

Abstract
NMR chemical shifts are exquisitely sensitive probes for conformation and dynamics in molecules and supramolecular assemblies. While isotropic chemical shifts are easily measured with high accuracy and precision in conventional NMR experiments, they remain challenging to calculate quantum mechanically, particularly in inherently dynamic biological systems. Using a model benchmark protein, the 133-residue agglutinin from Oscillatoria agardhii (OAA), which has been extensively characterized by us previously, we have explored the integration of X-ray crystallography, solution NMR, MAS NMR, and quantum mechanics/molecular mechanics (QM/MM) calculations for analysis of 13Cα and 15N isotropic chemical shifts. The influence of local interactions, quaternary contacts, and dynamics on the accuracy of calculated chemical shifts is analyzed. Our approach is broadly applicable and expected to be beneficial in chemical shift analysis and chemical-shift-based structure refinement for proteins and protein assemblies.
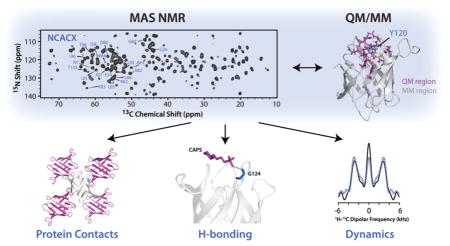
Graphical abstract
INTRODUCTION
Spectroscopic methods are the premier techniques employed in the analysis of molecular structure. Amongst these, NMR can provide atomic-level detail on conformation and dynamics for a wide range of molecular systems, either in solution, or in the solid state, even in whole cells.1–4 NMR is uniquely positioned to yield atom-specific dynamics data on large systems, information which is inaccessible by other structural-biological techniques. Although solution NMR has a long history in biomolecular structure elucidation, over the last decade, magic angle spinning (MAS) solid state NMR spectroscopy has come to the forth as the method of choice for the analysis of crystalline or amorphous biological assemblies.2, 5–8
Despite the fact that chemical shifts can be measured in NMR experiments with great precision and accuracy, their exquisite dependency on geometry, electronic and dynamic properties have made chemical shift calculations a considerable challenge9–11. For biological molecules, such as proteins, most of the chemical shift based structural analyses have focused on isotropic shifts and rely on semi-empirical database approaches, such as CSI, SHIFTX, and others,12–17 aimed at incorporating chemical shift data into the structure determination process. Recently, CS-ROSETTA18 and CAMSHIFT19 approaches have been proposed for deriving 3D protein model structures, based on isotropic chemical shifts alone, without any need for other distance and angle constraints. While these approaches are promising, their general applicability, particularly with respect to local conformational details remains to be established.
The prediction of NMR chemical shifts from quantum mechanical (QM) methods is an alternative approach20–22 and has undergone major advancements within the last decade, including improvements in the basis sets and the extension of higher levels of theory, as well as Density Functional Theory (DFT) techniques.9, 23–26 DFT calculations are accurate enough to yield good agreements between chemical shift and molecular structure for medium-sized molecules,27–28 and have also been explored for protein structure validation and refinement.23, 29 While this approach has been rarely used for proteins in the past, given the associated computational demands, it currently is starting to gain momentum due to the rapidly increasing availability of sufficiently powerful computational resources and robust software. The most ubiquitous protocol employs DFT in the cluster mode or a mixed quantum mechanics/molecular mechanics (QM/MM) mode. An automated fragmentation quantum mechanics/molecular mechanics (AF-QM/MM) approach was introduced by Merz and colleagues, and permits QM/MM calculations on large proteins.30–31 Other groups successfully employed this strategy for NMR chemical shift calculations, to fragment a protein sequence and define the appropriate regions to be treated at QM vs. MM level; they benchmarked the methods and the basis sets, which are both accurate and relatively inexpensive in terms of computational resources.24, 32 Case and coworkers demonstrated, using 20 proteins, that accurate isotropic shifts for non-labile 1H and 13C nuclei can be calculated following the prescribed protocol, while for amide 15N, and even more pronounced, for amide 1H chemical shifts, larger discrepancies between experimental and calculated shifts remain.32
In contrast to small organic molecules, proteins are inherently dynamic polymers, and the presence of conformational dynamics will grossly compromise results of QM/MM chemical shift calculations based on a single static geometry. For such systems, a combination of molecular dynamics (MD) simulations with QM/MM is a superior choice33–37, as we recently demonstrated for HIV-1 capsid assemblies38. In that study, we achieved excellent agreement between the chemical shift tensors, extracted from MAS solid state spectra and their predicted values, using a combined MD/QM/MM approach, even for highly dynamic residues.
Being able to accurately and reliably predict chemical shifts for proteins in the future would be of considerable value, since these shifts could potentially be used for iterative structure refinement. The present work was motivated by the aspiration to derive a robust protocol to reliably calculate protein 13Cα and 15N isotropic chemical shifts by QM/MM, using a model system for which experimental solution isotropic chemical shifts are known, for which a high-resolution X-ray structure is available, and which can be prepared in the same state for MAS NMR studies.
We selected as our model system the 133 residue protein agglutinin from Oscillatoria agardhii (OAA). This protein exhibits an unusual beta-barrel fold (Figure 1A).39, 40 The amino acid sequence of OAA contains two repeats (Figure 1B), which, in the structure, are related by a pseudo 2-fold symmetry. The equivalent symmetry-related residues exhibit different chemical shifts in solution. OAA is comprised predominantly of β-strands, linked by loops. These loops create the two ligand binding sites for mannose sugars: the lower-affinity site 1 comprises the loops connecting strands β1-β2 and β9-β10, and the higher-affinity site 2 is formed by the loop connecting strands β4-β5 and β6-β7. Our laboratory has solved the X-ray structure of OAA at 1.4 Å resolution (PDB: 3OBL)39, and we have an extensive body of solution NMR information, including complete chemical shift assignments (BMRB: 25306)39–42. Furthermore, as demonstrated here, microcrystalline OAA yields excellent-quality MAS NMR spectra. These permitted resonance assignments to be obtained for a large portion of the sequence. The extracted 1H-13C/15N dipolar order parameters indicate the presence of dynamics on the nano-to microsecond timescales for residues in two ligand binding sites. By integrating X-ray crystallography, solution NMR, MAS NMR, and QM/MM calculations, we have addressed the influence of the following factors on the accuracy of QM/MM chemical shift calculations: i) crystal contacts; ii) dynamics; iii) hydrogen bonding. The results indicate that calculated 13Cα chemical shifts give excellent agreement with experimental shifts, while calculated 15NH chemical shifts display bigger deviations with experiment due to their sensitivity to factors including hydrogen bonding, local electrostatics, and dynamics. Our work also reveals that crystal contacts have a major influence on local conformation, and hence chemical shifts. The effects of dynamics are smaller, but not negligible. We propose that accounting for these effects can improve DFT-based chemical shift calculations. The approach presented here is broadly applicable to analysis of chemical shifts in various proteins and protein assemblies beyond OAA.
Figure 1.

A: Ribbon representation of the X-ray structure (left) and amino acid sequence (right) of O. agardhii agglutinin. Residues whose backbone resonances were assigned are shown in bold. Identical residues in the sequence repeats are shown in pink, linker residues between the repeats are shown in green. β-strands are indicated by arrows above the sequence, and colored blue and gray for repeat 1 and 2, respectively, both in the structure and sequence. B: MAS NMR spectra of OAA microcrystals: NCOCX (top right), CORD (middle right), NCACX (bottom right). Assignments for selected resonances are shown and labeled with residue name and number. Expansions of selected areas in the CORD and NCACX spectra for the Thr, Ser, and Gly regions (magenta) are provided at the left side with black and magenta contours representing the spectra acquired at 14.1 and 19.96 T, respectively. Note that equivalent residues belonging to the two sequence repeats cannot be fully resolved at 14.1 T, but are distinct in the 19.96 T spectra A sample backbone walk is provided in the supporting information.
