2.
Results from genotyping 24 samples with the three groups of SNPs
| Average detection rate (%)
|
Accuracy (%) based on
|
|||||||
|---|---|---|---|---|---|---|---|---|
| Group | Chromosomal location | Average spacing (kb) | Number of SNPs | AG | CT | Both | Method Ia | Method II |
| I | 1, 2 | 500 | 1068 | 97.83 | 98.32 | 96.24 ± 1.02 | 99.68 | 99.92 |
| II | 6, 18 | 222 | 1172 | 98.54 | 98.85 | 97.58 ± 0.52 | 99.91 | 99.98 |
| III | 13-17 | 500 (250 for Chr. 17) | 1102 | 97.74 | 98.05 | 96.30 ± 0.56 | 99.84 | 99.96 |
| Average | — | — | — | 98.04 | 98.41 | 96.71 ± 1.71 | 99.82 | 99.96 |
Method used in Hardenbol et al. (2003).
Validation and application of the multiplex genotyping system
The three multiplex groups of SNPs were used to analyze 24 DNA samples of unrelated human individuals from four ethnic groups, African American, American Indian, Asian, and Caucasian. Genotypes were obtained independently by using ssDNA and respective probes in two directions.
Detection rates, concordance, and accuracy
Table 2 summarizes the results of typing the 24 samples. As shown, the average detection rates were 97.83%, 98.54%, and 97.74% for the three groups when the AG probes were used. Comparable rates (98.32%, 98.85%, and 97.74%) were obtained by using the CT probes. When both probes were used, 96.24%, 97.58%, and 96.30% of SNPs were detectable in both directions with an average rate of 96.71%. The small SDs in Table 2 also indicate that the reproducibility of the three groups was very high. The concordance rates were obtained by comparing the results from using probes in different directions, and were 94.38% 97.06%, and 95.99% for Groups I to III, respectively. A scatter plot for the intensities of an SNP in Group II from the 24 samples is shown in Figure 2C to demonstrate the reproducibility.
The accuracy of our approach was first determined with the method described by Hardenbol et al. (2003). The error rate for probes in one direction was treated as
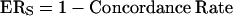 |
(1) |
where ERS is the error rate for probes in a single direction. The accuracy for dual probes is therefore calculated as
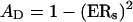 |
(2) |
where AD is the accuracy for dual probes. As shown in Table 2, ADs were calculated to be 99.68%, 99.91%, and 99.84% for the concordant genotypes from Groups I to III, respectively.
However, a lack of concordance does not necessarily mean incorrect genotyping. Because two colors were used in our system, it is reasonable to expect that approximately half of these genotypes were correct and the other half incorrect. To test this hypothesis, 47 genotypes with inconsistent results were further analyzed by RFLP and/or sequencing methods. In all, 23 genotypes obtained with the AG probes and 24 obtained with the CT probes were consistent with those obtained by RFLP and/or sequencing, indicating that ∼50% of the inconsistent genotypes were correct with respect to one probe. Therefore, the above formula (2) needs to be revised as
 |
(3) |
The accuracies were recalculated as 99.92%, 99.98%, and 99.96% for the concordant genotypes in the three groups.
To further validate our dual-probe approach, a DNA sample was genotyped with the three multiplex groups of SNPs. A panel of 1282 SNPs with concordant genotypes was found to be natural RFLPs and was reanalyzed with the RFLP method. We found that 25 (1.95%) resulting genotypes were inconsistent with those from microarray. All 25 genotypes were determined by sequencing, and all were found to be consistent with the microarray results, none with the RFLP results.
Sensitivity of the multiplex genotyping system—genotype determination of single human spermatozoa
Although the ability to genotype single sperm was first reported in 1988 (Li et al. 1988), markers that can be simultaneously genotyped have been limited to very few. We show here, for the first time, that >1000 SNPs may be included in such an analysis and that the genetic recombination events along the entire chromosomes can be analyzed and revealed directly without invoking statistical approaches. The SNPs in Group II were used for this purpose. Single sperm cells were prepared by flow cytometry as described previously (Pramanik and Li 2002). Then 15 single sperm samples were analyzed with probes in two directions. When the probes in one direction were used, the average detection rate was 91.88%, comparable to the rates from the previous studies in which very few SNPs were used (Cui et al. 1989; Goradia et al. 1991), indicating that our multiplex genotyping system is sensitive enough for this purpose. When sperm samples were analyzed with probes in both directions, the average concordance was 95.43%, which translates into an accuracy of 99.79% and 99.95%, respectively, by the two methods described above. A microarray image from single sperm analysis with Group II SNPs is shown in Figure 3.
