

Abstract
The key components of a transcriptional regulatory network are the connections between trans-acting transcription factors and cis-acting DNA-binding sites. In spite of several decades of intense research, only a fraction of the estimated ∼300 transcription factors in Escherichia coli have been linked to some of their binding sites in the genome. In this paper, we present a computational method to connect novel transcription factors and DNA motifs in E. coli. Our method uses three types of mutually independent information, two of which are gleaned by comparative analysis of multiple genomes and the third one derived from similarities of transcription-factor-DNA-binding-site interactions. The different types of information are combined to calculate the probability of a given transcription-factor-DNA-motif pair being a true pair. Tested on a study set of transcription factors and their DNA motifs, our method has a prediction accuracy of 59% for the top predictions and 85% for the top three predictions. When applied to 99 novel transcription factors and 70 novel DNA motifs, our method predicted 64 transcription-factor-DNA-motif pairs. Supporting evidence for some of the predicted pairs is presented. Functional annotations are made for 23 novel transcription factors based on the predicted transcription-factor-DNA-motif connections.
To a large extent, gene expression programs depend on the recognition of specific promoter sequences (transcription-factor-binding sites) by transcriptional regulatory proteins (transcription factors, TFs). Studies designed to identify these binding sequences and regulatory proteins, and determine the correct connections between them, provide the data necessary to build a model of the transcriptional regulatory network of an organism (Shen-Orr et al. 2002; Bar-Joseph et al. 2003; Liao et al. 2003; Kao et al. 2004). While traditional experimental methods (e.g., electrophoretic mobility shift and nuclease protection assays) have identified only a fraction of the transcription regulatory interactions of Escherichia coli (Martinez-Antonio and Collado-Vides 2003), modern high-throughput methods such as chromatin immunoprecipitation coupled with promoter microarrays (ChIP-chip experiments) (Ren et al. 2000) have the potential to rapidly associate many TFs with their cognate binding sites in the genome, and thus provide the genome-scale interaction data necessary to model the E. coli regulatory network. However, in order for ChIP-chip to work, the growth conditions under which the TFs are active need to be known before experiments are conducted, and determining these growth conditions will undoubtedly be a challenging task. Furthermore, cloning a large number of epitope-tagged TFs (including many novel factors) remains a labor-intensive process that would likely face unforeseeable technical difficulties. Another approach was described by Segal et al. (2003), who addressed the problem of connecting a group of TFs with their regulated genes in yeast using only microarray expression data. Their method relies on the assumption that TFs are themselves transcriptionally regulated such that their expression profiles correlate with their target genes. This assumption probably holds true for many TFs, but is violated in cases where other types of regulation are involved, such as post-translational modification and binding of small molecule effectors. In addition, the expression level of some transcription factors may be too low to be reliably detected with current microarray technology.
Fueled by the ever-increasing number of completely sequenced genomes, comparative genomics has proven to be a powerful tool to address a large variety of biological questions (Marcotte et al. 1999; Overbeek et al. 1999; Pellegrini et al. 1999; Korf et al. 2001; McCue et al. 2001, 2002; Rivas and Eddy 2001; Blanchette and Tompa 2002; Rajewsky et al. 2002; Ji et al. 2004). In particular, computational methods such as phylogenetic footprinting have been applied to the E. coli genome, allowing the discovery of many novel TF-binding sites (McCue et al. 2001, 2002; Rajewsky et al. 2002). Furthermore, clustering of phylogenetic footprints has yielded inferences on the sets of coregulated genes (regulons), generating DNA motif models for unknown TFs as well as many previously characterized TFs (van Nimwegen et al. 2002; Qin et al. 2003). Complete genomic sequence has also allowed the prediction of the full repertoire of TFs in E. coli using standard computational sequence analysis techniques (Perez-Rueda and Collado-Vides 2000; Babu and Teichmann 2003). Thus, computational predictions have already provided substantial data sets for each of the two necessary components of a regulatory network for E. coli: the binding sites and the TFs. In this paper, we describe an in silico approach that harnesses the power of comparative genomics to make connections between TFs with their cognate binding sites in the E. coli genome.
We hypothesize that information concerning the connection of a TF to its cognate DNA motif is carried in the genomes and can be extracted by comparing multiple genomic sequences and available structural data. Specifically, our method takes advantage of three types of information in order to assign a DNA-binding motif to a given TF: (1) a distance constraint between a TF and its closest binding site in the genome (Dmin information), (2) the phylogenetic correlation between TFs and their regulated genes (PC information), and (3) a binding specificity constraint for TFs having structurally similar DNA-binding domains (FMC information). For a given TF and DNA motif, the three types of information are combined to calculate the probability that such a TF ↔ DNA-motif pair is a true pair. We demonstrate the method using a study set of known TFs and their cognate DNA-binding motifs, and further apply the method to predict connections between novel TFs and DNA motifs from E. coli. In addition, for functionally unannotated TFs, we are able to infer their target cellular processes based on the overrepresented functional categories of their regulons. Our results demonstrate the value of combining heterogeneous data types to solve a challenging computational problem.
