

Abstract
We used arrays of 2069 BACs (1303 nonredundant autosomal clones) to map sequence variation among Mus spretus (SPRET/Ei and SPRET/Glasgow) and Mus musculus (C3H/HeJ, BALB/cJ, 129/J, DBA/2J, NIH, FVB/N, and C57BL/6) strains. We identified 80 clones representing 74 autosomal loci of copy number variation (|log2ratio| ≥ 0.4). These variant loci distinguish laboratory strains. By FISH mapping, we determined that 63 BACs mapped to a single site on C57BL/6J chromosomes, while 17 clones mapped to multiple chromosomes (n = 16) or multiple sites on one chromosome (n = 1). We also show that small ratio changes (Δ log2ratio ∼ 0.1) distinguish homozygous and heterozygous regions of the genome in interspecific backcross mice, providing an efficient method for genotyping progeny of backcrosses.
Natural evolutionary forces and selective inbreeding have given rise to many different mouse strains that exhibit specific characteristics and traits (Beck et al. 2000). The publication of the mouse genome sequence has accelerated investigation of genomic variation among different types of mice. Indeed, genome-wide single-nucleotide polymorphism (SNP) comparison studies across different inbred strains of mice have not only elucidated haplotype structure (Wade et al. 2002; Wiltshire et al. 2003), but have also provided markers for mapping phenotypic differences between strains. Sequence analysis has also identified segmental duplications in genomes, which are defined as stretches of DNA sequence ≥1-5 kb in length with ≥90% sequence conservation that are present in more than one location in the genome. They make up ∼5% of the human genome (Bailey et al. 2002) and lesser proportions of rodent genomes, 2.92% in the rat (Tuzun et al. 2004) and 1%-1.2% in the mouse (Cheung et al. 2003). These studies have found that both intrachromosomal and interchromosomal or transchromosomal segmental duplications are mosaics of sequences duplicated from within one chromosome or nonhomologous chromosomes, respectively. Transchromosomal duplications are predominantly located in regions close to the centromere and/or telomere and contain few functional genes, whereas intrachromosomal segmental duplications harbor functional genes and gene families and are predominantly found in euchromatic regions (Eichler et al. 1996; Jackson et al. 1999; Horvath et al. 2001; Cheung et al. 2003; Tuzun et al. 2004). Since both types appear to be recent evolutionary events (Cheung et al. 2001), they are likely to contribute to the generation of phenotypic diversity among laboratory mice.
In the course of application of BAC array comparative genomic hybridization (array CGH) to characterize tumors in mouse models, we noticed large strain-specific ratio variations for many BACs, indicating probable germ-line copy number variations. Since these relative increases and decreases in copy number at particular loci could underlie certain strain specific phenotypes, we undertook a systematic study to detect and map variant loci among laboratory mouse genomes. This approach provides complementary information to genome sequencing, because large-scale copy number differences are not easily identified by sequence analysis (Locke et al. 2003), and it allows investigation of mouse genomes that have not been scheduled for whole-genome sequencing. Here, we report on the substantial copy number variation we found among mouse strains.
Careful analysis of the results of our interstrain comparisons indicated that the BAC arrays also provided the ability to detect small ratio variations that presumably reflect nucleotide level variation among the strains. We show that this capability can be used to distinguish chromosomal regions of heterozygosity and homozygosity in interspecific backcross mice. These regions differ by log2ratio ∼ 0.1. Given the high density of genome coverage possible on BAC arrays and the rate at which hybridizations can be performed, this novel application of array CGH provides a highly efficient means to genotype progeny of interspecific backcross mice.
Results
Mapping large-scale variation in mouse genomes
We used arrays comprised of 2069 mouse BAC clones (1303 nonredundant, nonoverlapping autosomal clones) to map loci of segmental DNA copy number variation involving autosomes in multiple different individuals from seven inbred Mus musculus strains, including C3H/HeJ (n = 5), BALB/cJ (n = 4), 129P3/J (n = 4), DBA/2J (n = 4), NIH (n = 4), FVB/N (n = 4), and C57BL/6J (n = 9), as well as inbred (SPRET/EiJ; n = 8) and outbred (SPRET/Glasgow; n =4) Mus spretus individuals using genomic DNA from a single FVB/N individual as the reference (Supplemental Tables A-J). We identified loci with variant copy number by selecting autosomal clones that gave adequate hybridization signals in at least 95% of all hybridizations, and their log2ratios were ≥0.4 or ≤-0.4 in at least three individuals independent of strain in which they were identified. Eighty clones, corresponding to 74 regions of copy number variation relative to FVB/N, met these criteria (Fig. 1; Supplemental Fig. A). We observed that the variant loci represented both strain-specific (n = 5) and shared (n = 69) ratio differences across multiple strains or species. Agglomerative hierarchical clustering of all individuals from all strains based on these 74 polymorphic regions (Fig. 2) was consistent with divergence of the strains and species estimated by other criteria and breeding history (Beck et al. 2000). Previous reports of copy number polymorphisms in the major histocompatibility locus (Amadou et al. 2003) and in a long-range repeat on Chromosome 1 (Agulnik et al. 1993) have shown similar phylogenetically related differences among strains and species.
Figure 1.

Array CGH profiles. (A) Normalized genome-wide DNA copy number profiles of mouse strain DBA/2J using FVB/N genomic DNA as a reference. BACs are ordered by position on the genome starting with Chromosome 1 and ending with Chromosome X. Vertical bars indicate chromosome boundaries. Each array (MouseArray 3.1) contained 2069 BAC clones, 1848 of which have been mapped onto the draft sequence of the mouse genome (October 2003 freeze). We highlighted in red data points corresponding to a number of the 75 polymorphic clones, which in this individual showed |log2ratio| > 0.25. (B,C) Normalized DNA copy number profiles of Chromosomes 13 and 17 after hybridization with SPRET/Glasgow and C57BL/6J genomic DNA, respectively, using FVB/N genomic DNA as a reference. Note the high-level DNA copy number polymorphism on the proximal arm of Chromosome 13 showing a gain (log ratio ∼ 1) spanning three overlapping BAC clones. The proximal part of Chromosome 17 shows 2a lower-level DNA copy number polymorphism (log2ratio ∼ 0.5) encompassing one BAC clone.
Figure 2.

Agglomerative hierarchical clustering of 74 regions of sequence variation. We used a Euclidian metric and Ward method to cluster both samples and clones. For overlapping BACs in contigs, we used the one clone from the contig for which the greatest number of observations met the threshold (clone denoted by an asterisk). Note that all mice cluster in two separate branches according to species in agreement with accepted phylogeny. Within the M. spretus cluster, all SPRET/Ei mice cluster together and are more closely related to one another than to the outbred SPRET/Glasgow mice. In the M. musculus branch, all individual mice from each of the strains cluster together. Furthermore, FVB/N and NIH are closest to each other in the clustering dendrogram, which again agrees with the strain phylogeny.