MATERIALS AND METHODS
Protein expression, purification, and crystallization of OAA were performed as described previously39. For all MAS NMR experiments, 30 mg of crystals were packed into 3.2 mm thin-wall Bruker rotors.
MAS NMR experiments were carried out on a 14.1 T narrow bore Bruker AVIII spectrometer outfitted with a 3.2 mm HCN EFree MAS probe. Larmor frequencies were 599.8 MHz (1H), 150.8 MHz (13C), and 60.8 MHz (15N). Several 2D data sets were collected on a 19.96 T narrow bore Bruker AVIII spectrometer using a 3.2 mm HCN EFree MAS probe; Larmor frequencies were 850.4 MHz (1H), 213.9 MHz (13C), and 86.2 MHz (15N). The MAS frequency was set at 14 kHz for all experiments, and was controlled to within ± 5 Hz by a Bruker MAS III controller. KBr was used as an external temperature sensor43, and the actual temperature of the sample (corrected for rotational heating) was maintained at 4°C using the Bruker BCU temperature controller, which was stable to ± 0.1°. 13C and 15N chemical shifts were referenced with respect to the external standards adamantane (CH2 group referenced to DSS at 40.76 ppm44) and NH4Cl (39.27 ppm45), respectively. Typical 90° pulse lengths were 2.8 μs (1H), 4.0 μs (13C), and 4.8 μs (15N), and the contact time of 1H-15N/13C cross polarization (CP) was 2.2/1.8 ms. 1H-15N/13C CP employed a 95–105% linear amplitude ramp on the 1H channel with the center Hartmann-Hahn matched to the first spinning side band. The band-selective magnetization transfer from 15N to 13Cα was performed through a 4.5 ms SPECIFIC-CP46 with a tangent amplitude ramp on the 15N channel (49 kHz rf field center) and a constant rf field on the 13C channel (35 kHz). High-power 1H continuous wave (CW) decoupling (89 kHz) was applied during the SPECIFIC-CP period, and SPINAL-64 decoupling47 (89 kHz) was applied during the direct (t2) and indirect (t1) acquisition periods. In 1H-13C RN-DIPSHIFT and 1H-15N RN-PARS 3D experiments,48–49 R1214-based symmetry sequence was used to reintroduce the 1H-15N/13C dipolar interaction during the t1 evolution period, and the phase-alternated rf field irradiation (84 kHz) was applied on the 15N/13C channel. Simultaneous π pulses were applied on the 13C channel at the center of every two rotor periods to decouple 15N-13C dipolar interactions.
Processing of NMR data was carried out in NMRpipe50; the spectra were analyzed with both Sparky and CCPN.51–52 In all 2D and 3D datasets, 30° or 60° shifted sine bell apodization was followed by Lorentz-to-Gaussian transformation. The R-symmetry 1H-X dipolar correlation data sets were evaluated as the real-FT of the corresponding indirect dimension zero-filled to 256 points prior to FT.
Numerical simulations of 1H-15N/13C dipolar lineshapes were performed using the Minuit package in SIMPSON53 versions 1.1.2. To produce a powder average, 320 pairs of α, β angles were generated according to the REPULSION54 algorithm, and 16 γ angles (resulting in a total of 5, 120 angle triplets) were used for all simulations. The rigid limit was taken to be 11.3455 and 22.756 kHz for 1H-15N and 1H-13C respectively. NMR parameters in the experiment matched those used during the fitting routine.
QM/MM calculations of 13C and 15N isotropic chemical shifts were carried out in Gaussian09,57 at the OLYP58/tzvp59 level for the quantum mechanical region, using the scripts generated in AFNMR,60 and using chain A of PDB ID: 3OBL as initial input. The structure was minimized using the Amber FF99SB molecular mechanics force field and referenced to ubiquitin (PDB ID: 1D3Z) calculated at the same level of theory (1H =32.0 ppm, 13C=182.5, and 15N= 237.8 ppm.)32 Intermolecular interfaces were identified by PISA.61
RESULTS
MAS NMR spectroscopy of OAA crystals
2D MAS NMR spectra of microcrystalline OAA exhibit excellent resolution and are of high sensitivity (Figure 1B). Several data sets, including 13C-13C 2D CORD62, 2D and 3D NCACX and NCOCX, and 3D CONCA spectra were acquired at 14.1 T, and assignments for 107 residues were obtained. While for proteins of similar size the magnetic field strength of 14.1 T and conventional heteronuclear dipolar-based 2D and 3D experiments would be sufficient for complete resonance assignments, given the sequence repeats in OAA, significant resonance overlap is present. Therefore, we could not distinguish between stretches of residues from the two sequence repeats. In solution, resonances from equivalent residues in the two sequence repeats could be unequivocally identified on the basis of their distinct 1H chemical shifts. To obtain proton chemical shifts in the MAS NMR experiments would require either deuteration of non-exchangeable protons in conjunction with full or partial amide back exchange, fast MAS (60–110 kHz) conditions, or both.
Gratifyingly, 2D MAS spectra recorded at 19.96 T resolved most of the overlapping resonances and individual peaks are observed in the 2D CORD and NCACX spectra of OAA for equivalent residues in the sequence repeats (Figure 1B). This demonstrates that by using high magnetic fields it is possible to carry out a complete structural analysis without the need for additional experiments. While further fast MAS experiments as well as 3D experiments at 19.96 T are currently ongoing in our laboratory, sufficient residue-specific resonance assignments for a large number of residues were available for an in-depth analysis of 13C and 15N chemical shifts and dipolar order parameters, as discussed below.
Analysis of 13C and 15N solution and MAS isotropic chemical shifts of OAA
The correlations between the experimental solution and two sets of calculated 13Cα and 15NH chemical shifts of OAA are provided in Figure 2. The shifts were calculated using SHIFTX214 as well as by DFT at the QM/MM level in Gaussian09 for the X-ray structure of OAA (PDB ID 3OBL). As can be appreciated, the correlations between the experimental 13Cα shifts and those computed either semi-empirically or quantum mechanically are good to excellent, with the SHIFTX2-based predictions exhibiting better agreement with the experimental values. This result is consistent with previous reports from several groups12, 32, 63 and is not surprising, as SHIFTX2-based calculations by nature of their data-base driven properties take into account dynamic averaging of isotropic shifts for mobile regions. In addition, the agreement between experiment and calculations for 15NH shifts is generally worse than that for the 13Cα shifts, exhibiting a considerable degree of scatter. The reasons why calculations of 15NH shifts are fraught with difficulties are the extreme sensitivity of nitrogen chemical shifts to hydrogen bonding and local electrostatic effects, as well as solvent and dynamics influences. Interestingly, for both 13Cα and 15NH, better agreement is obtained between experimental solution NMR shifts and calculated shifts than between SHIFTX2 and QM/MM predicted shifts. Results based on SPARTA+12, another chemical shift prediction algorithm, were also evaluated. No significant differences in correlations based on the use of SPARTA+ or SHIFTX2 were observed. Fit parameters for linear correlations based upon SPARTA+ chemical shift prediction are included in Table 1. Linear correlation plots based on SPARTA+ chemical shift prediction are provided in the Supporting Information (Figure S2). 13Cβ chemical shifts had similar high quality linear correlations as seen for 13Cβ chemical shifts between MAS and solution NMR, SHIFTX2, and DFT. 13Cβ linear correlations are shown in the Supporting Information (Figure S3).