Results
Distance constraint between a TF and its closest binding site in the genome, Dmin information
Bacterial TFs are often autoregulatory; 55% (58 of 105) of all known E. coli TFs in the database RegulonDB are autoregulated (Martinez-Antonio and Collado-Vides 2003). Besides autoregulation, it has been noticed in many cases (Dickson et al. 1975; Eichler et al. 1996; Palacios and Escalante-Semerena 2000; Torres et al. 2003) that TFs and the genes they regulate are near each other in the genome. Both phenomena imply a distance constraint between the TF and its closest binding site in the genome. In the first case, the distance constraint is due to the requirement for autoregulation. In the second case, the distance constraint may be the result of horizontal gene transfer because it would not be advantageous to acquire a new operon conferring a new function if the operon is not expressed correctly (Lawrence 1999; Martinez-Antonio and Collado-Vides 2003). To use this distance constraint information, we introduced the quantity Dmin. Two types of Dmins can be considered: Dmin_self is the distance between a TF gene and its closest binding site in the genome. Dmin_cross is the distance between a TF gene and the closest binding site for a different TF. Owing to the existence of a distance constraint between a TF and its own closest site in the genome, we expect Dmin_self to be smaller than Dmin_cross for most other TFs. In Figure 1A, we plot the distributions of Dmin_self and Dmin_cross for the study set of 35 TFs. The mean of the Dmin_self distribution is significantly smaller than the mean of the Dmin_cross distribution (p =4.8 × 10-7). Thus, we can use distance constraint information to connect novel TFs with their cognate DNA motifs. For any pair of transcription factor TFj and DNA motif Mi, we can calculate the probability that they are a true pair given their Dmin value 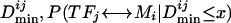 (Supplemental Fig. 4A).
(Supplemental Fig. 4A).
Figure 1.

Three types of information. (A) Two types of normalized D distributions. Dmin_self: the distance between a TF gene and its closest binding site in the genome. Dmin_cross: the distance between a TF gene and the closest site for a different TF. (B) Two types of phylogenetic correlation (PC) distributions. PC_true: PC for true TF-DNA-motif pairs. PC_false: PC for false TF-DNA-motif pairs. (C) Distribution of average similarity scores for motifs from the same family and from different families. Motifs were aligned using an ungapped Smith-Waterman algorithm and scored using the ALLR statistic.
Phylogenetic correlation between TFs and their regulated genes (regulons), PC information
TFs and their regulated genes tend to evolve concurrently (Mironov et al. 1999; Gelfand et al. 2000; Tan et al. 2001). Thus, we can connect TFs and DNA motifs through correlation between their occurrences in a comparative analysis of multiple species. The “regulon” refers to the set of genes directly regulated by a common TF. If the TF is not known, such as in our problem, the “regulon” can also be defined as the set of genes controlled by a common DNA motif belonging to the as yet unknown TF. For a given DNA motif Mi and a species Gk, we can build a  vector (see Methods) to represent the putative regulon controlled by the DNA motif Mi in species Gk. Each element of
vector (see Methods) to represent the putative regulon controlled by the DNA motif Mi in species Gk. Each element of  denotes the probability that a gene is controlled by Mi. An alternative to
denotes the probability that a gene is controlled by Mi. An alternative to 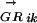 is a binary vector whose elements indicate whether or not orthologs in species Gk are controlled by Mi by imposing a binding site cutoff. However, a binary vector contains no information about the number or quality of the binding sites in front of a gene (regulation strength by Mi), whereas the
is a binary vector whose elements indicate whether or not orthologs in species Gk are controlled by Mi by imposing a binding site cutoff. However, a binary vector contains no information about the number or quality of the binding sites in front of a gene (regulation strength by Mi), whereas the 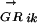 vector does. In pilot studies, we found that
vector does. In pilot studies, we found that 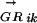 gave a better performance than binary vectors (data not shown). We then computed the Pearson's correlation coefficient between the E. coli
gave a better performance than binary vectors (data not shown). We then computed the Pearson's correlation coefficient between the E. coli 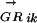 vector (k = E. coli) and the
vector (k = E. coli) and the 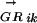 vector for each of the other species considered. This measures the degree of conservation of the E. coli regulon determined by Mi in species Gk. When multiple species are considered, the
vector for each of the other species considered. This measures the degree of conservation of the E. coli regulon determined by Mi in species Gk. When multiple species are considered, the 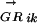 vector correlation coefficients form a new vector,
vector correlation coefficients form a new vector,  , which described the evolutionary changes of the E. coliMi regulon in other species. We then compared the
, which described the evolutionary changes of the E. coliMi regulon in other species. We then compared the 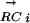 vector with the
vector with the  vector that describes the phylogenetic profile of TFj in the various genomes. We expect that there will be no strong correlation in those species that have lost TFj. For example, using the TF trpR, Table 1 shows the vector for its conservation
vector that describes the phylogenetic profile of TFj in the various genomes. We expect that there will be no strong correlation in those species that have lost TFj. For example, using the TF trpR, Table 1 shows the vector for its conservation 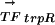 and the vector for its regulon conservation
and the vector for its regulon conservation 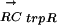 in 13 γ-proteobacteria. As can be seen, only those species that have an ortholog to E. coli trpR (with a 1 in the TF column) also have a large positive value in the
in 13 γ-proteobacteria. As can be seen, only those species that have an ortholog to E. coli trpR (with a 1 in the TF column) also have a large positive value in the 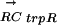 vector, meaning conservation of the regulon. We can use this phylogenetic correlation information to connect novel TFs with their cognate DNA motifs based on the difference in PC values between true and false TF-DNA-motif pairs (p = 2.12 × 10-4) (Fig. 1B). For any pair of transcription factor TFj and DNA motif Mi, we can calculate the probability that they are a true pair given their PCij value
vector, meaning conservation of the regulon. We can use this phylogenetic correlation information to connect novel TFs with their cognate DNA motifs based on the difference in PC values between true and false TF-DNA-motif pairs (p = 2.12 × 10-4) (Fig. 1B). For any pair of transcription factor TFj and DNA motif Mi, we can calculate the probability that they are a true pair given their PCij value 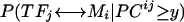 (Supplemental Fig. 4B).