Organization of variant loci in the genome
Analysis of duplication content in human, mouse, and rat genomes has revealed both a nonrandom chromosomal distribution and a bias toward clustering of segmental duplications at pericentromeric and subtelomeric regions (Eichler et al. 1996; Jackson et al. 1999; Horvath et al. 2001; Cheung et al. 2003; Thomas et al. 2003; Tuzun et al. 2004). Therefore, to determine whether these murine variant loci identified by array CGH map to particular chromosomal regions, we mapped all 80 polymorphic BACs on C57BL/6J metaphase spreads using FISH. We assessed the distribution of the variant loci by classifying the BAC clones representing these loci as mapping to the proximal 1/3, middle 1/3, or distal 1/3 of the chromosome based on end-sequence mapping and FISH position. For this analysis, we considered BACs with overlapping positions in the genome sequence as representative of a single variant locus. We observed that 63 clones mapped to a single site on C57BL/6J chromosomes (Supplemental Fig. B), while 17 clones hybridized to multiple places in the genome (Supplemental Fig. C).
Two clones, RP23-263M19 and RP23-206D2 mapped to multiple centromeric sites on C57BL/6J chromosomes and showed large ratio variation when M. spretus genomic DNA was hybridized using M. musculus genomic DNA as a reference (log2ratio < -3 and log2ratio < -0.5, respectively). FISH mapping revealed that these ratio variations, which are consistent either with deletion of the region in M. spretus or amplification of the sequence in M. musculus, corresponded to reduced representation of sequences in M. spretus compared to M. musculus (Supplemental Fig. C). We determined that RP23-263M19 contains sequence homologous to the M. musculus major satellite consensus sequence (Horz and Altenburger 1981) by sequencing small genomic subclones of RP23-263M19, whereas BAC end sequencing indicated that RP23-206D2 contains at least a portion of the ribosomal DNA sequence. Furthermore, RP23-206D2 hybridized to the centromeric regions of Chromosomes 12, 15, 16, 18, and 19 in C57BL/6J (M. musculus) metaphases, whereas it hybridized to the telomeric regions of Chromosomes 4, 13, and 19 in SPRETUS/Ei (Supplemental Fig. C), consistent with the known distribution of the rDNA in the genomes of these species and inbred strains (Henderson et al. 1974; Elsevier and Ruddle 1975; Dev et al. 1977; Eicher and Shown 1993; Kurihara et al. 1994).
Fifteen other clones that mapped to multiple sites by FISH (excluding the highly repetitive clones RP23-263M19 and RP23-206D2) showed different patterns of ratio variation among the different strains (Fig. 3). In C57BL/6J, we found these BAC clones hybridized to 21 sites in the proximal 1/3, 15 sites in the middle 1/3, and only seven sites in the distal 1/3 of the chromosomes, with an average number of three signals per BAC (range 2-8). We also observed that the distribution of hybridization signals was nonrandom. Moreover, we found that several BACs hybridized to similar sites on the same chromosomes. For example, we observed hybridization more frequently to Chromosomes 1 (distal), 5 (middle), 7 (proximal), 9 (proximal and middle), 12 (proximal and middle), 14 (proximal), and 17 (proximal/middle). In some cases, BACs hybridized to more than one of these sites (Fig. 4). The fact that most of these BACs hybridize near pericentromeric regions in addition to one or more distal sites on the chromosome is similar to the reported distribution of segmental duplications in other mammalian genomes (Eichler et al. 1996; Jackson et al. 1999; Horvath et al. 2001; Cheung et al. 2003; Thomas et al. 2003; Tuzun et al. 2004).
Figure 3.

Copy number differences among mouse strains compared to FVB/N for BACs mapping to multiple sites by FISH. We display the average log2ratio computed over all individuals from each mouse strain or species.
Figure 4.
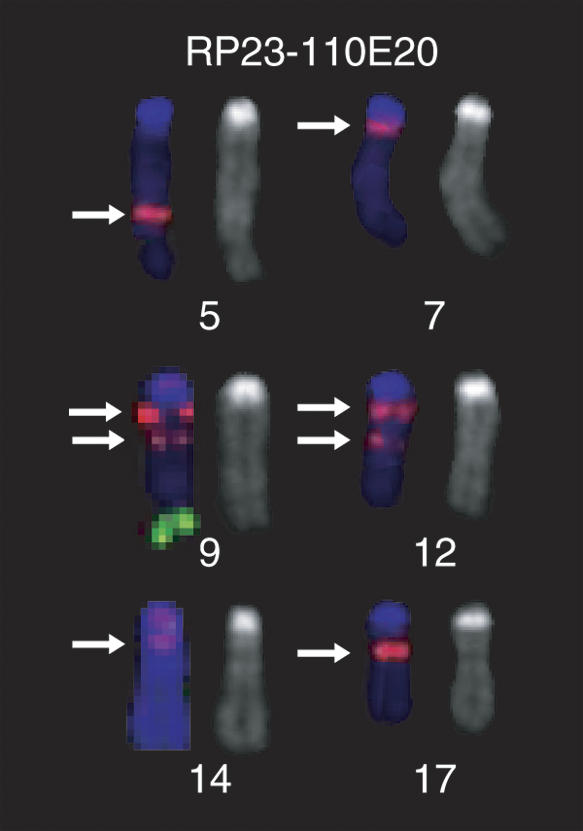
Cytogenetic mapping of a multisite BAC. Distribution of hybridization signals on C57BL/6J metaphase spreads from clones RP23-110E20 (red) and RP23-106H14 (green), marker for Chromosome 9. RP23-110E20 was selected from the RP23 library by an STS that mapped to 21.6 Mb on Chromosome 9. We observed hybridization signals at two sites on Chromosome 9 in addition to regions on Chromosomes 5, 7, 12 (2 loci), 14, and 17. Note that at least four out of a total of eight hybridization loci showing hybridization with RP23-110E20 are located in the proximal 1/3 of the corresponding chromosomes. The arrows indicate the positions of the RP23-110E20 hybridization signals.
We found that the set of 60 single-site BACs that were mapped with respect to the May 2004 freeze identified loci on all chromosomes except 9, 10, and 16. However, we found that there was not enough evidence to reject the null hypothesis of a uniform distribution of these clones across all chromosomes (Supplemental Fig. D). We observed no bias in the position on the chromosome for these clones. Most often, we observed only one hybridization signal by FISH, even in those cases in which the clone showed a ratio difference between C57BL/6J and FVB/N, indicating that it is likely that the ratio deviation between the strains reflects intrachromosomal differences in the number of copies of a sequence at the locus (i.e., too close together to be resolved by FISH). Indeed, 14/44 BAC end sequences showed multiple overlapping alignments on the genome sequence, consistent with local sequence duplications (Supplemental Fig. B). On the other hand, in some cases when we mapped BACs on chromosomes from different strains or species, we observed different numbers of FISH signals, consistent with the ratio variations measured by array CGH (data not shown).