Figure 3.

A microarray image from the analysis of single sperm with Group II SNPs. Each probe was printed twice as neighboring spots on the microarray. Spots in red and green, homozygous; yellow, heterozygous; white, pink, and light green, spots with strong signal that have exceeded the linear range; and dark, low signal but not necessarily mean no signal or too low for genotype calls. Yellow spots are either from SNPs that were not real because of the presences of a small portion of SNPs consisting of paralogous sequence variants in the databases (Cheung et al. 2003; Fredman et al. 2004), or from a low level (∼5%) of contamination as demonstrated in the previous studies (Cui et al. 1989; Goradia et al. 1991), which has been shown to be from oligonucleotides synthesized by the current hemi-open-oligonucleotide synthesis system. Note that heterozygous SNPs are treated as uninformative in genetic analyses with single sperm.
Discussion
Compared with other existing high-throughput approaches, our system has the following advantages:
Simplified experimental procedures: Our system does not require the attachment of universal tails to the amplicons.
High sensitivity: For regular genetic analysis, 5 ng of human genomic DNA can be routinely used in comparison with the hundreds of nanograms or even 2 μg of genomic DNA required by other methods. The present paper is the first report on genotyping of >1000 SNPs from single haploid cells. Our method can also be used to analyze a small amount of paraffin-embedded tissue isolated by microdissection (data not shown). Such success has made it possible to analyze, on a genomic scale, proliferative lesions that are usually microscopic in size in tumor samples, and to understand cancer development in a comprehensive way.
DNA extraction is not required: With our system, genomic DNA released from single or very few cells can be used directly for amplification.
Cost-effective: Since multiplex amplification with our system requires only PCR and ssDNA generation, less expense is needed for oligonucleotides and enzymes.
No demand for specialized equipment: Since our approach uses the widely available microarray facility for genotype determination, it may easily be used by the researchers.
Highly flexible: Users may tailor their multiplex groups to a desirable size and content without depending on commercial customization.
A high SNP conversion rate: Primers for multiplex amplification may be flexibly selected in a large zone flanking the polymorphic sites (300 bp), and, therefore, it is possible to have a high assay conversion rate. As described above, results from our simulation analysis indicate that primers can be designed for ∼90% of SNPs.
Compared with the system described by Kennedy et al. (2003) and Matsuzaki et al. (2004), which uses a single primer for multiplex amplification (not single probe for detection), our method requires specific primers for each SNP. When a large number of SNPs are included in a study, the cost of oligonucleotide synthesis becomes significant. This issue can be partially addressed by developing commonly shared SNP panels and by establishing oligonucleotide distribution facilities so that oligonucleotide stock may be shared by many research groups. However, the use of specific primers makes our system more flexible so that users can easily tailor the size and content of the multiplex groups based on their need.
By using dual probes, we have observed a small fraction of inconsistent genotypes (see above). One possible cause for such inconsistencies could be unknown polymorphisms and mutations located in the probe regions. Because SNPs with a minor allele frequency of 10% or higher can be found in each 600 bp in the human genome (Kruglyak and Nickerson 2001; Sachidanandam et al. 2001), and because the average length of the probes used in the present study was 29 bases, ∼9.7% of SNPs may have probe regions containing other SNPs, some of which may have significantly contributed to the inconsistencies. This raises the question about the limitation of single-probe-based SNP scoring methods, and necessitates the use of dual probes for high accuracy.
Differences in detection rate and consistency were seen among the three multiplex groups, which is a reflection of variability in the incorporation of improvements to the SNP selection process. The rates for Group II were generally better than those for Groups I and III mainly because SNPs in group II were selected from a newer version of the database in which more errors were corrected and more SNPs were submitted so that closely located SNPs could be avoided when the primers and probes were designed.