Figure 2.

Summary of experimental solution NMR and calculated isotropic chemical shifts for 13Cα and 15NH in O agardhii agglution. A–F: Correlations between experimental solution 13 Cα and 15NH chemical shifts and QM/MM (A and D) or SHIFTX2 calculated shifts (B and E) as well as the correlation between SHIFTX2 and QM/MM calculated chemical shifts (C and F), using the X-ray structure of OAA (PDB ID 3OBL) as the model. G: Differences between calculated (QM/MM) and experimental (solution NMR) isotropic chemical shifts along the linear amino acid sequence. No trends with respect to secondary structure, ligand-binding sites or residue type are noted.
Table 1.
Slope (m) and variance (R2) for linear correlations of 13Cα and 15NH MAS NMR chemical shifts with solution NMR, SHIFTX2, SPARTA+, and QM/MM-derived chemical shifts for subsets of residues.
| Leu-47 and Leu-114 are treated as crystal contacts
| ||||
|---|---|---|---|---|
| All residues | Static Residues | Non-Interface Residues | Static and Non-Interface Residues | |
| 13Cα vs solution NMR | m = 0.99 | m = 0.98 | m = 1 | m = 1 |
| R2 = 0.97 | R2=0.97 | R2 = 0.99 | R2 = 0.99 | |
| 13Cα vs QM/MM | m = 0.86 | m = 0.83 | m = 0.83 | m = 0.83 |
| R2 = 0.92 | R2 = 0.92 | R2 = 0.94 | R2 = 0.95 | |
| 13Cα vs SHIFTX2 | m = 0.97 | m = 0.97 | m = 0.96 | m = 0.97 |
| R2 = 0.95 | R2 = 0.95 | R2 = 0.97 | R2 = 0.98 | |
| 13Cα vs SPARTA | m = 0.98 | m = 0.97 | m = 0.97 | m = 0.98 |
| R2 = 0.95 | R2 = 0.96 | R2 = 0.97 | R2 = 0.97 | |
| 15NH vs solution NMR | m = 0.93 | m = 0.92 | m = 1 | m = 1 |
| R2 = 0.88 | R2 = 0.86 | R2 = 0.97 | R2 = 0.97 | |
| 15NH vs QM/MM | m = 1.07 | m = 1.05 | m = 1.06 | m = 1.04 |
| R2 = 0.78 | R2 = 0.74 | R2 = 0.74 | R2 = 0.74 | |
| 15NH vs SHIFTX2 | m = 0.76 | m = 0.82 | m = 0.81 | m = 0.84 |
| R2 = 0.70 | R2 = 0.76 | R2 = 0.80 | R2 = 0.82 | |
| 15NH vs SPARTA | m = 0.76 | m = 0.80 | m = 0.80 | m = 0.83 |
| R2 = 0.70 | R2 = 0.74 | R2 = 0.75 | R2 = 0.82 | |
|
| ||||
| Leu-47 and Leu-114 are not treated as crystal contacts | ||||
|
| ||||
| All residues | Static Residues | Non-Interface Residues | Static and non-interface | |
| 13 Cα vs solution NMR | m = 0.99 | m = 0.98 | m = 0.99 | m = 0.99 |
| R2 = 0.97 | R2=0.97 | R2 = 0.98 | R2 = 0.98 | |
| 13Cα vs QM/MM | m = 0.86 | m = 0.83 | m = 0.82 | m = 0.83 |
| R2 = 0.92 | R2 = 0.92 | R2 = 0.92 | R2 = 0.93 | |
| 13Cα vs SHIFTX2 | m = 0.97 | m = 0.97 | m = 0.95 | m = 0.96 |
| R2 = 0.95 | R2 = 0.95 | R2 = 0.96 | R2 = 0.97 | |
| 13Cα vs SPARTA | m = 0.98 | m = 0.97 | m = 0.96 | m = 0.97 |
| R2 = 0.95 | R2 = 0.96 | R2 = 0.96 | R2 = 0.96 | |
| 15NH vs solution NMR | m = 0.93 | m = 0.92 | m = 0.89 | m = 0.89 |
| R2 = 0.88 | R2 = 0.86 | R2 = 0.87 | R2 = 0.86 | |
| 15NH vs QM/MM | m = 1.07 | m = 1.05 | m = 0.94 | m = 0.92 |
| R2 = 0.78 | R2 = 0.74 | R2 = 0.65 | R2 = 0.65 | |
| 15NH vs SHIFTX2 | m = 0.76 | m = 0.82 | m = 0.78 | m = 0.81 |
| R2 = 0.70 | R2 = 0.76 | R2 = 0.79 | R2 = 0.80 | |
| 15NH vs SPARTA | m = 0.76 | m = 0.80 | m = 0.75 | m = 0.77 |
| R2 = 0.70 | R2 = 0.74 | R2 = 0.73 | R2 = 0.79 | |
|
| ||||
| Leu-47 and Leu-114 are excluded from analysis altogether | ||||
|
| ||||
| All residues, 47/114 removed | Static Residues 47/114 removed | Non-Interface Residues, 47/114 removed | Static and Non-Interface, 47/114 removed | |
| 13Cα vs solution NMR | m = 0.99 | m = 0.99 | m = 1 | m = 1 |
| R2 = 0.98 | R2 = 0.98 | R2 = 0.99 | R2 = 0.99 | |
| 13Cα vs QM/MM | m = 0.86 | m = 0.83 | m = 0.83 | m = 0.83 |
| R2 = 0.93 | R2 = 0.93 | R2 = 0.94 | R2 = 0.95 | |
| 13Cα vs SHIFTX2 | m = 0.98 | m = 0.97 | m = 0.96 | m = 0.97 |
| R2 = 0.96 | R2 = 0.96 | R2 = 0.97 | R2 = 0.98 | |
| 13Cα vs SPARTA | m = 0.98 | m = 0.97 | m = 0.97 | m = 0.98 |
| R2 = 0.96 | R2 = 0.96 | R2=0.97 | R2 = 0.97 | |
| 15NH vs solution NMR | m = 1 | m = 0.99 | m = 1 | m = 1 |
| R2 = 0.93 | R2 = 0.92 | R2 = 0.97 | R2 = 0.97 | |
| 15NH vs QM/MM | m = 1.13 | m = 1.15 | m = 1.06 | m = 1.04 |
| R2 = 0.82 | R2 = 0.80 | R2 = 0.74 | R2 = 0.74 | |
| 15NH vs SHIFTX2 | m = 0.78 | m = 0.85 | m = 0.81 | m = 0.84 |
| R2 = 0.69 | R2 = 0.76 | R2 = 0.80 | R2 = 0.82 | |
| 15NH vs SPARTA | m = 0.79 | m = 0.83 | m = 0.80 | m = 0.83 |
| R2 = 0.70 | R2 = 0.75 | R2 = 0.75 | R2 = 0.82 | |
In order to evaluate whether the differences between experimental and computed shifts pertain to any specific subsets of residues (e.g., loops, binding sites or specific residue types), we have plotted the differences between the experimental and QM/MM computed shift vs. residue number. No obvious trends emerged, however (Figure 2G).