Small-scale sequence variation between mouse species allows mapping of genomic content in interspecific backcrosses
In addition to the clones with highly variant ratios representing segmental copy number variation, we observed small ratio variations among all of the clones on the array. Since the magnitude of the variation appeared to depend on which strain was being compared to the FVB/N reference, it seemed unlikely that the variation was due entirely to random fluctuations. Therefore, to quantify the difference among strains, we removed clones with absolute log2ratio ≥ 0.3 to eliminate large ratio variants from the analysis and calculated the average median absolute deviation (MAD) of all clones for all strains (Supplemental Fig. E). We observed a range in MAD from 0.07 for FVB/N and NIH strains to 0.12 for SPRET/Ei. We interpret the observed low MAD for the FVB/N and NIH strains as due to the fact that NIH is closely related to FVB/N, which was used as the reference for hybridization. The increase in MAD in the SPRET/Ei, and to a lesser extent in the other strains, is likely attributable to differences in sequence composition, ranging from DNA copy number variations comprising a BAC clone down to single nucleotide differences that affect hybridization efficiency.
Given this observation, we reasoned that BAC array CGH would provide the capability to distinguish strain-specific components of complex genomes, such as those present in interspecific backcross progeny. Therefore, we obtained genomic DNA from three previously characterized mice, which were the progeny of the cross (NIH × SPRET/Glasgow) F1 × NIH as well as the (NIH × SPRET/Glasgow) F1 parent. Owing to recombination in meiosis, the chromosomes of the backcross mice will have extended regions that are either homozygous NIH or heterozygous NIH/SPRET. The ratio profiles and a statistical analysis for the hybridizations of three of these backcross mice using NIH genomic DNA as the reference are shown in Figure 5A,B,C. We observed that there appeared to be two ratio states differing by log2ratio ∼ 0.1, with each of the backcross progeny mice having a unique constellation of subtle ratio changes. Hybridization using DNA from the (NIH × SPRET/Glasgow) F1 individual with NIH as the reference revealed no ratio changes (Fig. 5D). We compared the observed ratio changes to available microsatellite data for ∼100 markers across the genome for all three of the backcross mice and found very high concordance between ratio state and genome composition. Specifically, we found that the higher ratio state corresponded to regions in the genome homozygous for NIH, while regions in the genome showing a lower ratio corresponded to regions heterozygous for NIH (Fig. 5A,B,C). Together these observations indicate that array CGH offers a new method for genotyping the progeny of interspecific backcrosses. It allows genome-wide mapping to be accomplished in a single experiment at high resolution, which depends on the number and spacing of clones on the array. For example, since genotype information is obtained for each clone on the array, then arrays of ∼3000 clones would provide 1 Mb mapping resolution across the genome.
Figure 5.

Array CGH mapping of interspecific backcrosses. (A,B,C) Genome-wide DNA copy number profiles of three different individuals from the cross (NIH × SPRET/Glasgow) F1 × NIH using NIH genomic DNA as a reference. We normalized these ratios relative to the ratios from the hybridization of the (NIH × SPRET/Glasgow) F1 parent versus NIH as a reference, in order to minimize fluctuation in ratios due to technical sources. Note the small deviations in copy number ratio around zero (average log2ratio < 0.2). The panels on the right show the smoothed genome-wide DNA copy number profiles and microsatellite mapping data for ∼100 markers across the genome for each of the different mice. Blue indicates regions determined to be homozygous and red regions heterozygous for NIH by microsatellite mapping. We confirmed these observations by carrying out two more hybridizations. First, we repeated the hybridizations for all three mice and found all previously observed ratio changes, and second, we did a dye-swap experiment, which resulted in the expected inversion of the ratio changes (data not shown). Microsatellite mapping agrees with the observed subtle changes in the log2ratio and indicates that relative increases in the log ratio are regions homozygous for NIH, while regions of the genome that exhibit a relative decrease in the log2ratio correspond to regions heterozygous for NIH. (D) Genome-wide DNA copy number profile (left) and smoothed genome-wide DNA copy number profile (right) of the (NIH × SPRET/Glasgow) F1 parent using NIH as a reference. Note the absence of subtle copy number deviations as observed in A,B,C.
Discussion
Here we have described large- and small-scale DNA copy number variants, measured by array CGH, that distinguish the genomes of closely related mouse strains. We observed that 6% of the BAC clones (74 BACs representing polymorphic regions/1303 nonre-dundant BACs on the array) identified large-scale copy number variations in M. musculus and M. spretus strains relative to FVB/N, with the highest frequency of loci identifying probable intrachromosomal copy number variants (i.e., those identified by single-site BACs). However, we expect that these variant sequences make up much less than 6% of the genome sequence, because the magnitude of the ratio changes, which ranged from our defined threshold of |log2ratio| = 0.4 to |log2ratio| > 3, indicates that only a portion of a BAC is involved in most of the variants. (For example, if the entire BAC were included in a duplicated or deleted region, then one would expect a ratio of 0.8-1.0 for the duplication and <-3 for the deletion, since the regions would be homozygous in one strain compared to another.) We also note that ratios on other BACs did not exceed the thresholds, indicative of even smaller segmental duplications, although there is an increased possibility of spurious observations when small ratio changes are measured.
We also found that only 17 of the BACs representing the 74 large-scale polymorphic regions hybridized to multiple places in the genome by FISH, which may be because of the nature of the copy number variant loci detectable by array CGH or the composition of the array (Locke et al. 2004). It is likely that regions of high repeat sequence density have been underrepresented on this array, and such regions may be sources of sequence variation between mouse strains and species, which arguably might be more likely to be distributed at multiple sites in the genome. It is also possible that BACs that map to multiple sites by FISH may be chimeric, containing genomic fragments from different parts of the genome. However, while a low level of chimerism is present in the RP23 library (estimated to be ∼1%) (Osoegawa et al. 2000), we believe that ratio variation among strains is not due to clone chimerism for several reasons. First, chimerism would not affect ratios. Second, we would not have observed that strains and species clustered if the variant loci were randomly formed chimeric clones (Fig. 2). Third, BAC end-sequencing confirmed the integrity of many of the clones that detected strain-specific variation.