Development of the biomedical field has generated a strong demand for understanding biological processes on a large or genome scale. Systematic and comprehensive genetic analysis is critical for many projects such as identification of genes responsible for complex diseases, the exhaustive identification of genetic alterations in the cancer genome after microdissection, and for the understanding of many biological processes such as aging and drug metabolism. Our progress in developing the high-throughput genotyping system can significantly facilitate largescale analyses and to perform many studies, such as genomescale analysis of microscopic lesions in cancer with paraffinembedded tissue, which may not have been feasible in the past.
Methods
SNP selection and primer design
A computer program was written for SNP selection. It extracts SNP data from XML files downloaded from the NCBI dbSNP database (ftp://ftp.ncbi.nih.gov/snp/human/chr_rpts/) and filters out sequences containing more than one SNP within a userdefined interval (130 bases on each side of the polymorphic sites in the present study) to avoid possible complications in primer design. The program also excludes sequences containing any consecutive stretches of 10 or more mononucleotides, nine or more dinucleotide repeating units, or sequences containing five or more trinucleotide (or more) repeating units. Users may define the CG content (25%-75% for the present study) for the selected sequences. Candidate SNP sequences were submitted to the NCBI and UCSC Web sites for BLAST (http://www.ncbi.nlm.nih.gov/BLAST) and BLAT (http://www.genome.ucsc.edu/cgi-bin/hgBlat?db=hg8) searches to eliminate possible false SNPs caused by repetitive sequences.
To select sequence frames for primers, another computer program was written. The candidate sequence frames were first selected based on a user-defined melting temperature range (55°C to 78°C in this application) within a user-defined sequence range surrounding the polymorphic sites (150 bp in the present study). Further selection was then performed on qualified frames based on the following criteria: (1) fewer than four consecutively complementary bases between the 3′-ends of any frames; (2) fewer than eight but one consecutively complementary bases between the 3′-ends of any frames; (3) fewer than 10 consecutively complementary bases between the 3′-end of any frame and anywhere in all the others; (4) fewer than 12 but one consecutively complementary bases between the 3′-end of any frame and anywhere in all others; (5) complementary bases fewer than 75% anywhere between any two frames; and (6) complementary bases fewer than 13 bases between the 3′-end of any frame and any amplicon sequence.
Multiplex PCR and ssDNA preparation
The procedures for multiplex amplification and genotype determination are illustrated in Figure 1. First, multiplex PCR (Fig. 1A) was performed in 30 μL of PCR mix containing 1× PCR buffer (50 mM KCl, 100 mM Tris-HCl at pH 8.3, 1.5 mM MgCl2, and 100 μg/mL gelatin), four dNTPs (200 μM each; Invitrogen), primers (20 nM each) for all SNPs in the multiplex group, 6 units of HotStar Taq DNA polymerase (QIAGEN), and 5 ng of DNA (Coriell Institute for Medical Research). The samples were first heated to 94°C for 15 min to activate the Taq DNA polymerase followed by 40 PCR cycles. Each PCR cycle consisted of 40 sec at 94°C for denaturation and 2 min at 55°C followed by 5 min of ramping from 55°C to 70°C for annealing and extension. A final extension step was carried out at 72°C for 3 min at the end of the 40-th cycle. PCR amplifications were performed with thermal cyclers capable of ramping as slow as 0.01°C/sec, including the PTC100 Programmable Thermal Controller (MJ Research), T3 Thermocycler (Biometra), and PxE Thermal Cycler (Thermo Electron). ssDNA was generated (Fig. 1B) in both directions by using the same conditions for multiplex PCR except: (1) 1-2 μL of product from the multiplex PCR was used as templates, (2) only one primer (one of the primer-probes) for each SNP; and (3) 45 PCR cycles. The correlation between primers and probes is illustrated in Figure 1 and explained in more detail in the Results section.
Genotype determination by microarray
Preparation of microarray slides
Gold Seal Micro slides (Becton Dickson) were soaked in 30% bleach with shaking for 1-2 h followed by rinsing five times with deionized H2O and three times with MilliQ H2O. The slides were then sonicated in 15% Fisher brand Versa-Clean Liquid Concentrate with heat on for 1-2 h, and then rinsed with shaking in deionized H2O 10 times and five times with MilliQ H2O. Slides were dried by centrifugation at 1000 rpm for 5 min in a GS-6 Beckman Centrifuge. The slides were then baked at 140°C in a vacuum oven (Fisher Scientific Model 280A) for 4-6 h.