(Supplemental Fig. 4B).
Table 1.
An example of phylogenetic correlation between the transcription factor trpR and its regulon
| Species | TF | Regulon conservation |
|---|---|---|
| E. coli | 1 | 1.0000 |
| H. influenzae | 1 | 0.3953 |
| P. multocida | 1 | 0.5237 |
| P. aeruginosa | 0 | −0.0399 |
| P. putida | 0 | 0.0297 |
| P. syringae | 0 | 0.0507 |
| S. oneidensis | 0 | 0.0866 |
| S. typhi | 1 | 0.9721 |
| V. cholerae | 1 | 0.9664 |
| V. parahaemolyticus | 1 | 0.3588 |
| V. vulnificus | 1 | 0.8106 |
| X. campestris | 0 | −0.0495 |
| Y. pestis | 1 | 0.2105 |
“1” indicates the existence of a trpR ortholog in the given species, “0” otherwise. The degree of regulon conservation is calculated using E. coli as the reference species (see Methods for calculation). The phylogenetic correlation is PCtrpR = 0.8016.
Binding specificity constraint for TFs having structurally similar DNA-binding domains, FMC information
TFs that are more similar to one another are expected to bind to sites that are more similar to each other than to dissimilar pairs. For instance, the DNA motifs of many TF family members are often similar (Luscombe et al. 2000; Rigali et al. 2002; Sandelin and Wasserman 2004). Using the study set of 35 TFs, we examined this issue in more detail by studying the relationship of the DNA-binding domain (DBD) of TFs within a family and similarities in their DNA motifs. The database SUPERFAMILY (Madera et al. 2004) contains hidden Markov models (HMMs) for DBDs belonging to different structural families. We used the database to classify the 35 TFs into seven structural families based on the similarity between our query DBDs and the DBD HMMs in the database. For these seven TF families, we calculated the average pairwise similarities of their corresponding DNA motifs. Figure 1C shows the distributions of average pairwise similarity scores for motifs belonging to the same family and motifs from different families. The mean intrafamily pairwise similarity score is significantly larger than the mean interfamily pairwise similarity score (p = 0.003). This result further confirmed the hypothesis that TFs with similar DBDs tend to have similar DNA-binding motifs. To use this binding constraint information, we introduced the quantity FMC (familial motif conservation), which measures the average similarity between a query DNA motif and a family of DNA motifs. FMC serves as an estimate of the membership of a query motif to a family of motifs. For any pair of transcription factor TFj and DNA motif Mi, we can calculate the FMC between Mi and the DNA motif family of which TFj's motif is a member. We can then calculate the probability that they are a true pair given their FMC value FMCij, P(TFj ↔ Mi| FMCij ≥ z (Supplemental Fig. 4C).
Making TF → DNA-motif connections by combining three types of information
Given a set of TFs and DNA motifs, we calculate the probability that a TF↔DNA-motif pair is true for all possible pairs. We can calculate three types of probabilities for each pair based on the three types of information described before: 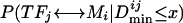 P(TFj ↔ Mi|PCij ≥ y), and P(TFj ↔ Mi|FMCij ≥ z). Since the different types of information are independent of each other, the joint probability
P(TFj ↔ Mi|PCij ≥ y), and P(TFj ↔ Mi|FMCij ≥ z). Since the different types of information are independent of each other, the joint probability 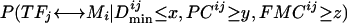 , PCij ≥ y, FMCij ≥ z) considering all types of information can be calculated using the three conditional probabilities mentioned above (see Methods). By combining different information, the probabilities of true connections could be raised much higher above the background probabilities of false connections. This increase in signal-to-noise ratio can be illustrated using the P_true/P_avg ratio (Table 2), the probability ratios between the true connections (P_true) and the average of all connections for a given TF (P_avg). On average, the P_true/P_avg ratios were 2.18, 4.03, and 6.48 after one, two, and three types of information were used, respectively.