Further research is needed to characterize the large-scale variant loci among the strains, although we did identify previously described polymorphisms (Amadou et al. 2003; Cheung et al. 2003) such as olfactory receptor genes (e.g., RP23-85A8) and the major histocompatibility locus on Chromosome 17 (e.g., RP23-178C19 and RP23-143P23). Some of the other BACs that map to single sites by FISH are also likely to identify regions that contain gene families and/or low copy number repeats. For example, Akr1b3, Akr1b8, and Akr1b7 map within the region spanned by the contig of three polymorphic BACs on Chromosome 6. Similarly, several BAC end sequences align at two or more overlapping positions on the genome sequence (UCSC Mouse Genome May 2004; Supplemental Figs. B and C), indicative of intrachromosomal duplication. Such low-copy repeated sequences may have rendered the region more prone to unequal crossover events and thus generated the copy number difference between strains. In the human genome, rearrangements involving segmental duplications are associated with several diseases or developmental anomaly syndromes (Ji et al. 2000; Emanuel and Shaikh 2001; Shaw and Lupski 2004). Similarly, unequal crossing over between repeated sequences appears to play a major role in the evolution of yeasts grown under selective pressure in the laboratory (Dunham et al. 2002) and in adaptive evolution of sherry wine (flor) yeasts in which copy number variations may comprise as much as 38% of the genome (Infante et al. 2003).
Sequence variations found within an inbred mouse strain include those that arise de novo, such as the highly polymorphic pseudoautosomal region (Kipling et al. 1996) and the hypervariable mouse minisatellite locus, Ms6-hm (Kelly et al. 1991). Polymorphisms also are maintained in some inbred strains in spite of many generations of inbreeding (Yuan et al. 1996). We looked for ratio variations within strains for the clones that we initially identified by their variation among strains. However, although we observed some clones showing variation within a strain, we could not rule out technical artifacts. Thus, we conclude that the large-scale variations we detected by array CGH are generally stable in a particular strain.
We also distinguished small-scale variations among mouse strains and species that allowed us to apply array-based CGH to differentiate regions of heterozygosity and homozygosity in interspecific backcross (NIH × SPRET/Glasgow) mice. M. spretus diverged from M. musculus 3 million years ago, but the two species still remain capable of interbreeding. It has been possible to map a large number of genetic traits in backcrosses using these parental strains (Nagase et al. 1995; Staelens et al. 2002; Ewart-Toland et al. 2003; Guenet and Bonhomme 2003), because highly divergent strain combinations such as these offer the greatest opportunity to detect phenotypic differences and so maximize success of identifying genes that control specific phenotypes. We note that the approaches described in this report should be applicable to other crosses involving distantly related strains such as Castaneous or Molossinus, and possibly even to crosses between M. musculus strains. Furthermore, genotype information is obtained for each clone on the array, which provides high-resolution mapping in a single experiment.
In the backcross experiments, we observed that regions heterozygous for NIH showed a decrease in log2ratio of ∼0.1 compared to homozygous NIH regions in hybridizations using NIH as the reference genome. We propose that the observed higher ratio in regions homozygous for NIH is due to greater sequence conservation between NIH genomic DNA and the C57BL/6J BAC DNA in the array spots compared to the SPRET/Glasgow genomic DNA. On a linear scale, the relative ratio of the heterozygous to homozygous portions of the genome is ∼0.93, implying that the SPRET/Glasgow genomic DNA binds to the array elements with ∼85%-90% the efficiency of NIH. While detection of single nucleotide differences by array hybridization is routine when using short oligonucleotides for array elements, it might at first seem surprising that one can detect such sequence variations on BAC arrays. However, we suggest that it is possible because of a nonuniform distribution of the sequence differences in the genome, resulting in a significant reduction in hybridization to a small proportion of the DNA fragments of a BAC. For example, sequencing data (Wade et al. 2002; Wiltshire et al. 2003) and estimates of the degree of sequence divergence compatible with breeding (Sidman and Shaffer 1994) indicate that the average sequence divergence between the two genomes is on the order of somewhat less than 1%. If we assume that the average length of base pairing for binding to a DNA fragment in an array spot is ∼100 bases, then for an average sequence divergence of 1%, one expects ∼25% of the hybridization sites will have ≥2% divergence and 8% of the fragments will have ≥3% divergence if the differences are randomly distributed. It is also possible that the sequence variation may be even more unequally distributed for functional reasons. The fact that the ratios are always lower in the regions heterozygous for NIH and M. spretus further supports the view that the subtle ratio variations are due to sequence differences distributed over the genome, rather than resulting from copy number changes of large genomic segments, which, as we have shown, result in both ratio increases and decreases in comparisons between two species. However, it is very likely that there is a continuum of sequence differences between these two types of genomic variation, each with its characteristic effect on comparative hybridizations. Thus, we expect that some of what appears to be “noise” in these measurements will eventually be understood in terms of more subtle genomic characteristics.
Methods
Mouse strains
We obtained DNA from inbred M. musculus and M. spretus mice from the Jackson Laboratory and outbred M. spretus (SPRET/Glasgow) from a colony maintained by the Balmain laboratory at the University of California San Francisco.
DNA extraction
We either obtained genomic DNA from different inbred strains of mice from the Jackson Laboratories or isolated genomic DNA from spleens dissected from mice. To isolate DNA, we first froze the spleens on dry ice, and then thinly sliced them. We placed the tissue into a buffer containing 10 mM Tris, 1 mM EDTA, 100 mM NaCl, and 1 mg/mL proteinase K, and incubated it overnight at 56°C with shaking. Next, we added RNase to a concentration of 0.25 mg/mL, incubated the mixture at 37°C for 1 h, and then incubated the sample again in proteinase K (1 mg/mL) at 56 ° C for 2 h. We extracted the preparation with an equal volume of buffer-saturated phenol, collected the aqueous phase, and extracted with an equal volume of chloroform-isoamyl alcohol (24: 1). We recovered the final aqueous phase; added sodium acetate to a final concentration of 0.3 M followed by two volumes of ice-cold ethanol, and inverted the mixture gently until the DNA precipitate appeared. We collected the precipitate by spooling the DNA, washed it in 70% ethanol, and resuspended it in TE. We measured the DNA concentration using fluorometry.
Preparation of mouse BAC arrays
We selected BACs covering the genome from the RP23 C57BL/6J library by two routes: (1) by screening libraries with overgo probes for mapped STS markers as described previously (Cheung et al. 2001) and (2) by identifying BACs with particular STS markers using published information (Cai et al. 1998; Hodgson et al. 2001) or information from the mouse genome sequencing program as the sequencing and radiation hybrid mapping projects progressed. Given the available resources at the time we assembled the array, this procedure resulted in redundant clones and a nonrandom distribution of BACs among the different chromosomes. In the assembly of the human BAC arrays we reported previously (Snijders et al. 2001), we attempted to minimize the number of potentially polymorphic or multisite clones on the array by screening using both FISH (Cheung et al. 2001) and array CGH with known aneuploid cell lines (Snijders et al. 2001). However, for the mouse array, we included all selected BACs on the array and subsequently carried out FISH mapping to identify multisite clones or potentially erroneously mapped clones. Thus, while there is some bias in the representation of the chromosomes on the arrays, we have not intentionally selected against BAC clones representing regions of sequence variation among strains. Information on the clone set is provided at http://microarrays.roswellpark.org. We prepared DNA spotting solutions from the BACs by ligation-mediated PCR as described previously for assembly of human BAC arrays (Snijders et al. 2001). We printed each BAC clone in triplicate, with spots on 130-μm centers in 12 × 12 mm area on chromium surfaces using a custom robot.