Microarray preparation
One volume of probe was mixed with 4 vol of microarray printing solution, EZ'nBrite (distributed by GenBase Biosciences Corp.), for a final concentration of 40 μM for each probe in the wells of the 384-well plates. Probes were then spotted onto the washed glass slides by using the microarray spotter, OminGrid Accent (GeneMachines), under a humidity of 50% to 55% and temperature of 22°C to 25°C.
Hybridization
Hybridization was done with 1× hybridization solution (5× Denhart's solution, 0.5% SDS, 5× SSC, 20 μL of ssDNA/1000 microarray spots) in a Hybridization Chamber (Corning) at 56°C for 2.5-4 h. Chambers were emerged in iced water for ∼30 sec before opening. The slide was washed at 56°C with 1× SSC and 0.1% SDS for 10 min, twice with 0.5× SSC for 30 sec, and twice with 0.2× SSC for 30 sec.
Labeling probes by single-base extension
Probes were labeled in 25 μL of labeling solution containing  volume of Sequenase buffer (supplied by the vendor), 0.5 units/μL Sequenase (Amersham Pharmacia Biosciences), Cy3-ddATP and Cy5-ddGTP (PE Biosystems) for AG probes, and Cy3-ddUTP and Cy5-ddCTP for CT probes (750 nM each). The reaction was incubated at 70°C for 10 min. The slide was washed under conditions specified for the washing after hybridization as described above.
volume of Sequenase buffer (supplied by the vendor), 0.5 units/μL Sequenase (Amersham Pharmacia Biosciences), Cy3-ddATP and Cy5-ddGTP (PE Biosystems) for AG probes, and Cy3-ddUTP and Cy5-ddCTP for CT probes (750 nM each). The reaction was incubated at 70°C for 10 min. The slide was washed under conditions specified for the washing after hybridization as described above.
Data analysis
Microarrays were scanned with GenePix 4000B (Axon Instruments). The resulting images were analyzed with either the GenePix Pro (Axon Instruments) or ImaGene (BioDiscovery) software. Genotypes were determined by using the computer program, AccuTyping, developed in our laboratory as described in the text.
Acknowledgments
We thank Michael Reiss for his comments on the manuscript. This work was supported by grants from the National Human Genome Research Institute (R01 HG02094), and the National Cancer Institute (R01 CA77363 and R33 CA 96309) to H.L. The core technology described in the present paper is covered by a pending PCT patent application (PCT/US03/31874).
Footnotes
[Supplemental material is available online at www.genome.org and http://www2.umdnj.edu/lilabweb/Publications/Multiplex3G.]
Article and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.2885205.
References
- Brennan, M.D. 2001. High throughput genotyping technologies for pharmacogenomics. Am. J. Pharmacogenomics 1: 295-302. [DOI] [PubMed] [Google Scholar]
- Brentani, H., Caballero, O.L., Camargo, A.A., Da Silva, A.M., Da Silva Jr., W.A., Neto, E.D., Grivet, M., Gruber, A., Guimaraes, P.E., Hide, W., et al. 2003. The generation and utilization of a cancer-oriented representation of the human transcriptome by using expressed sequence tags. Proc. Natl. Acad. Sci. 100: 13418-13423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butcher, L.M., Meaburn, E., Liu, L., Fernandes, C., Hill, L., Al-Chalabi, A., Plomin, R., Schalkwyk, L., and Craig, I.W. 2004. Genotyping pooled DNA on microarrays: A systematic genome screen of thousands of SNPs in Large samples to detect QTLs for complex traits. Behav. Genet. 34: 549-555. [DOI] [PubMed] [Google Scholar]