, PCij ≥ y, FMCij ≥ z) considering all types of information can be calculated using the three conditional probabilities mentioned above (see Methods). By combining different information, the probabilities of true connections could be raised much higher above the background probabilities of false connections. This increase in signal-to-noise ratio can be illustrated using the P_true/P_avg ratio (Table 2), the probability ratios between the true connections (P_true) and the average of all connections for a given TF (P_avg). On average, the P_true/P_avg ratios were 2.18, 4.03, and 6.48 after one, two, and three types of information were used, respectively.
Table 2.
Probability increase for true TF-DNA motif pairs using different combinations of information
| Type of information | Average P_true/P_avg |
|---|---|
| No information | 1 |
| FMC | 2.48 |
| PC | 1.33 |
| Dmin | 2.74 |
| FMC-Dmin | 5.38 |
| FMC-PC | 3.21 |
| PC-Dmin | 3.49 |
| FMC-PC-Dmin | 6.48 |
Probabilities calculated using the study set of TFs and their DNA motifs. P_true: the probability of the true TF-DNA motif pair. P_avg: the average probabilities of all TF-DNA motif pairs for a given TF.
Assessment of the algorithm using the study set
To test the performance of our algorithm, we assembled a set of 35 well-characterized transcription factors and their corresponding DNA-binding motifs. These 35 TFs regulate a large variety of cellular processes in response to many different stimuli. Based on the number of binding sites, information content, and E-value of the motif models (Supplemental Fig. 5), these 35 DNA motifs are fairly representative of all DNA motifs discovered in E. coli so far. The study set of 35 TFs and their DNA motifs was used to evaluate the prediction accuracy of our algorithm in three different ways. First, we use only the study set of 35 motifs and TFs and we determine how accurately we make correct assignments using different combinations of information. In Figure 2, the cumulative frequency of the true connection ranks was plotted. As can be seen, combining different information improves prediction accuracy. By combining the three types of information, 63% of the true connections were ranked first out of the 35 possible connections for each TF, and 85% of the true connections were ranked in the top three. In comparison, only 3% of the true connections would be ranked first and 9% ranked in the top three if we didn't have any information and made our predictions by random guessing.
Figure 2.

Prediction accuracy on the study set using different combinations of information. Size of search space: 35 TFs × 35 motifs. The connections are ranked according to P(TF ↔ M).
In a second test we evaluated our prediction accuracy on the study set after combining the study set with the set of novel TFs and DNA motifs. There are 134 TFs and 105 DNA motifs in this combined set. Now, the probability of success by random guessing is three times smaller than using the study set alone. As illustrated in Figure 3, the prediction accuracy only dropped slightly (59% of the true connections were ranked first, and 81% of the true connections were ranked in the top three), suggesting that our algorithm still works well in a larger search space.
Figure 3.

Prediction accuracy on the study set using all three types of information. Size of search space: 134 TFs × 105 motifs. The connections are ranked according to P(TF ↔ M).
In the third test we did a fivefold cross-validation with the combined study set and novel set, as described for the previous test. For each cross-validation, 28 TF and DNA motif pairs from the study set were used to derive the probability distributions, and the remaining seven TF and DNA motif pairs were used to assess the accuracy. As described above, we combined the test set of seven pairs with the novel set of TFs and DNA motifs when making predictions. The prediction accuracy measurement is the same as described in Figure 1 (fractions of true pairs ranked in the top three). The average prediction accuracy based on the five rounds of cross-validation is similar to that shown in Figure 1: 64.7% of the true pairs ranked as number one, 15.2% of the true pairs ranked as number two, and 10.4% of the true pairs ranked as number three. This demonstrates that the accuracy remains high for known pairs that were not used to determine the three probability distributions.
Predicted connections between novel TFs and DNA motifs
For all 6930 TF-DNA-motif pairs in the novel set, we calculated their probabilities of being a true pair using all three types of information. From these posterior probabilities, we can make two types of predictions. The first type of prediction is the top three DNA motifs for each TF ranked by their probabilities, the top-three-pick (TTP) method. However, since there are more TFs than DNA motifs, this forces DNA motifs to be associated with TFs whose cognate motifs are not present in the set of motifs considered. This also allows two or more TFs to share motifs. Although it is possible for paralogous TFs to have highly similar DBDs and DNA motifs, we expect such cases to be rare. To resolve those issues, we resort to the maximal weighted matching algorithm (MWM) to make a set of unique predictions (Cary and Stormo 1995). Using the table of probabilities as input, the MWM algorithm predicted 64 unique TF-DNA-motif pairs, leaving six motifs with no associated TFs (Supplemental Table 7). Most of the predictions by MWM are ranked in the top three out of the 70 possible motifs that can be associated with each TF, indicating a good overlap between the MWM predictions and the TTP predictions. Specifically, among the MWM predictions, 28 TFs have a predicted DNA motif ranked as number one, 11 TFs have a motif ranked as number two, and 13 TFs have a motif ranked as number three. In total, 81% of the 64 TFs have a predicted DNA motif ranked in the top three.