DNA labeling by random priming
For each labeling reaction, we used ∼300 ng each of test or reference genomic DNA in a volume of 15 μL containing 10 mM Tris-1 mM EDTA (pH 7.5); 0.2 mM unlabeled dATP, dCTP, and dGTP; 0.1 mM unlabeled dTTP; 1× random primer (Bioprime DNA labeling system; Invitrogen); and 0.4 mM Cyanine-3 (test) or Cyanine-5 (reference) conjugated dUTP (Amersham). We denatured the DNA in a thermal cycler at 100°C for 15 min and cooled it to 4°C before adding 12 units of Klenow fragment (Bioprime DNA labeling system; Invitrogen). Using a thermal cycler we incubated the reaction at 16°C for 10 min and 37°C for 20 min for 30-40 cycles. We removed unincorporated nucleotides from the labeling reaction using a Sephadex G-50 spin column and visually assessed the labeling efficiency by the intensity of the color of the flow-through.
Array hybridization
For each hybridization, we combined Cy3-dUTP-labeled test DNA and Cy5-dUTP-labeled reference DNA with 40 μg of mouse Cot-1 DNA (Invitrogen) and precipitated the mixture using sodium acetate at 0.3 M and two volumes of ice-cold ethanol. We chilled the mixture at -20°C for 20 min and collected the precipitate by centrifugation at 17,400g for 25 min. We removed the supernatant, wicked dry the pellet with a tissue, and resuspended the pellet in a 40-μL hybridization solution consisting of 50% formamide, 10% dextran sulfate (molecular weight ∼ 500,000), 2× SSC, 4% SDS, and water. To denature the probe mixture, we heated it at 75°C for 20 min and then incubated it for several hours at 37°C to allow preannealing of the Cot-1 DNA to the probe DNA.
We applied a ring of rubber cement around the perimeter of each array to contain the hybridization mixture. We added 50 μL of hybridization solution containing no probe DNA within the perimeter of the rubber cement to pre-wet the array for 15 min. We aspirated the wetting solution and added the hybridization mixture to the array and then placed the slide into a polyethylene slide-mailing container containing 40 μL of 50% formamide, 2× SSC to maintain humidity, sealed the container with Parafilm, and incubated it with rocking at 37°C for 60 h. After hybridization, we rinsed off excess hybridization mixture using PN buffer (0.1 M Na2HPO4/NaH2PO4 at pH 8.5, 0.1% NP-40) and washed the arrays at 45°C in the following order: 50% formamide, 2× SSC for 20 min, 2× SSC for 10 min, 0.1× SSC for 10 min, PN buffer for 10 min, and 0.1× SSC for 10 min. We removed the rubber cement borders and drained off excess liquid. We wet-mounted the arrays under a coverslip in a solution containing 90% glycerol, 10% PBS, and 1 μM DAPI. We stabilized the coverslip using a border of fingernail polish along two edges.
Imaging and analysis
We acquired 16-bit 1024 × 1024 pixel DAPI, Cy3 and Cy5 images using a custom-built CCD camera system as described previously (Pinkel et al. 1998) and used “UCSF SPOT” software (Jain et al. 2002) to automatically segment the spots based on the DAPI images, perform local background correction, and to calculate various measurement parameters, including log2ratios of the total integrated Cy3 and Cy5 intensities for each spot. We used a second custom program SPROC to obtain averaged ratios of the triplicate spots for each clone, standard deviations of the triplicates, and plotting position for the BACs on the October 2003 freeze of the mouse genome sequence (http://genome.ucsc.edu). We edited the data files to remove ratios on clones for which only one of the triplicates remained after SPROC analysis and/or the standard deviation of the log2ratios of the triplicates was >0.2. We averaged the values for replicate clones and removed exact replicates from the data set. We created two different versions of the data set. For the copy number polymorphism analysis, we included only autosomal clones that were present in at least 95% of the hybridizations. We included clones that were present in at least 75% of the samples when plotting ratios along the genome and in the analysis of heterozygosity and homozygosity in backcross animals.
Statistical methods
We identified BAC clones representing putative copy number variants using a conservative threshold of |log2ratio| = 0.4 in at least three samples. When contiguous clones were identified, we retained the one clone from the contig for which the greatest number of observations met the threshold. We used the resulting set of clones to cluster the samples using an agglomerative hierarchical clustering algorithm with Euclidean metric and Ward method. We reordered the samples within each branch according to their means in such a way that tree topology was preserved.
We estimated the variability of the individual hybridizations by computing the Median Absolute Deviation (MAD) of all clones for a given sample with |log2ratio| ≤ 0.3. We considered the clones that had absolute mean value ≥0.3 in a given strain and ≤0.1 in the remaining strains to represent possible copy number polymorphisms specific to that strain, even though they might not meet the more conservative criteria described above. We tested the uniformity of the distribution of the mapped clones and of the single-site polymorphism clones among chromosomes using the χ2 goodness of fit test (Snedecor and Cochran 1989). Specifically, we tested the equality of the number of the mapped clones per megabase on different chromosomes, as well as the equality of the proportion of the single-site polymorphic clones among clones mapped on the genome sequence of different chromosomes.
To identify heterozygous and homozygous regions in the genomes of backcross animals, we first normalized each hybridization to the F1 hybridization in order to minimize systematic ratio fluctuations due to technical sources. We then applied a modification of the unsupervised Hidden Markov Model (HMM) state-fitting method described previously (Fridlyand et al. 2004). Here, we restricted the maximum number of states to be two and fit an HMM to all chromosomes simultaneously by concatenating the neighboring chromosomes and assigning values to either state 1 or 2. We selected the number of states (1 or 2) using a BIC criterion with δ = 1.5 (Schwarz 1978; Broman and Speed 2002a,b). We merged the states if their median values were <0.05 apart. For each hybridization, we plotted the smoothed value of all clones that were not aberrations or outliers. The smoothed value is computed as the median value of the corresponding state for a given clone. In the case of one state only, we used the median value of all clones for a given hybridization. We note that the unsupervised HMM procedure used to assign individual BACs to a given state (Fig. 5) does so by assigning a probability to each BAC of being in a given state and then allocates each BAC to the state with the largest probability (Fridlyand et al. 2004). Thus, a natural output of the procedure is a measure of the confidence with which a BAC is assigned as heterozygous or homozygous, which allowed us to determine that the majority of BACs were assigned to their respective states with >95% confidence.