The comparison between MAS, solution, and computed 13Cβ and 15NH shifts is presented in Figure 3. The experimental shifts recorded on microcrystalline OAA and OAA in solution are in excellent agreement, with only a few outliers (Figure 3a). The residues for which solution and solid-state NMR shifts exhibit large differences are those that are involved in crystal contacts (Figure 4A) or interactions with CAPS molecules (3-cyclohexyl-1-propylsulfonic acid, Figure 4B) in the crystallization buffer. Naturally, differences would be expected for these residues. We examined the interaction surfaces in the crystals and classified them into several subsets: 1) residues that participate in intermolecular protein-protein hydrogen bonds; 2) other buried residues at the protein-protein interfaces that do not explicitly participate in hydrogen bonds; 3) residues that form direct hydrogen bonds with the CAPS molecule; and 4) other buried residues at the protein-ligand interfaces. The complete list of residues involved in intermolecular contacts is provided in the Supporting Information (Table S1). No trends emerged as to which contacts resulted in larger chemical shift differences, and all four were treated as a single group in the subsequent analysis. Large differences are observed for both, 15NH and 13Cα chemical shifts of residue G20 located in β2; for residue T46, on the other hand, large differences are only present for the 15NH amide shift. We attribute these differences to intermolecular contacts formed in microcrystalline OAA that are absent in solution. Overall, comparing the solution NMR structure of OAA64 (2MWH) with the crystal structure (3OBL) reveals that many residues that exhibit chemical shift differences also show conformational differences between the two structures (Figure 4C,D).
Figure 3.

Summary of experimental solution and MAS shifts and calculated isotropic chemical shifts for 13Cα and 15NH in O agardhii agglttinin A–F: Correlations between experimental between experimental solution 13Cα and 15NH chemical shifts, calculated QM/MM shifts, calculated SHIFTX2 shifts and experimental MAS 13Cα and 15NH chemical shifts using the X-structure of OAA (PDB ID 3OBL) as the model. In panels A and D, outliers in the correlation are labled with residue name and number. G–I: Backbone RMS chemical shift deviations Δδiso = ((δCαSS−δCαSoln,QM/MM,SHIFTX2)2 + 1/2(δNHSS − δNHSoln,QM/MM/SHIFTX2)2)1/2 between MAS NMR chemical shifts and experimental solution NMR isortopic chemical shifts (G), DFT-calcuted chemical shifts (H), and SHIFTX2 predicted chemical shifts (I) along the liner amino acid sequence. 15N chemical shifts were given a weighting of 0.5 relative to 13C chemical shifts to reflect the relative sensitivities of the two nuclei to changes in the electronic environment79–80. Residues involved in protein-protein or protein-ligand intermolecular contacts in the crystal structure of OAA are marked by grey shading. Magenta coloring indicates repeat regions of the sequence for which MAS NMR chemical shifts are identical for both repeats. In contrast to MAS NMR, in the solution NMR data sets, chemical shifts were not assumed to be identical for the 2 repeats. Rather the resolved solution NMR chemical shifts, which can be resolved in the proton dimension, were used.
Figure 4.

Intermolecular interactions in the OAA crystals. A: Protein-protein crystal contacts determined by PISA. B: A CAPS molecule forms a hydrogen bond with Gly-124. C,D: Superposition of the X-ray (gray) and solution NMR (blue) structures of OAA, illustrating structural differences introduced by crystal packing at Gly-20 (C) and Thr-46 (D) between two adjacent molecules (grey and magenta) in the crystal lattice.
The quality of the correlations as well as the numerical parameters of the linear fits for experimental vs. computed shifts for microcrystalline OAA are very similar to those that were obtained for OAA in solution, discussed above, and all are summarized in Table 1. For 13Cα, the agreement between experiment and theory is generally best, while, for 15NH, considerable scatter is observed. For all correlations but one, removing the data for residues that are involved in intermolecular contacts improves the agreement between MAS NMR chemical shifts and solution NMR, SHIFTX2, and QM/MM chemical shifts. The one exception in the correlation of 15NH shifts with QM/MM most likely is the result of increased scatter by the reduction in the number of data points. The same effect was noted when data for mobile residues were removed from the correlation. For correlations of MAS NMR chemical shifts with solution NMR or DFT-derived shifts, removing the data for mobile residues alone had no statistically significant effect on the quality of correlations. If the dynamics of OAA are similar in its crystalline and solution states, it is not surprising that the MAS vs solution NMR chemical shift correlation exhibits no dependence on dynamic residues. With respect to the correlation with DFT-derived chemical shifts, the absence of a dynamics-dependence may seem to be more surprising. We have demonstrated that dynamics can have dramatic effect on chemical shift tensors in HIV-1 protein capsid assemblies.38, 65 In CA capsid protein, the CypA binding loop has remarkably low order parameters, and the CSA tensors undergo dynamic averaging. In contrast, the dynamics of OAA are very modest, with the smallest order parameters on the order of 0.75 (1H-15N) to 0.8 (1H-13C). Therefore, it appears that in OAA there are no significant changes in the electronic environment as a result of these modest dynamics. Therefore, the isotropic chemical shifts are not influenced to a large extent by dynamics within the sensitivity of MAS NMR chemical shifts. Whether this observation holds for the chemical shift anisotropy and asymmetry parameters remains to be investigated.
In some correlations, namely 13Cα MAS NMR shifts vs QM/MM and SHIFTX2 predicted shifts, removing the data from both mobile and intermolecular contact residues resulted in modest improvement, compared to removal of data from contact residues alone, suggesting that dynamics compromises the agreements to a certain degree. Taken together, our results indicate that the most significant contribution to the differences between MAS NMR, solution NMR and calculated chemical shifts arises from intermolecular contacts in the crystalline state, which are not present in solution. This naturally will also affect the calculated chemical shifts, since these were based on a monomeric OAA molecule. Carrying QM/MM calculations out on larger crystalline assemblies may capture the electronic environment at the contact sides better, although the differences between MAS NMR and QM/MM shifts appear to be small, at least in comparison to the differences between MAS and solution NMR chemical shifts (Figure 3H).
Surprisingly, the largest chemical shift backbone RMSDs are observed for Leu-47 and Leu-114 (both, 13Cα and 15NH). While the sidechains of these two residues are buried in the interior of the OAA β-barrel, the backbone 13Cα and 15NH are surface-exposed and thus may exhibit chemical shift perturbations due to changes in the local electronic environment, associated with the formation of crystal contacts. Table 1 summarizes the correlation parameters when Leu-47 and Leu-114 are: i) treated as crystal contacts (i.e. removed from the statistical analysis along with all other residues identified by PISA as interface residues); ii) not treated as crystal contacts, iii) removed from the analysis altogether. The results indicate that the same trends hold when Leu-47/Leu-114 are either treated as crystal contacts or discarded. When these residues are not treated as crystal contacts, their substantial deviations skew the linear correlations, and in multiple cases “swamp out” statistical improvements due to the removal of other intermodular contacts or dynamic residues.
It may be of interest to note that, in contrast to the solution 15NH chemical shifts, which exhibited a better correlation with the shifts predicted by SHIFTX2 than those calculated by QM/MM, the experimental MAS NMR 15NH chemical shifts are in better agreement with QM/MM calculated chemical shifts than those predicted by SHIFTX2 (variances of 0.78 and 0.70, respectively). Further studies are required to determine if this difference generally holds for other systems as well.