Assessment of the algorithm on the novel predictions
The three TFs in the novel set with fewer than five known binding sites (ntrC, pdhR, xylR) provided a means to assess the performance of our algorithm. Both pdhR and xylR were correctly associated with their known DNA motifs, but ntrC was left unassociated by the MWM algorithm. However, ntrC's motif was ranked in the top three by the TTP method. This result is very interesting because it provides an independent validation for our algorithm. We also searched the literature for additional supporting evidence for our predictions. Of the 64 predicted TFs, 15 have known target genes discovered via genetic analysis, but no binding sites have been reported. If our method makes correct predictions, there should be high-scoring sites for the predicted motif in the regulatory region for the known target genes. We searched the regulatory regions of these genes for binding sites of the predicted DNA motif for each TF. For nine out of these 15 cases, a binding site (scored above the site cutoff) was identified in the regulatory region of the target gene (Table 3), eight predicted by both the TTP and MWM algorithm and one by the TTP algorithm alone. For all reported sites in Table 3, the probability of occurring by chance is significantly smaller than 0.05. Thus, it is likely that those nine DNA motifs are truly recognized by the predicted TFs.
Table 3.
Predicted TF-DNA motif pairs with known target genes.
All pairs were predicted by both MWM and TTP except for dsdC and ntrC (labeled with a star) which are predicted only by TTP. P-value of a site having the observed score was calculated as the tail probability of a normal distribution for all possible scores in the genome.

Cellular target processes of novel transcription factors
Once a TF is connected to a DNA motif, we can infer the target cellular process of the TF by studying the enrichment of functional categories of the regulated genes. We made use of 21 COG (Tatusov et al. 1997) functional categories assigned to the 4279 genes in the E. coli K-12 genome (Blattner et al. 1997). The 32 DNA motifs (nine known, 23 novel) whose regulons contain at least one overrepresented COG functional category are listed in Table 4 along with their corresponding TFs. Of the 13 known DNA motifs in the McCue et al. (2002) prediction set, nine have regulons with enriched COG functional categories. All enriched functional categories are consistent with the known cellular process controlled by the associated TFs. We also associated target cellular process(es) for 10 TFs whose DNA motifs were unknown before this study but some knowledge about their target genes were known. Some of the associated processes are consistent with the current knowledge about the TFs. For instance, the TF baeR regulates several transmembrane proteins (mdtABC) that form a hetero-multimeric drug efflux pump (Nagakubo et al. 2002). This is consistent with the target cellular process predicted by functional category enrichment: cell envelope biogenesis, outer membrane. Another example is the TF ascG, which regulates genes involved in carbon source metabolism (Postma et al. 1993). The remaining 13 predictions are for putative TFs without any functional annotation. Thus, our study provided the first description of the cellular processes that these putative TFs may regulate.
Table 4.
Enrichment of regulons for genes within COG functional categories
| DNA motif ID | TF | COG functional category | −log10 (P-value) |
|---|---|---|---|
| Known TF-DNA motif pairs | |||
| c1373_e | crp | Carbohydrate transport and metabolism; energy production and conversion | 10.03, 3.38 |
| c2225_o | fadR | Lipid metabolism | 5.03 |
| c594_e | fnr | Energy production and conversion | 6.06 |
| c577_e | fruR | Carbohydrate transport and metabolism | 8.06 |
| c583_e | lexA | DNA replication, recombination and repair; cell division and chromosome partitioning | 10.54, 4.53 |
| c590_e | metJ | Amino acid transport and metabolism | 5.06 |
| c591_e | mlc | Energy production and conversion; carbohydrate transport and metabolism | 3.42, 3.19 |
| c582_e | purR | Nucleotide transport and metabolism | 9.28 |
| c595_e | trpR | Amino acid transport and metabolism | 4.27 |
| Novel TF-DNA motif pairs | |||
| c4574 | ascG | Carbohydrate transport and metabolism | 5.44 |
| c352_o | baeR | Cell envelope biogenesis, outer membrane | 3.33 |
| c580_e | cadC | Energy production and conversion; nucleotide transport and metabolism | 3.60, 2.65 |
| c489_o | hydG | Transcription | 3.33 |
| c6105_o | idnR | Energy production and conversion | 5.14 |
| c7434_o | lrhA | Amino acid transport and metabolism | 3.12 |
| c645_o | nlp | Nucleotide transport and metabolism; translation, ribosomal structure and biogenesis | 4.82, 5.14 |
| c646_o | ntrC | Amino acid transport and metabolism | 3.11 |
| c648_o | pdhR | Energy production and conversion | 5.55 |
| c647_o | xylR | Carbohydrate transport and metabolism | 4.47 |
| c417_e | yagA | Carbohydrate transport and metabolism | 3.25 |
| c419_o | ycfQ | Signal transduction mechanisms | 2.74 |
| c428_o | ydcN | Energy production and conversion | 4.35 |
| c428_e | ydfH | Energy production and conversion | 2.73 |
| c6271_e | ydhB | Energy production and conversion; cell envelope biogenesis, outer membrane | 2.91, 3.19 |
| c571_e | yfeR | Lipid metabolism; cell envelope biogenesis, outer membrane | 3.51, 3.00 |
| c581_e | yfeT | Nucleotide transport and metabolism | 2.77 |
| c477_o | yfhH | Energy production and conversion | 2.79 |
| c3037_o | yhjC | Translation, ribosomal structure and biogenesis | 9.94 |
| c498_o | yidZ | Translation, ribosomal structure and biogenesis | 2.65 |
| c474_o | yjfQ | Translation, ribosomal structure and biogenesis | 3.39 |
| c467_o | ynfL | Coenzyme metabolism | 3.32 |
| c641_o | yqhC | DNA replication, recombination and repair | 3.06 |
P-values were calculated as the tail probability of the hypergeometric distribution for finding at least k genes from a particular COG functional category in a regulon of size n. Since we considered 21 functional categories, 2.4 × 10−3 was chosen as the P-value cutoff to give an overall significance level of 0.05 for each regulon. Associations of TFs and DNA motifs are made according to the MWM method.