BAC end-sequencing analysis
In order to verify the identity of the variant loci, we obtained BAC end sequences for 54 clones. We subjected the end sequences to BLAT analysis on the May 2004 freeze of the mouse genome (UCSC Genome Browser). We considered a BAC to be properly aligned on the genome sequence if the sequence pairs from both end sequences from any one BAC were on the same chromosome, no more than 300 kb apart on opposite strands. We determined that 63 of the clones mapped correctly based on concordance of our FISH and/or BAC end-sequence data with position in the May 2004 freeze as assigned by BAC end pairs or STS position. However, we found that the positions of 11 clones were discrepant, five were mapped on random segments and another one was not mapped on the May 2004 freeze. We found no evidence of chimerism based on the positions of the BAC end sequences on the May 2004 freeze.
Fluorescent in situ hybridization (FISH)
In order to verify the identity of the variant loci, we mapped all 80 polymorphic BACs on C57BL/6J metaphase spreads using FISH. We carried out two-color FISH using two different BAC DNAs, one labeled with Cy3-dUTP and the other with fluorescein-dCTP, so that a control probe hybridizing to a known chromosomal region could be incorporated into the same hybridization as the test probe. We labeled 20 ng of BAC DNA in a 10-μL reaction containing 0.2 mM each of unlabeled dATP, dCTP, and dGTP and 0.1 mM dTTP if cy3-dUTP was the label; or 0.2 mM unlabeled dATP, dGTP, and dTTP and 0.1 mM dCTP if fluorescein-dCTP was the label; 1× random primer (Bioprime DNA labeling system, Invitrogen); and 0.4 mM Cy3-dUTP or fluorescein-dCTP. We denatured the BAC DNA along with random primers and nucleotides at 100°C for 15 min and then cooled the mixture to 4°C after which we added 20 units of Klenow fragment (Bioprime DNA labeling kit; Invitrogen) and incubated the reaction at 37°C for ∼16 h. We combined 5 μL each of a Cy3 and a fluorescein labeling reaction with 6 μg of mouse Cot-1 DNA and precipitated the mixture using sodium acetate at 0.3 M and two volumes of ice-cold ethanol. We removed the supernatant and wicked dry the pellet with a tissue and then resuspended it in 10 μL of a hybridization solution consisting of 50% formamide, 10% dextran sulfate (molecular weight ∼ 500,000), 2× SSC, and 2% SDS. We then denatured the hybridization solution at 75°C for 20 min.
To prepare metaphases for hybridization, we washed microscope slides containing metaphase spreads from M. musculus or M. spretus embryonic fibroblasts in 2× SSC at 37°C for 30 min, dehydrated them in an ethanol series, and allowed them to air dry. We then denatured the metaphase spreads in 70% formamide, 2× SSC (pH 7.0) at 75°C for 4 min, dehydrated them again in an ethanol series, and allowed them to air dry. After warming the metaphase spreads to 37°C, we applied the denatured probe to the metaphases. We hybridized the probes under a coverslip in a humidified chamber at 37°C for 40 h. We then removed the coverslip and excess hybridization solution by immersing the slide in PN buffer and washed the arrays at 45°C in the following order: 50% formamide, 2× SSC for 20 min, 2× SSC for 10 min, 0.1× SSC for 10 min, PN buffer for 10 min, and 0.1× SSC for 10 min. After removing excess liquid we coverslip mounted the slide in Vectashield (Vector Laboratories) and used a CCD-based imaging system to capture three color images.
Preparation and sequencing of small genomic subclones of RP23-263M19
We fragmented RP23-263M19 DNA by sonication, blunt-end-cloned the DNA into pBluescript II KS+ linearized by SmaI digestion, and transfected the plasmids into competent Escherichia coli (TOP10; Invitrogen). We picked 189 colonies into microtiter plates and then prepared colony blots by seeding bacteria from the microtiter plates onto positively charged membranes. In order to investigate which sequences in RP23-263M19 were over-represented in M. musculus compared to M. spretus, we lysed the colonies, fixed the DNA, and hybridized the membrane with a mixture of biotin labeled M. musculus genomic DNA and excess unlabeled M. spretus genomic DNA to suppress homologous sequences. After chemiluminescent detection of the hybridized labeled DNA using streptavidin coupled to alkaline phosphatase, we picked 10 clones showing strong hybridization signals and sequenced these plasmids unidirectionally. We performed sequence alignment using CLUSTAL W.
Note added in proof
While this paper was under review, Li et al. (2004) reported similar findings using BAC arrays comprised of only clones mapped on the mouse physical map as reported in 2002 (Gregory et al. 2002). A comparison of the 74 polymorphic loci reported here with a list of 346 clones from the Li et al. publication revealed no obvious overlap between the two sets of clones. We suggest two possible reasons for the lack of concordance. First, different selection criteria may have been used, but we cannot determine whether this is true, because the ratio data and cutoffs for calling polymorphic clones were not published by Li et al. (2004). Second, clones for our arrays were selected with STS markers prior to extensive mouse genome sequencing. By end sequence and STS content, several of our BACs identifying polymorphisms were mapped to sequence contigs that are yet to be included in the assembly (e.g., RP23-199A7 and RP23-104D1; Supplemental Table A and Supplemental Fig. B). Some others did not map to any sites on the current assembly. In addition, FISH mapping of our polymorphic clones revealed that several map to multiple sites in the mouse genome including pericentromeric regions. Arguably, clones harboring copy number polymorphisms might be more likely to pose problems for sequence assembly, but will in time be placed on the genome as the sequence nears completion. Nevertheless, taken together this publication and the Li et al. (2004) report indicate an abundance of copy number polymorphisms in mouse genomes.
Acknowledgments
We thank members of the Albertson and Pinkel laboratories, who participated in the assembly and printing of the mouse BAC arrays; Gillian Hirst for providing mouse embryo fibroblasts used for preparation of metaphase spreads; Lawrence Hon for help with BAC end-sequence analysis; and Karen Kimura and the UCSF Cancer Center Informatics Core for maintenance of the microarray database. We also thank Susan Deveau of the Jackson Laboratory for her assistance in obtaining DNA from mouse strains. Gregg Magrane and the UCSF Immunohistochemistry and Molecular Pathology Core assisted with FISH mapping of some of the array clones. This work was supported by NIH/NCI grants CA84118 and P30 CA16056 and a Supplement P02137165CN from the NCI Mouse Models of Human Cancer Consortium.
Footnotes
Article and publication are at http://www.genome.org/cgi/doi/10.1101/gr.2902505.
[Supplemental material is available online at www.genome.org.]