- Cheung, J., Estivill, X., Khaja, R., MacDonald, J.R., Lau, K., Tsui, L.C., and Scherer, S.W. 2003. Genome-wide detection of segmental duplications and potential assembly errors in the human genome sequence. Genome Biol. 4: R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui, X.F., Li, H.H., Goradia, T.M., Lange, K., Kazazian Jr., H.H., Galas, D., and Arnheim, N. 1989. Single-sperm typing: Determination of genetic distance between the G γ-globin and parathyroid hormone loci by using the polymerase chain reaction and allele-specific oligomers. Proc. Natl. Acad. Sci. 86: 9389-9393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards, M.C. and Gibbs, R.A. 1994. Multiplex PCR: Advantages, development, and applications. PCR Methods Appl. 3: S65-S75. [DOI] [PubMed] [Google Scholar]
- Ewing, B. and Green, P. 2000. Analysis of expressed sequence tags indicates 35,000 human genes. Nat. Genet. 25: 232-234. [DOI] [PubMed] [Google Scholar]
- Fan, J.-B., Oliphant, A., Shen, R., Kermani, B.G., Garcia, F., Gunderson, K.L., Hansen, M., Steemers, F., Butler, S.L., Deloukas, P., et al. 2004. Highly parallel SNP genotyping. In Cold Spring Harbor symposia on quantitative biology, pp. 69-78. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. [DOI] [PubMed]
- Fredman, D., White, S.J., Potter, S., Eichler, E.E., Den Dunnen, J.T., and Brookes, A.J. 2004. Complex SNP-related sequence variation in segmental genome duplications. Nat. Genet. 36: 861-866. [DOI] [PubMed] [Google Scholar]
- Goradia, T.M., Stanton Jr., V.P., Cui, X.F., Aburatani, H., Li, H.H., Lange, K., Housman, D.E., and Arnheim, N. 1991. Ordering three DNA polymorphisms on human chromosome 3 by sperm typing. Genomics 10: 748-755. [DOI] [PubMed] [Google Scholar]
- Hardenbol, P., Baner, J., Jain, M., Nilsson, M., Namsaraev, E.A., Karlin-Neumann, G.A., Fakhrai-Rad, H., Ronaghi, M., Willis, T.D., Landegren, U., et al. 2003. Multiplexed genotyping with sequence-tagged molecular inversion probes. Nat. Biotechnol. 21: 673-678. [DOI] [PubMed] [Google Scholar]
- Kennedy, G.C., Matsuzaki, H., Dong, S., Liu, W.M., Huang, J., Liu, G., Su, X., Cao, M., Chen, W., Zhang, J., et al. 2003. Large-scale genotyping of complex DNA. Nat. Biotechnol. 21: 1233-1237. [DOI] [PubMed] [Google Scholar]
- Kruglyak, L. and Nickerson, D.A. 2001. Variation is the spice of life. Nat. Genet. 27: 234-236. [DOI] [PubMed] [Google Scholar]
- Kwok, P.Y. and Chen, X. 2003. Detection of single nucleotide polymorphisms. Curr. Issues Mol. Biol. 5: 43-60. [PubMed] [Google Scholar]
- Lander, E.S., Linton, L.M., Birren, B., Nusbaum, C., Zody, M.C., Baldwin, J., Devon, K., Dewar, K., Doyle, M., FitzHugh, W., et al. 2001. Initial sequencing and analysis of the human genome. Nature 409: 860-921. [DOI] [PubMed] [Google Scholar]
- Li, H.H., Gyllensten, U.B., Cui, X.F., Saiki, R.K., Erlich, H.A., and Arnheim, N. 1988. Amplification and analysis of DNA sequences in single human sperm and diploid cells. Nature 335: 414-417. [DOI] [PubMed] [Google Scholar]
- Lin, Z., Cui, X., and Li, H. 1996. Multiplex genotype determination at a large number of gene loci. Proc. Natl. Acad. Sci. 93: 2582-2587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindblad-Toh, K., Tanenbaum, D.M., Daly, M.J., Winchester, E., Lui, W.O., Villapakkam, A., Stanton, S.E., Larsson, C., Hudson, T.J., Johnson, B.E., et al. 2000. Loss-of-heterozygosity analysis of small-cell lung carcinomas using single-nucleotide polymorphism arrays. Nat. Biotechnol. 18: 1001-1005. [DOI] [PubMed] [Google Scholar]
- Markoulatos, P., Siafakas, N., and Moncany, M. 2002. Multiplex polymerase chain reaction: A practical approach. J. Clin. Lab. Anal. 16: 47-51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuzaki, H., Loi, H., Dong, S., Tsai, Y.Y., Fang, J., Law, J., Di, X., Liu, W.M., Yang, G., Liu, G., et al. 2004. Parallel genotyping of over 10,000 SNPs using a one-primer assay on a high-density oligonucleotide array. Genome Res. 14: 414-425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mullis, K.B. and Faloona, F.A. 1987. Specific synthesis of DNA in vitro via a polymerase-catalyzed chain reaction. Methods Enzymol. 155: 335-350. [DOI] [PubMed] [Google Scholar]