Discussion
One of the grand goals for post-genomic research is to understand the organization of the transcriptional regulatory network in an organism. Fundamental requirements for this goal are the delineation of a network's architecture, its wiring diagram, and estimation of the parameters of its components. To do this, one can take a bottom-up approach in which the parts list of TFs and DNA motifs in a genome can be identified first. Subsequently, these two sets of elements can be connected, resulting in a set of primary connections in the genetic network. These primary connections then can serve as a scaffold upon which the entire regulatory network containing higher-order interactions can ultimately be built. Shen-Orr et al. (2002) has taken the first step to identify higher-order connections in the regulatory network of E. coli based on a collection of known primary connections. They found three types of network motifs (higher-order connections) that are significantly overrepresented in the E. coli network. However, this network is far from complete because of our limited knowledge about the connections between TFs and DNA-binding sites. Thus, identifying primary connections between TFs and DNA-binding sites represents a major bottleneck for modeling transcriptional regulatory networks. In this paper, we have taken the first step to address this problem through computational means.
We hypothesize that information concerning the connection of a TF to its DNA motif is carried in the genome sequences, and we can extract this information by comparing multiple genomic sequences. TFs and their binding sites are often in similar genomic locations (Dmin information), and they tend to evolve concurrently with their regulated genes (PC information). We explored both types of information to identify TF-DNA-motif connections. Both Dmin and PC information were derived from a comparative analysis of multiple genomes. Using a comparative genomics approach has both advantages and drawbacks. On one hand, it increases our confidence in the predictions as our inference is based on reinforced signals from multiple genomes. On the other hand, this also means the success of our method depends on a wise choice of species. It has been observed that phylogenetic distance, similarity of habitat, genome size, and the number of shared genes are important factors in the selection of species for phylogenetic footprinting (McCue et al. 2002). Since we are also studying transcriptional regulatory systems using comparative genomics, we expect the same factors to be important for species selection. However, we have the additional concern that the species should have enough variation in their regulatory networks to provide our method with the signals necessary to distinguish true DNA-motif ↔ TF connections from background. Taking both considerations into account, we chose the seven species used by McCue et al. (2001, 2002) for discovering novel DNA motifs in E. coli and added six more γ-proteobacterial species (Supplemental Table 5). This set of species provides a good balance of conservation and variation for our analyses.
We also took advantage of the observation that TFs from the same structural family tend to have similar DNA motifs. The 248 TFs in E. coli fall into 12 families based on the structural similarity of their DBDs. Based on binding data from RegulonDB and DPinteract, DNA motif models can be constructed for 77 TFs in these 12 families. Some of the motif models are less accurate since they are built from a very small number of known sites (<5). As the amount of binding site data increases for various families of TFs, the utility and accuracy of the FMC information will be enhanced.
We have shown the value of combining heterogeneous information to connect novel TFs and DNA motifs. All three types of information provided evidence for particular connections between TFs and DNA motifs. None of these three types of information alone is highly specific and still maintains reasonable sensitivity. But by combining the evidence of each type we were able to achieve good prediction accuracy, as shown in Figures 2 and 3. In addition, the flexibility of our approach allows new and diverse types of information to be incorporated easily because the order of the information is not important. With the availability of multiple finished genomes for many bacterial groups, our approach can also be applied to other bacterial species with large numbers of neighbors, such as the Gram positive bacterium Bacillus subtilis and the environmentally significant bacterium Shewanella oneidensis.
Methods
Genomic sequences
All Genomic sequences were downloaded from the NCBI RefSeq database (Pruitt and Maglott 2001). The genomes used in our study are E. coli K-12 MG1655, Haemophilus influenzae Rd, Pasteurella multocida, Pseudomonas aeruginosa PAO1, Pseudomonas putida KT2440, Pseudomonas syringae pv. tomato, Shewanella oneidensis MR-1, Salmonella typhi CT-18, Vibrio cholerae El Tor, Vibrio parahaemolyticus RIMD 2210633, Vibrio vulnificus CMCP6, Xanthomonas campestri pv. campestri, and Yersinia pestis CO92.
Two gene sequences were deemed orthologous if they satisfied the following three criteria simultaneously (Huynen and Bork 1998; Tan et al. 2001): (1) They were the most similar sequences for each other between the two genomes. (2) Their BLASTP E-value was lower than 10-10. (3) Their BLASTP alignment extended to at least 60% of one of the sequences.