References
- Agulnik, S., Plass, C., Traut, W., and Winking, H. 1993. Evolution of a long-range repeat family in chromosome 1 of the genus Mus. Mamm. Genome 4: 704-710. [DOI] [PubMed] [Google Scholar]
- Amadou, C., Younger, R.M., Sims, S., Matthews, L.H., Rogers, J., Kumanovics, A., Ziegler, A., Beck, S., and Lindahl, K.F. 2003. Co-duplication of olfactory receptor and MHC class I genes in the mouse major histocompatibility complex. Hum. Mol. Genet. 12: 3025-3040. [DOI] [PubMed] [Google Scholar]
- Bailey, J.A., Yavor, A.M., Viggiano, L., Misceo, D., Horvath, J.E., Archidiacono, N., Schwartz, S., Rocchi, M., and Eichler, E.E. 2002. Human-specific duplication and mosaic transcripts: The recent paralogous structure of chromosome 22. Am. J. Hum. Genet. 70: 83-100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beck, J.A., Lloyd, S., Hafezparast, M., Lennon-Pierce, M., Eppig, J.T., Festing, M.F., and Fisher, E.M. 2000. Genealogies of mouse inbred strains. Nat. Genet. 24: 23-25. [DOI] [PubMed] [Google Scholar]
- Broman, K.W. and Speed, T.P. 2002a. Discussion on the meeting on `Statistical modelling of genetic data.' J. R. Stat. Soc. Ser. B 64: 737-775. [Google Scholar]
- ____. 2002b. A model selection approach for the identification of quantitative trait loci in experimental crosses. J. R. Stat. Soc. Ser. B 64: 641-656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai, W.W., Reneker, J., Chow, C.W., Vaishnav, M., and Bradley, A. 1998. An anchored framework BAC map of mouse chromosome 11 assembled using multiplex oligonucleotide hybridization. Genomics 54: 387-397. [DOI] [PubMed] [Google Scholar]
- Cheung, V.G., Nowak, N., Jang, W., Kirsch, I.R., Zhao, S., Chen, X.N., Furey, T.S., Kim, U.J., Kuo, W.L., Olivier, M., et al. 2001. Integration of cytogenetic landmarks into the draft sequence of the human genome. Nature 409: 953-958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung, J., Wilson, M.D., Zhang, J., Khaja, R., MacDonald, J.R., Heng, H.H., Koop, B.F., and Scherer, S.W. 2003. Recent segmental and gene duplications in the mouse genome. Genome Biol. 4: R47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dev, V.G., Tantravahi, R., Miller, D.A., and Miller, O.J. 1977. Nucleolus organizers in Mus musculus subspecies and in the RAG mouse cell line. Genetics 86: 389-398. [PMC free article] [PubMed] [Google Scholar]
- Dunham, M.J., Badrane, H., Ferea, T., Adams, J., Brown, P.O., Rosenzweig, F., and Botstein, D. 2002. Characteristic genome rearrangements in experimental evolution of Saccharomyces cerevisiae. Proc. Natl. Acad. Sci. 99: 16144-16149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eicher, E.M. and Shown, E.P. 1993. Molecular markers that define the distal ends of mouse autosomes 4, 13, and 19 and the sex chromosomes. Mamm. Genome 4: 226-229. [DOI] [PubMed] [Google Scholar]
- Eichler, E.E., Lu, F., Shen, Y., Antonacci, R., Jurecic, V., Doggett, N.A., Moyzis, R.K., Baldini, A., Gibbs, R.A., and Nelson, D.L. 1996. Duplication of a gene-rich cluster between 16p11.1 and Xq28: A novel pericentromeric-directed mechanism for paralogous genome evolution. Hum. Mol. Genet. 5: 899-912. [DOI] [PubMed] [Google Scholar]
- Elsevier, S.M. and Ruddle, F.H. 1975. Location of genes coding for 18S and 28S ribosomal RNA within the genome of Mus musculus. Chromosoma 52: 219-228. [DOI] [PubMed] [Google Scholar]
- Emanuel, B.S. and Shaikh, T.H. 2001. Segmental duplications: An `expanding' role in genomic instability and disease. Nat. Rev. Genet. 2: 791-800. [DOI] [PubMed] [Google Scholar]
- Ewart-Toland, A., Briassouli, P., de Koning, J.P., Mao, J.H., Yuan, J., Chan, F., MacCarthy-Morrogh, L., Ponder, B.A., Nagase, H., Burn, J., et al. 2003. Identification of Stk6/STK15 as a candidate low-penetrance tumor-susceptibility gene in mouse and human. Nat. Genet. 34: 403-412. [DOI] [PubMed] [Google Scholar]
- Fridlyand, J., Snijders, A.M., Pinkel, D., Albertson, D.G., and Jain, A.N. 2004. Hidden Markov models approach to the analysis of array CGH data. J. Mulitvariate Anal. 90: 132-153. [Google Scholar]
- Gregory, S.G., Sekhon, M., Schein, J., Zhao, S., Osoegawa, K., Scott, C.E., Evans, R.S., Burridge, P.W., Cox, T.V., Fox, C.A., et al. 2002. A physical map of the mouse genome. Nature 418: 743-750. [DOI] [PubMed] [Google Scholar]
- Guenet, J.L. and Bonhomme, F. 2003. Wild mice: An ever-increasing contribution to a popular mammalian model. Trends Genet. 19: 24-31. [DOI] [PubMed] [Google Scholar]
- Henderson, A.S., Eicher, E.M., Yu, M.T., and Atwood, K.C. 1974. The chromosomal location of ribosomal DNA in the mouse. Chromosoma 49: 155-160. [DOI] [PubMed] [Google Scholar]
- Hodgson, G., Hager, J.H., Volik, S., Hariono, S., Wernick, M., Moore, D., Nowak, N., Albertson, D.G., Pinkel, D., Collins, C., et al. 2001. Genome scanning with array CGH delineates regional alterations in mouse islet carcinomas. Nat. Genet. 29: 459-464. [DOI] [PubMed] [Google Scholar]
- Horvath, J.E., Bailey, J.A., Locke, D.P., and Eichler, E.E. 2001. Lessons from the human genome: Transitions between euchromatin and heterochromatin. Hum. Mol. Genet. 10: 2215-2223. [DOI] [PubMed] [Google Scholar]
- Horz, W. and Altenburger, W. 1981. Nucleotide sequence of mouse satellite DNA. Nucleic Acids Res. 9: 683-696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Infante, J.J., Dombek, K.M., Rebordinos, L., Cantoral, J.M., and Young, E.T. 2003. Genome-wide amplifications caused by chromosomal rearrangements play a major role in the adaptive evolution of natural yeast. Genetics 165: 1745-1759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson, M.S., Rocchi, M., Thompson, G., Hearn, T., Crosier, M., Guy, J., Kirk, D., Mulligan, L., Ricco, A., Piccininni, S., et al. 1999. Sequences flanking the centromere of human chromosome 10 are a complex patchwork of arm-specific sequences, stable duplications and unstable sequences with homologies to telomeric and other centromeric locations. Hum. Mol. Genet. 8: 205-215. [DOI] [PubMed] [Google Scholar]