Nano- to microsecond timescale dynamics in OAA
It is well known that chemical shifts are dynamically averaged when the atoms are undergoing nano- to microseconds timescale motions.66, 33 It, therefore, seemed prudent to evaluate whether the differences between experimental and computed chemical shifts could arise from dynamics in OAA. Consequently, we needed to identify which residues in OAA are mobile. This was achieved by recording 1H-15N and 1H-13Cα dipolar lineshapes by MAS NMR. Lineshapes for representative residues and dipolar order parameters, as well as crystallographic B factors, are provided in Figure 5.
Figure 5.

MAS NMR experimental 1H-15N and 1H-13Cα dipolar line shapes, order parameters and crystallographic B-factors of crystalline O. agardhii agglutinin. A and B: experimental (black) and best fit (blue) 1H-15N and 1H-13Cα dipolar lineshapes, respectively, recorded in RN-PARS and RN-DIPSHIFT MAS NMR experiments. C–E: 1H-15 dipolar order parameters, crystallographic B factors, and 1H-13Cα dipolar order parameters vs. residue number. Regions of reduced order parameters, which are indicative of motions occurring on ns-μs timescales, coincide with regions of increased B factors and are shaded in magenta. F, G: Correlation between B factors and 1H-15N and 1H-13Cα dipolar order parameters, respectively.
As can be noted, both 1H-15N and 1H-13C dipolar order parameters and B factors are correlated, with large B factors corresponding to low order parameters (Figure 5C,D,E). This is particularly interesting in light of the fact that, in addition to static disorder in the crystal, the B factors report on conformational disorder in the structures and, indirectly, on motions occurring over a much wider range of time scales than those captured by dipolar order parameters. This finding is in line with previous observations in other microcrystalline proteins, such as E. coli thioredoxin.66–70
Another intriguing observation is that OAA appears to exhibit more motion in the crystal (Figure 5) than one might anticipate, based on its seemingly rigid β-barrel type structure. The largest 1H-13C order parameters are 0.95, lower than the typical values recorded for rigid residues in other proteins by us and by others, including HIV-1 capsid protein assemblies.65, 70–72 While we cannot entirely rule out that these lower order parameters in OAA are due to systematic differences in experimental conditions, this is unlikely. Residues 10–16 and 78–83, connecting β1 to β2 and β6 to β7, respectively exhibit the lowest order parameters and highest B factors. These loops are involved in forming the sugar binding sites 1 (weak) and 2 (strong), respectively. Remarkably, for these residues the solution and MAS NMR chemical shifts are in excellent agreement, suggesting that the associated loop motions in OAA are similar in solution and in the solid state. In light of this observation it will be interesting in the future to examine the dynamics of OAA in complex with bound sugar.
DISCUSSION
As outlined earlier, OAA represents a potentially ideal benchmark system for comparing solution and solid-state NMR parameters with those predicted computationally, given that the protein is well folded with the majority of the residues residing in β-strands, is thermodynamically stable and crystallizes readily. Therefore, the same crystal form can be used for X-ray diffraction and MAS NMR experiments and influences from crystal contacts can be discerned in the MAS NMR shifts.
Our original expectation was that the best correlation would be observed between the experimental MAS NMR and QM/MM calculated 13Cα shifts. The better agreement was found between the experimental solution and MAS NMR 13Cα shifts, as well as between experimental solution and SHIFTX2-calculated 13Cα values. For 15NH shifts, the correlations are generally worse; however, the best agreement is again observed between the experimental solution and MAS NMR isotropic shift values. It is well known that hydrogen bonding and the associated electronic effects influence the quantum mechanical calculation of 15NH shifts significantly 9, 24. Since H atoms were added to the crystal coordinates by standard approaches to generate the input model for the QM/MM calculations and OAA is a β-barrel protein with the β-strands participating in an extensive network of hydrogen bonds, the accuracy of the calculated 15NH shifts may have been compromised to some degree, if the energy-minimized proton positions do not reflect the true proton positions in the crystal.
Not unexpectedly, dramatic differences between the solution and MAS NMR chemical shifts are detected for residues involved in crystal contacts (Figure 3G). On the other hand, dynamic residues, such as in the binding loops, appear to not introduce differences between the chemical shifts in solution vs. the solid state, suggesting that the dynamics are very similar in both states. Crystal contacts are a significant contributor to differences between MAS NMR chemical shifts and calculated shifts (DFT or SHIFTX2), but there appear to be other factors at play as well, indicated by overall higher RMSDs for calculated/predicted vs MAS NMR chemical shifts (Figure 3H,I).
CONCLUSIONS AND FUTURE OUTLOOK
Using the protein OAA as a model benchmark system, we have evaluated the accuracy of chemical shift calculations. Our study indicates that for 13Cα shifts, reasonably accurate predictions can be made, whereas much work remains in order to establish reliable protocols for 15NH isotropic chemical shift calculations. Crystal contacts, dynamics, hydrogen bonding, and other local interactions modulate the chemical shifts and need to be carefully taken into account. It is anticipated that future studies focused on the analysis of CSA tensors, rather than isotropic shifts only, are necessary, and that reliable incorporation of shift tensors into structure/dynamics characterization protocols will further improve structural characterization of proteins and large macromolecular assemblies.
Unlike isotropic chemical shifts, chemical shift anisotropy tensors can be accurately calculated at the DFT level. We recently reported this for 15NH chemical shift anisotropy tensors of HIV-1 CA assemblies.38 Since isotropic shifts are an average over three principal components of the tensor, the resulting errors are large, relative to those in the full CSA tensor calculations.73–77 Moreover, integration of MD simulations and DFT enables accurate calculation of dynamically averaged 15NH CSA tensors and these tensors are readily accessible from contemporary MAS NMR experiments.78 Acquisition of the corresponding data sets, together with the data required for resonance assignments and structural characterization, is beneficial as these yield an immediate probe of nano- to microsecond timescale motions. To improve isotropic chemical shift calculations, the CSA tensors can then be used in conjunction with isotropic shifts and other NMR parameters in atomic-resolution structural/dynamics characterization of the system of interest.
Supplementary Material
Acknowledgments
We thank David A. Case for the AFNMR scripts. This work was supported by the National Institutes of Health (NIH Grants GM080642 and GM082251). We acknowledge the support of the National Science Foundation (NSF Grant CHE0959496) for the acquisition of the 850 MHz NMR spectrometer at the University of Delaware and of the National Institutes of Health (NIH Grants P30GM103519 and P30GM110758) for the support of core instrumentation infrastructure at the University of Delaware. CMQ acknowledges the support of the National Institutes of Health Postdoctoral Fellowship grant F32GM113452. This paper was written to honor Klaus Schulten on his 70th birthday. Sadly and tragically, he did not live to see this issue of JPC come to light. We therefore dedicate our contribution to his memory.