Study set transcription factors and their DNA-binding motifs
TF-binding sites obtained through footprinting experiments were extracted from the databases RegulonDB v3.2 (Salgado et al. 2004) and DPinteract (http://arep.med.harvard.edu/dpinteract/index.html). After removing redundant sites for the same TF, 35 TFs have at least five experimentally verified binding sites. The average number of binding sites per TF is 20. Weight matrix models for DNA-binding sites were constructed for these 35 TFs using the multiple sequence alignment program CONSENSUS (Hertz and Stormo 1999). In this paper, DNA motifs refer to these weight matrix models. The TFs are araC, arcA, argR, cpxR, crp, deoR, dnaA, fadR, flhD, fnr, fruR, fur, galR, glpR, gntR, hipB, ilvY, lexA, lrp, malT, marA, metJ, metR, mlc, modE, nagC, narL, narP, ompR, phoB, purR, soxS, torR, trpR, and tyrR.
Full sets of TFs and DNA motifs in E. coli
Two groups have conducted computational surveys to identify the repertoire of TFs in E. coli. Using a combination of sequence homology search and literature mining, Perez-Rueda and Collado-Vides (2000) identified a total of 314 TFs in E. coli. By looking for signature protein domains present in TFs, Babu and Teichmann (2003) independently identified a total of 273 TFs in E. coli. We took the intersection of the two sets as the set of TFs in E. coli, which contains 248 proteins. McCue et al. (2002) conducted a whole-genome phylogenetic footprinting study of E. coli and six additional genomes, resulting in thousands of palindromic DNA motif predictions that consist of cross-species site alignments. Clustering a set of statistically significant (P < 0.05) motifs from this study yielded predicted regulons (Qin et al. 2003). For this study we used as input a set of 113 motifs (predicted regulons) that resulted from clustering a less stringent set of statistically significant (P < 0.2) phylogenetic footprint motifs (L.A. McCue, unpubl.; data available at http://www.wadsworth.org/resnres/bioinfo/).
Novel sets of TFs and DNA motifs in E. coli
From the full set of 248 TFs and 113 DNA motifs, we identified a set of novel TFs and DNA motifs to make connections. They were selected via the following procedures. For DNA motifs, we removed all motifs containing E. coli sites that overlap with verified TF-binding sites for any of the 35 TFs in the study set, reported RNA secondary structures, and intergenic repeats. We ended up with 70 novel DNA motifs. Among them, three motifs contain binding sites for known TFs (ntrC, pdhR, xylR) that are not included in our study set because they have fewer than five verified binding sites. These motifs remained in our novel set as an additional way to evaluate the performance of our algorithm because the TF-DNA-motif associations are known in these cases. For TFs, we removed those with documented sites in RegulonDB and/or DPinteract, including the study set TFs and TFs with known nonpalindromic DNA motifs. Again, ntrC, pdhR, and xylR were retained for performance evaluation. In addition, since the set of DNA motifs was derived from alignments having sites from E. coli and at least one more genome that is not S. typhi (owing to the close phylogenetic distance between E. coli and S. typhi, alignments having sites from only these two genomes may not represent functional DNA elements), we removed TFs that only occur in E. coli or in E. coli and S. typhi. This procedure resulted in 99 novel TFs (Supplemental Table 6).
Calculation of normalized Dmin (minimal distance)
The study set and novel DNA motifs in this study represent partial regulons; in some cases a DNA motif may contain only one or two binding sites from a given species like E. coli. Thus, we needed to more fully delineate the regulons in order to calculate a minimal distance between a TF-encoding gene and a binding site. Specifically, for a given DNA motif Mi, all intertranscription unit regions in the genome were scored against the weight matrix representing Mi (constructed from clustered sites as described in above). Sites scoring above the mean minus 1.5 standard deviations of the training set scores were regarded as likely binding sites of Mi. Determining the cutoff score for binding sites is a hard problem and inevitably involves some arbitrary decision. Previously, the mean minus one or two standard deviations, or the lowest score of training sequences, have each been used as site cutoffs (Robison et al. 1998; Gelfand et al. 2000; De Wulf et al. 2002). The cutoff used in this study is a compromise that we have found yields many true binding sites without too many false positives (Tan et al. 2001).
For a given transcription factor TFj, we then calculate a 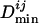 by the following procedure. The binding site closest to the translation start of TFj is used to calculate the unnormalized minimal distance
by the following procedure. The binding site closest to the translation start of TFj is used to calculate the unnormalized minimal distance 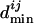 , which is the number of genes between the closest binding site and the translation start of TFj. Each
, which is the number of genes between the closest binding site and the translation start of TFj. Each 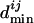 is divided by
is divided by 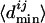 to give the normalized
to give the normalized 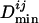 , where
, where 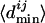 denotes the mean of all
denotes the mean of all 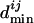 's between Mi and the set of TFs under consideration. The normalization is needed to account for the difference between motifs (having low information content) that occur very frequently and motifs (having high information content) that occur very infrequently. We calculate a
's between Mi and the set of TFs under consideration. The normalization is needed to account for the difference between motifs (having low information content) that occur very frequently and motifs (having high information content) that occur very infrequently. We calculate a 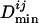 for each genome and the final
for each genome and the final 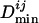 is the average of all
is the average of all 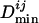 's for species having an ortholog of TFj.