- Pastinen, T., Kurg, A., Metspalu, A., Peltonen, L., and Syvanen, A.C. 1997. Minisequencing: A specific tool for DNA analysis and diagnostics on oligonucleotide arrays. Genome Res. 7: 606-614. [DOI] [PubMed] [Google Scholar]
- Pastinen, T., Raitio, M., Lindroos, K., Tainola, P., Peltonen, L., and Syvanen, A.C. 2000. A system for specific, high-throughput genotyping by allele-specific primer extension on microarrays. Genome Res. 10: 1031-1042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pennisi, E. 2003. Human genome. A low number wins the GeneSweep Pool. Science 300: 1484. [DOI] [PubMed] [Google Scholar]
- Pramanik, S. and Li, H. 2002. Direct detection of insertion/deletion polymorphisms in an autosomal region by analyzing high-density markers in individual spermatozoa. Am. J. Hum. Genet. 71: 1342-1352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roest Crollius, H., Jaillon, O., Bernot, A., Dasilva, C., Bouneau, L., Fischer, C., Fizames, C., Wincker, P., Brottier, P., Quetier, F., et al. 2000. Estimate of human gene number provided by genome-wide analysis using Tetraodon nigroviridis DNA sequence. Nat. Genet. 25: 235-238. [DOI] [PubMed] [Google Scholar]
- Sachidanandam, R., Weissman, D., Schmidt, S.C., Kakol, J.M., Stein, L.D., Marth, G., Sherry, S., Mullikin, J.C., Mortimore, B.J., Willey, D.L., et al. 2001. A map of human genome sequence variation containing 1.42 million single nucleotide polymorphisms. Nature 409: 928-933. [DOI] [PubMed] [Google Scholar]
- Saiki, R.K., Scharf, S., Faloona, F., Mullis, K.B., Horn, G.T., Erlich, H.A., and Arnheim, N. 1985. Enzymatic amplification of β-globin genomic sequences and restriction site analysis for diagnosis of sickle cell anemia. Science 230: 1350-1354. [DOI] [PubMed] [Google Scholar]
- Shumaker, J.M., Metspalu, A., and Caskey, C.T. 1996. Mutation detection by solid phase primer extension. Hum. Mutat. 7: 346-354. [DOI] [PubMed] [Google Scholar]
- Syvanen, A.C. 1999. From gels to chips: “Minisequencing” primer extension for analysis of point mutations and single nucleotide polymorphisms. Hum. Mutat. 13: 1-10. [DOI] [PubMed] [Google Scholar]
- Venter, J.C., Adams, M.D., Myers, E.W., Li, P.W., Mural, R.J., Sutton, G.G., Smith, H.O., Yandell, M., Evans, C.A., Holt, R.A., et al. 2001. The sequence of the human genome. Science 291: 1304-1351. [DOI] [PubMed] [Google Scholar]
- Yeakley, J.M., Fan, J.B., Doucet, D., Luo, L., Wickham, E., Ye, Z., Chee, M.S., and Fu, X.D. 2002. Profiling alternative splicing on fiber-optic arrays. Nat. Biotechnol. 20: 353-358. [DOI] [PubMed] [Google Scholar]
- Zhou, X., Mok, S.C., Chen, Z., Li, Y., and Wong, D.T. 2004. Concurrent analysis of loss of heterozygosity (LOH) and copy number abnormality (CNA) for oral premalignancy progression using the Affymetrix 10K SNP mapping array. Hum. Genet. 115: 327-330. [DOI] [PubMed] [Google Scholar]
WEB SITE REFERENCES
- ftp://ftp.ncbi.nih.gov/snp/human/chr_rpts/; National Center for Biotechnology Information Human Chromosome Reports.
- http://www.genome.ucsc.edu/cgi-bin/hgBlat?db=hg8; University of California Santa Cruz Human BLAT Search.
- http://www.hapmap.org; HapMap project.
- http://www.ncbi.nlm.nih.gov/BLAST; National Center for Biotechnology Information BLAST Search.
- http://www2.umdnj.edu/lilabweb/Publications/Multiplex3G; sequences for the SNPs used in the present study.