Footnotes
Sequential assignment walk for three stretches of OAA residues; correlations between experimental and predicted shifts using different programs; intermolecular interfaces in OAA as identified by PISA. This information is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Goldbourt A. Biomolecular Magic-Angle Spinning Solid-State NMR: Recent Methods and Applications. Curr Opin Biotech. 2013;24:705–715. doi: 10.1016/j.copbio.2013.02.010. [DOI] [PubMed] [Google Scholar]
- 2.Yan S, Suiter CL, Hou GJ, Zhang HL, Polenova T. Probing Structure and Dynamics of Protein Assemblies by Magic Angle Spinning NMR Spectroscopy. Acc Chem Res. 2013;46:2047–2058. doi: 10.1021/ar300309s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Byeon IJL, Ahn J, Mitra M, Byeon CH, Hercik K, Hritz J, Charlton LM, Levin JG, Gronenborn AM. NMR Structure of Human Restriction Factor APOBEC3A Reveals Substrate Binding and Enzyme Specificity. Nat Commun. 2013;4 doi: 10.1038/ncomms2883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wickham JR, Mason RN, Rice CV. Solid-State NMR Studies of The Crystalline and Amorphous Domains Within PEO and PEO: LiTf Systems. Solid State Nucl Magn Reson. 2007;31:184–192. doi: 10.1016/j.ssnmr.2007.05.001. [DOI] [PubMed] [Google Scholar]
- 5.Comellas G, Rienstra CM. Protein Structure Determination By Magic-Angle Spinning Solid-State NMR, and Insights into the Formation, Structure, And Stability of Amyloid Fibrils. Annu Rev Biophys. 2013;42:515–36. doi: 10.1146/annurev-biophys-083012-130356. [DOI] [PubMed] [Google Scholar]
- 6.Loquet A, Habenstein B, Lange A. Structural Investigations of Molecular Machines by Solid-State NMR. Acc Chem Res. 2013;46:2070–9. doi: 10.1021/ar300320p. [DOI] [PubMed] [Google Scholar]
- 7.Quinn CM, Lu M, Suiter CL, Hou GJ, Zhang HL, Polenova T. Magic Angle Spinning NMR of Viruses. Prog NMR Spectrosc. 2015;86–87:21–40. doi: 10.1016/j.pnmrs.2015.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Suiter CL, Quinn CM, Lu MM, Hou GJ, Zhang HL, Polenova T. MAS NMR of HIV-1 Protein Assemblies. J Magn Reson. 2015;253:10–22. doi: 10.1016/j.jmr.2014.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.He X, Zhu T, Wang X, Liu J, Zhang JZ. Fragment Quantum Mechanical Calculation of Proteins and its Applications. Acc Chem Res. 2014;47:2748–57. doi: 10.1021/ar500077t. [DOI] [PubMed] [Google Scholar]
- 10.Helgaker T, Jaszunski M, Ruud K. Ab Initio Methods for the Calculation of NMR Shielding and Indirect Spin-Spin Coupling Constants. Chem Rev. 1999;99:293–352. doi: 10.1021/cr960017t. [DOI] [PubMed] [Google Scholar]
- 11.de Dios AC, Jameson CJ. Recent Advances in Nuclear Shielding Calculations. In: Webb GA, editor. Annu Rep NMR Spectrosc. Vol. 77. Elsevier; London, United Kingdom: 2012. pp. 1–80. [Google Scholar]
- 12.Shen Y, Bax A. SPARTA Plus: A Modest Improvement in Empirical NMR Chemical Shift Prediction by Means of an Artificial Neural Network. J Biomol NMR. 2010;48:13–22. doi: 10.1007/s10858-010-9433-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hafsa NE, Arndt D, Wishart DS. CSI 3.0: A Web Server for Identifying Secondary and Super-Secondary Structure in Proteins Using NMR Chemical Shifts. Nucl Acids Res. 2015;43:W370–W377. doi: 10.1093/nar/gkv494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Han B, Liu Y, Ginzinger SW, Wishart DS. SHIFTX2: Significantly Improved Protein Chemical Shift Prediction. J Biomol NMR. 2011;50:43–57. doi: 10.1007/s10858-011-9478-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Neal S, Nip AM, Zhang HY, Wishart DS. Rapid And Accurate Calculation of Protein H-1, C-13 And N-15 Chemical Shifts. J Biomol NMR. 2003;26:215–240. doi: 10.1023/a:1023812930288. [DOI] [PubMed] [Google Scholar]
- 16.Shen Y, Delaglio F, Cornilescu G, Bax A. TALOS+: A Hybrid Method for Predicting Protein Backbone Torsion Angles from NMR Chemical Shifts. J Biomol NMR. 2009;44:213–23. doi: 10.1007/s10858-009-9333-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Meiler J. PROSHIFT: Protein Chemical Shift Prediction Using Artificial Neural Networks. J Biomol NMR. 2003;26:25–37. doi: 10.1023/a:1023060720156. [DOI] [PubMed] [Google Scholar]
- 18.Shen Y, Vernon R, Baker D, Bax A. De Novo Protein Structure Generation from Incomplete Chemical Shift Assignments. J Biomol NMR. 2009;43:63–78. doi: 10.1007/s10858-008-9288-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kohlhoff KJ, Robustelli P, Cavalli A, Salvatella X, Vendruscolo M. Fast and Accurate Predictions of Protein NMR Chemical Shifts from Interatomic Distances. J Am Chem Soc. 2009;131:13894–13895. doi: 10.1021/ja903772t. [DOI] [PubMed] [Google Scholar]
- 20.Xu XP, Case DA. Automated Prediction of (15)N, (13)C(Alpha), (13)C(Beta) and (13)C’Chemical Shifts in Proteins Using a Density Functional Database. J Biomol NMR. 2001;21:321–333. doi: 10.1023/a:1013324104681. [DOI] [PubMed] [Google Scholar]
- 21.Vila JA, Arnautova YA, Martin OA, Scheraga HA. Quantum-Mechanics-Derived C-13(Alpha) Chemical Shift Server (Cheshift) for Protein Structure Validation. Proc Natl Acad Sci USA. 2009;106:16972–16977. doi: 10.1073/pnas.0908833106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jacob CR, Visscher L. Calculation of Nuclear Magnetic Resonance Shieldings Using Frozen-Density Embedding. J Chem Phys. 2006;125 doi: 10.1063/1.2370947. [DOI] [PubMed] [Google Scholar]
- 23.Christensen AS, Linnet TE, Borg M, Boomsma W, Lindorff-Larsen K, Hamelryck T, Jensen JH. Protein Structure Validation and Refinement Using Amide Proton Chemical Shifts Derived from Quantum Mechanics. PloS One. 2013;8:e84123. doi: 10.1371/journal.pone.0084123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Frank A, Moller HM, Exner TE. Toward the Quantum Chemical Calculation of NMR Chemical Shifts of Proteins. 2. Level of Theory, Basis Set, and Solvents Model Dependence. J Chem Theory Comput. 2012;8:1480–92. doi: 10.1021/ct200913r. [DOI] [PubMed] [Google Scholar]
- 25.Fileti EE, Georg HC, Coutinho K, Canuto S. Isotropic and Anisotropic NMR Chemical Shifts in Liquid Water: A Sequential QM/MM Study. J Brazil Chem Soc. 2007;18:74–84. [Google Scholar]
- 26.Casabianca LB, De Dios AC. Ab Initio Calculations of NMR Chemical Shifts. J Chem Phys. 2008;128 doi: 10.1063/1.2816784. [DOI] [PubMed] [Google Scholar]
- 27.Hill DE, Vasdev N, Holland JP. Evaluating the Accuracy of Density Functional Theory for Calculating 1H and 13C NMR Chemical Shifts in Drug Molecules. Comput Theor Chem. 2015;1051:161–172. [Google Scholar]
- 28.Di Micco S, Chini MG, Riccio R, Bifulco G. Quantum Mechanical Calculation of NMR Parameters in the Stereostructural Determination of Natural Products. Eur J Org Chem. 2010:1411–1434.