- Jain, A.N., Tokuyasu, T.A., Snijders, A.M., Segraves, R., Albertson, D.G., and Pinkel, D. 2002. Fully automatic quantification of microarray image data. Genome Res. 12: 325-332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji, Y., Eichler, E.E., Schwartz, S., and Nicholls, R.D. 2000. Structure of chromosomal duplicons and their role in mediating human genomic disorders. Genome Res. 10: 597-610. [DOI] [PubMed] [Google Scholar]
- Kelly, R., Gibbs, M., Collick, A., and Jeffreys, A.J. 1991. Spontaneous mutation at the hypervariable mouse minisatellite locus Ms6-hm: Flanking DNA sequence and analysis of germline and early somatic mutation events. Proc. R Soc. Lond. B Biol. Sci. 245: 235-245. [DOI] [PubMed] [Google Scholar]
- Kipling, D., Salido, E.C., Shapiro, L.J., and Cooke, H.J. 1996. High frequency de novo alterations in the long-range genomic structure of the mouse pseudoautosomal region. Nat. Genet. 13: 78-80. [DOI] [PubMed] [Google Scholar]
- Kurihara, Y., Suh, D.S., Suzuki, H., and Moriwaki, K. 1994. Chromosomal locations of Ag-NORs and clusters of ribosomal DNA in laboratory strains of mice. Mamm. Genome 5: 225-228. [DOI] [PubMed] [Google Scholar]
- Li, J., Jiang, T., Mao, J.H., Balmain, A., Peterson, L., Harris, C., Rao, P.H., Havlak, P., Gibbs, R., and Cai, W.W. 2004. Genomic segmental polymorphisms in inbred mouse strains. Nat. Genet. 36: 952-954. [DOI] [PubMed] [Google Scholar]
- Locke, D.P., Segraves, R., Carbone, L., Archidiacono, N., Albertson, D.G., Pinkel, D., and Eichler, E.E. 2003. Large-scale variation among human and great ape genomes determined by array comparative genomic hybridization. Genome Res. 13: 347-357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Locke, D.P., Segraves, R., Nicholls, R.D., Schwartz, S., Pinkel, D., Albertson, D.G., and Eichler, E.E. 2004. BAC microarray analysis of 15q11-q13 rearrangements and the impact of segmental duplications. J. Med. Genet. 41: 175-182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagase, H., Bryson, S., Cordell, H., Kemp, C.J., Fee, F., and Balmain, A. 1995. Distinct genetic loci control development of benign and malignant skin tumours in mice. Nat. Genet. 10: 424-429. [DOI] [PubMed] [Google Scholar]
- Osoegawa, K., Tateno, M., Woon, P.Y., Frengen, E., Mammoser, A.G., Catanese, J.J., Hayashizaki, Y., and de Jong, P.J. 2000. Bacterial artificial chromosome libraries for mouse sequencing and functional analysis. Genome Res. 10: 116-128. [PMC free article] [PubMed] [Google Scholar]
- Pinkel, D., Segraves, R., Sudar, D., Clark, S., Poole, I., Kowbel, D., Collins, C., Kuo, W.L., Chen, C., Zhai, Y., et al. 1998. High resolution analysis of DNA copy number variation using comparative genomic hybridization to microarrays. Nat. Genet. 20: 207-211. [DOI] [PubMed] [Google Scholar]
- Schwarz, G. 1978. Estimating the dimension of a model. Annals of Statistics 6: 461-464. [Google Scholar]
- Shaw, C.J. and Lupski, J.R. 2004. Implications of human genome architecture for rearrangement-based disorders: The genomic basis of disease. Hum. Mol. Genet. 13 Suppl 1: R57-R64. [DOI] [PubMed] [Google Scholar]
- Sidman, C.L. and Shaffer, D.J. 1994. Large-scale genomic comparison using two-dimensional DNA gels. Genomics 23: 15-22. [DOI] [PubMed] [Google Scholar]
- Snedecor, G.W. and Cochran, W.G. 1989. Statistical methods. Iowa State University Press, Ames, IA.
- Snijders, A.M., Nowak, N., Segraves, R., Blackwood, S., Brown, N., Conroy, J., Hamilton, G., Hindle, A.K., Huey, B., Kimura, K., et al. 2001. Assembly of microarrays for genome-wide measurement of DNA copy number. Nat. Genet. 29: 263-264. [DOI] [PubMed] [Google Scholar]
- Staelens, J., Wielockx, B., Puimege, L., Van Roy, F., Guenet, J.L., and Libert, C. 2002. Hyporesponsiveness of SPRET/Ei mice to lethal shock induced by tumor necrosis factor and implications for a TNF-based antitumor therapy. Proc. Natl. Acad. Sci. 99: 9340-9345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas, J.W., Schueler, M.G., Summers, T.J., Blakesley, R.W., McDowell, J.C., Thomas, P.J., Idol, J.R., Maduro, V.V., Lee-Lin, S.Q., Touchman, J.W., et al. 2003. Pericentromeric duplications in the laboratory mouse. Genome Res. 13: 55-63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuzun, E., Bailey, J.A., and Eichler, E.E. 2004. Recent segmental duplications in the working draft assembly of the brown Norway rat. Genome Res. 14: 493-506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wade, C.M., Kulbokas III, E.J., Kirby, A.W., Zody, M.C., Mullikin, J.C., Lander, E.S., Lindblad-Toh, K., and Daly, M.J. 2002. The mosaic structure of variation in the laboratory mouse genome. Nature 420: 574-578. [DOI] [PubMed] [Google Scholar]
- Wiltshire, T., Pletcher, M.T., Batalov, S., Barnes, S.W., Tarantino, L.M., Cooke, M.P., Wu, H., Smylie, K., Santrosyan, A., Copeland, N.G., et al. 2003. Genome-wide single-nucleotide polymorphism analysis defines haplotype patterns in mouse. Proc. Natl. Acad. Sci. 100: 3380-3385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan, B., Shum-Siu, A., Lentsch, E.M., Hu, L.H., and Hendler, F.J. 1996. Frequent DNA polymorphisms exist in inbred CBA/J and C3H/HeN mice. Genomics 38: 58-71. [DOI] [PubMed] [Google Scholar]
WEB SITE REFERENCES
- http://genome.ucsc.edu; October 2003 freeze of the mouse genome sequence.
- http://microarrays.roswellpark.org; set of BAC clones used.