- 29.Vila JA, Aramini JM, Rossi P, Kuzin A, Su M, Seetharaman J, Xiao R, Tong L, Montelione GT, Scheraga HA. Quantum Chemical C-13(Alpha) Chemical Shift Calculations for Protein NMR Structure Determination, Refinement, and Validation. Proc Natl Acad Sci USA. 2008;105:14389–14394. doi: 10.1073/pnas.0807105105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.He X, Wang B, Merz KM., Jr Protein NMR Chemical Shift Calculations Based on the Automated Fragmentation QM/MM Approach. J Phys Chem B. 2009;113:10380–8. doi: 10.1021/jp901992p. [DOI] [PubMed] [Google Scholar]
- 31.Merz KM. Using Quantum Mechanical Approaches to Study Biological Systems. Acc Chem Res. 2014;47:2804–2811. doi: 10.1021/ar5001023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Swails J, Zhu T, He X, Case DA. AFNMR: Automated Fragmentation Quantum Mechanical Calculation of NMR Chemical Shifts for Biomolecules. J Biomol NMR. 2015;63:125–139. doi: 10.1007/s10858-015-9970-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Robustelli P, Stafford KA, Palmer AG. Interpreting Protein Structural Dynamics from NMR Chemical Shifts. J Am Chem Soc. 2012;134:6365–6374. doi: 10.1021/ja300265w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Li DW, Bruschweiler R. Certification of Molecular Dynamics Trajectories with NMR Chemical Shifts. J Phys Chem Lett. 2010;1:246–248. [Google Scholar]
- 35.Markwick PR, Cervantes CF, Abel BL, Komives EA, Blackledge M, McCammon JA. Enhanced Conformational Space Sampling Improves the Prediction of Chemical Shifts in Proteins. J Am Chem Soc. 2010;132:1220–1. doi: 10.1021/ja9093692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Scheurer C, Skrynnikov NR, Lienin SF, Straus SK, Bruschweiler R, Ernst RR. Effects of Dynamics and Environment on N-15 Chemical Shielding Anisotropy in Proteins. A Combination of Density Functional Theory, Molecular Dynamics Simulation, and NMR Relaxation. J Am Chem Soc. 1999;121:4242–4251. [Google Scholar]
- 37.Precechtelova J, Novak P, Munzarova ML, Kaupp M, Sklenar V. Phosphorus Chemical Shifts in a Nucleic Acid Backbone from Combined Molecular Dynamics and Density Functional Calculations. J Am Chem Soc. 2010;132:17139–17148. doi: 10.1021/ja104564g. [DOI] [PubMed] [Google Scholar]
- 38.Zhang H, Hou G, Lu M, Ahn J, Byeon IL, Langmead CJ, Perilla JR, Hung I, Gor’kov PL, Gan Z, et al. HIV-1 Capsid Function is Regulated by Dynamics: Quantitative Atomic-Resolution Insights by Integrating Magic-Angle-Spinning NMR, QM/MM, and MD. J Am Chem Soc. 2016 doi: 10.1021/jacs.6b08744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Koharudin LMI, Furey W, Gronenborn AM. Novel Fold and Carbohydrate Specificity of the Potent Anti-HIV Cyanobacterial Lectin from Oscillatoria agardhii. J Biol Chem. 2011;286:1588–1597. doi: 10.1074/jbc.M110.173278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Koharudin LMI, Kollipara S, Aiken C, Gronenborn AM. Structural Insights into the Anti-HIV Activity of the Oscillatoria agardhii Agglutinin Homolog Lectin Family. J Biol Chem. 2012;287:33796–33811. doi: 10.1074/jbc.M112.388579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Koharudin LM, Gronenborn AM. Structural Basis of the Anti-HIV Activity of the Cyanobacterial Oscillatoria Agardhii Agglutinin. Structure. 2011;19:1170–81. doi: 10.1016/j.str.2011.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Carneiro MG, Koharudin LM, Griesinger C, Gronenborn AM, Lee D. 1H13C And 15N Resonance Assignment of The Anti-HIV Lectin From Oscillatoria Agardhii. Biomol NMR Assign. 2015;9:317–9. doi: 10.1007/s12104-015-9600-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Thurber KR, Tycko R. Measurement of Sample Temperatures Under Magic-Angle Spinning from the Chemical Shift and Spin-Lattice Relaxation Rate of 79Br in KBr Powder. J Magn Reson. 2009;196:84–87. doi: 10.1016/j.jmr.2008.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Morcombe CR, Zilm KW. Chemical Shift Referencing in MAS Solid State NMR. J Magn Reson. 2003;162:479–86. doi: 10.1016/s1090-7807(03)00082-x. [DOI] [PubMed] [Google Scholar]
- 45.McDermott AE, Gu Z. Carbon and Nitrogen Chemical Shifts: Applications to Solid State Proteins. In: Grant DM, Harris RK, editors. Encyclopedia of Nuclear Magnetic Resonance. John Wiley and Sons; 1996. pp. 1137–1147. [Google Scholar]
- 46.Baldus M, Petkova AT, Herzfeld J, Griffin RG. Cross Polarization in the Tilted Frame: Assignment and Spectral Simplification in Heteronuclear Spin Systems. Mol Phys. 1998;95:1197–1207. [Google Scholar]
- 47.Fung BM, Khitrin AK, Ermolaev K. An Improved Broadband Decoupling Sequence for Liquid Crystals and Solids. J Magn Reson. 2000;142:97–101. doi: 10.1006/jmre.1999.1896. [DOI] [PubMed] [Google Scholar]
- 48.Hou G, Byeon IJL, Ahn J, Gronenborn AM, Polenova T. 1H–13C/1H–15N Heteronuclear Dipolar Recoupling by R-Symmetry Sequences under fast Magic Angle Spinning for Dynamics Analysis of Biological and Organic Solids. J Am Chem Soc. 2011;133:18646–18655. doi: 10.1021/ja203771a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hou G, Lu X, Vega AJ, Polenova T. Accurate Measurement of Heteronuclear Dipolar Couplings by Phase-Alternating R-Symmetry (PARS) Sequences in Magic Angle Spinning NMR Spectroscopy. J Chem Phys. 2014;141:104202. doi: 10.1063/1.4894226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, Bax A. NMRPipe - A Multidimensional Spectral Processing System Based On Unix Pipes. J Biomol NMR. 1995;6:277–293. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- 51.Vranken WF, Boucher W, Stevens TJ, Fogh RH, Pajon A, Llinas P, Ulrich EL, Markley JL, Ionides J, Laue ED. The CCPN Data Model For NMR Spectroscopy: Development of a Software Pipeline. Proteins. 2005;59:687–696. doi: 10.1002/prot.20449. [DOI] [PubMed] [Google Scholar]
- 52.Stevens TJ, Fogh RH, Boucher W, Higman VA, Eisenmenger F, Bardiaux B, van Rossum BJ, Oschkinat H, Laue ED. A Software Framework for Analysing Solid-State MAS NMR Data. J Biomol NMR. 2011;51:437–447. doi: 10.1007/s10858-011-9569-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bak M, Rasmussen JT, Nielsen NC. SIMPSON: A General Simulation Program for Solid-State NMR Spectroscopy. J Magn Reson. 2011;213:366–400. doi: 10.1016/j.jmr.2011.09.008. [DOI] [PubMed] [Google Scholar]
- 54.Bak M, Nielsen NC. REPULSION, A Novel Approach to Efficient Powder Averaging in Solid-State NMR. J Magn Reson. 1997;125:132–139. doi: 10.1006/jmre.1996.1087. [DOI] [PubMed] [Google Scholar]
- 55.Yao L, Vögeli B, Ying J, Bax A. NMR Determination of Amide N–H Equilibrium Bond Length from Concerted Dipolar Coupling Measurements. J Am Chem Soc. 2008;130:16518–16520. doi: 10.1021/ja805654f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Al-Karaghouli AR, Koetzle TF. Neutron Diffraction Study of l-Phenylalanine Hydrochloride. Acta Cryst B. 1975;31:2461–2465. [Google Scholar]
- 57.Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Scalmani G, Barone V, Mennucci B, Petersson GA, et al. Gaussian 09, Revision B.01. Wallingford CT: 2009. [Google Scholar]
- 58.Cohen AJ, Handy NC. Dynamic Correlation. Mol Phys. 2001;99:607–615. [Google Scholar]
- 59.Schäfer A, Huber C, Ahlrichs R. Fully Optimized Contracted Gaussian Basis Sets of Triple Zeta Valence Quality for Atoms Li to Kr. J Chem Phys. 1994;100:5829–5835. [Google Scholar]
- 60.Swails J, Zhu T, He X, Case DA. AFNMR: Automated Fragmentation Quantum Mechanical Calculation of NMR Chemical Shifts for Biomolecules. J Biomol NMR. 2015;63:125–39. doi: 10.1007/s10858-015-9970-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Krissinel E, Henrick K. Inference of Macromolecular Assemblies from Crystalline State. J Mol Biol. 2007;372:774–797. doi: 10.1016/j.jmb.2007.05.022. [DOI] [PubMed] [Google Scholar]
- 62.Hou G, Yan S, Trebosc J, Amoureux JP, Polenova T. Broadband Homonuclear Correlation Spectroscopy Driven by Combined R2nv Sequences under Fast Magic Angle Spinning for NMR Structural Analysis of Organic and Biological Solids. J Magn Reson. 2013;232:18–30. doi: 10.1016/j.jmr.2013.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Shen Y, Lange O, Delaglio F, Rossi P, Aramini JM, Liu GH, Eletsky A, Wu YB, Singarapu KK, Lemak A, et al. Consistent Blind Protein Structure Generation from NMR Chemical Shift Data. Proc Natl Acad Sci USA. 2008;105:4685–4690. doi: 10.1073/pnas.0800256105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Carneiro MG, Koharudin LMI, Ban D, Sabo TM, Trigo-Mourino P, Mazur A, Griesinger C, Gronenborn AM, Lee D. Sampling of Glycan-Bound Conformers by the Anti-HIV Lectin Oscillatoria agardhii agglutinin in the Absence of Sugar. Angew Chem Int Ed. 2015;54:6462–6465. doi: 10.1002/anie.201500213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Lu MM, Hou GJ, Zhang HL, Suiter CL, Ahn J, Byeon IJL, Perilla JR, Langmead CJ, Hung I, Gor’kov PL, et al. Dynamic Allostery Governs Cyclophilin A-HIV Capsid Interplay. Proc Natl Acad Sci USA. 2015;112:14617–14622. doi: 10.1073/pnas.1516920112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Yang J, Tasayco ML, Polenova T. Magic Angle Spinning NMR Experiments for Structural Studies of Differentially Enriched Protein Interfaces and Protein Assemblies. J Am Chem Soc. 2008;130:5798–807. doi: 10.1021/ja711304e. [DOI] [PubMed] [Google Scholar]
- 67.Reichert D, Zinkevich T, Saalwächter K, Krushelnitsky A. The Relation of the X-Ray B-Factor to Protein Dynamics: Insights from Recent Dynamic Solid-State NMR Data. J Biomol Struct Dyn. 2012;30:617–627. doi: 10.1080/07391102.2012.689695. [DOI] [PubMed] [Google Scholar]
- 68.Ivanir-Dabora H, Nimerovsky E, Madhu PK, Goldbourt A. Site-Resolved Backbone and Side-Chain Intermediate Dynamics in a Carbohydrate-Binding Module Protein Studied by Magic-Angle Spinning NMR Spectroscopy. Chem Eur J. 2015;21:10778–10785. doi: 10.1002/chem.201500856. [DOI] [PubMed] [Google Scholar]
- 69.Good DB, Wang SL, Ward ME, Struppe J, Brown LS, Lewandowski JR, Ladizhansky V. Conformational Dynamics of a Seven Transmembrane Helical Protein Anabaena Sensory Rhodopsin Probed by Solid-State NMR. J Am Chem Soc. 2014;136:2833–2842. doi: 10.1021/ja411633w. [DOI] [PubMed] [Google Scholar]
- 70.Yan S, Zhang H, Hou G, Ahmed S, Williams JC, Polenova T. Internal Dynamics of Dynactin CAP-Gly is Regulated By Microtubules and Plus End Tracking Protein EB1. J Biol Chem. 2015;290:1607–22. doi: 10.1074/jbc.M114.603118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Schanda P, Meier BH, Ernst M. Quantitative Analysis of Protein Backbone Dynamics in Microcrystalline Ubiquitin By Solid-State NMR Spectroscopy. J Am Chem Soc. 2010;132:15957–67. doi: 10.1021/ja100726a. [DOI] [PubMed] [Google Scholar]
- 72.Lorieau JL, Day LA, McDermott AE. Conformational Dynamics of an Intact Virus: Order Parameters for the Coat Protein of Pf1 Bacteriophage. Proc Natl Acad Sci USA. 2008;105:10366–10371. doi: 10.1073/pnas.0800405105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Jameson CJ, Dedios AC, Jameson AK. Nuclear Magnetic Shielding of Nitrogen in Ammonia. J Chem Phys. 1991;95:1069–1079. [Google Scholar]
- 74.Jameson CJ. Understanding NMR Chemical Shifts. Annu Rev Phys Chem. 1996;47:135–169. [Google Scholar]
- 75.Le HB, Oldfield E. Ab Initio Studies of Amide-N-15 Chemical Shifts in Dipeptides: Applications to Protein NMR Spectroscopy. J Phys Chem. 1996;100:16423–16428. [Google Scholar]
- 76.Sun HH, Sanders LK, Oldfield E. Carbon-13 NMR Shielding in the Twenty Common Amino Acids: Comparisons with Experimental Results in Proteins. J Am Chem Soc. 2002;124:5486–5495. doi: 10.1021/ja011863a. [DOI] [PubMed] [Google Scholar]
- 77.Wi S, Sun HH, Oldfield E, Hong M. Solid-State NMR and Quantum Chemical Investigations of C-13(Alpha) Shielding Tensor Magnitudes and Orientations In Peptides: Determining Phi and Psi Torsion Angles. J Am Chem Soc. 2005;127:6451–6458. doi: 10.1021/ja042935b. [DOI] [PubMed] [Google Scholar]
- 78.Hou G, Byeon IJL, Ahn J, Gronenborn AM, Polenova T. Recoupling of Chemical Shift Anisotropy by R-Symmetry Sequences in Magic Angle Spinning NMR Spectroscopy. J Chem Phys. 2012;137:134201. doi: 10.1063/1.4754149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Schumann FH, Riepl H, Maurer T, Gronwald W, Neidig KP, Kalbitzer HR. Combined Chemical Shift Changes and Amino Acid Specific Chemical Shift Mapping of Protein-Protein Interactions. J Biomol NMR. 2007;39:275–89. doi: 10.1007/s10858-007-9197-z. [DOI] [PubMed] [Google Scholar]
- 80.Williamson MP. Using Chemical Shift Perturbation to Characterize Ligand Binding. Prog NMR Spectrosc. 2013;73:1–16. doi: 10.1016/j.pnmrs.2013.02.001. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.