's for species having an ortholog of TFj.
Calculation of PC (phylogenetic correlation)
For a given DNA motif Mi and a transcription factor TFj, we calculate a PCij. The two vectors, 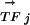 and
and 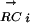 , are K-element vectors, where K is the number of species under consideration. Each element k of
, are K-element vectors, where K is the number of species under consideration. Each element k of 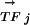 assumes the value 1 or 0, where 1 denotes that an ortholog of TFj was detected in species k, and 0 otherwise. Each element k of
assumes the value 1 or 0, where 1 denotes that an ortholog of TFj was detected in species k, and 0 otherwise. Each element k of 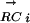 assumes a value that measures the degree of conservation between the E. coli regulon determined by Mi and its counterpart in species k. These values are calculated by using the gene regulation vector,
assumes a value that measures the degree of conservation between the E. coli regulon determined by Mi and its counterpart in species k. These values are calculated by using the gene regulation vector, 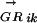 . For species k, the elements of
. For species k, the elements of 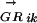 represent orthologs shared between E. coli and that species. The value of each element is the probability that the orthologous genes are controlled by the DNA motif Mi. Given a DNA motif Mi and the regulatory region S, the probability that this region is bound by a TF having the motif Mi can be approximated as:
represent orthologs shared between E. coli and that species. The value of each element is the probability that the orthologous genes are controlled by the DNA motif Mi. Given a DNA motif Mi and the regulatory region S, the probability that this region is bound by a TF having the motif Mi can be approximated as:
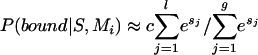 |
where l is the length of S and g is the length of the genome, Sj is the score of the sequence word starting at position j, and c is a constant depending on the concentration of the TF in the cell (Heumann et al. 1994; Workman and Stormo 2000). All genes in a transcription unit (TU) are assigned the same probability (of being regulated by Mi), which is calculated using the regulatory region of the TU. Transcription units are predicted using the method described in Salgado et al. (2000). The regulatory region of a TU is the intergenic sequence between the first gene of the TU and the last gene of the upstream TU. The Pearson's correlation coefficients between anE. coli 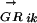 vector and
vector and 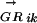 vectors for other genomes form the RCi vector, which describes the evolutionary changes of the regulon predicted to be controlled by the motif Mi. The phylogenetic correlation between the transcription factor TFj and the regulon controlled by Mi, PCij, is calculated as the Pearson's correlation coefficient between the
vectors for other genomes form the RCi vector, which describes the evolutionary changes of the regulon predicted to be controlled by the motif Mi. The phylogenetic correlation between the transcription factor TFj and the regulon controlled by Mi, PCij, is calculated as the Pearson's correlation coefficient between the 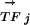 vector and the
vector and the  vector.
vector.
Calculation of FMC (familial motif conservation)
Given a DNA motif Mi and a transcription factor TFj belonging to the structural family α, FMCij is calculated as follows:
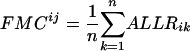 |
where n is the number of TFs in family α and ALLRik is the similarity score between motifs i and k. The majority of the DNA motifs have inverted or direct repeats; we can compare them by splitting the full matrices at the center of symmetry. This avoids the problem of allowing insertions/deletions in the alignment when comparing motifs with different length spacer regions between the most conserved positions. A few DNA motifs have asymmetric patterns, for them the full matrices are used. Now matrices can be aligned using an ungapped Smith-Waterman algorithm, modified for profile alignment instead of sequence alignment, which finds the highest scoring local alignment between them. The scoring function for the alignment between a pair of columns from two alignment matrices is the average log likelihood ratio (ALLR) (Wang and Stormo 2003):
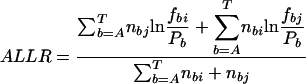 |
where i and j are two columns from the two alignment matrices respectively, nbi and nbj are count vectors for base b, fbi and fbj are frequency vectors for base b, and pb is the frequency of base b in the background model. The score of aligning two matrices is the sum of scores for all columns in the alignment.
Probability of a TF-DNA-motif connection
The posterior probability of a connection being true given one type of information can be calculated using Bayes' rule:
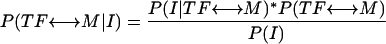 |
where I refers to one of the following three terms: Dmin ≤ x, PC ≥ y, or FMC ≥ z. We use a flat prior of 1/N, where N is the number of TFs or DNA motifs, whichever is smaller. Using Bayes' rule and the assumption of independence of the three types of information, the joint probability of a true TF ↔ DNA connection considering all types of information simultaneously, P(TF ↔ M|Dmin ≤ x, PC ≥ y, FMC ≥ z), is the product of the following four terms:
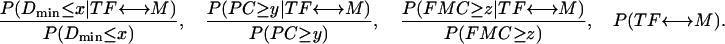 |
The first three terms represent the application of the three types of information independently, and the fourth term is the prior probability. Probability distributions calculated based on the study set are used to set the probabilities for the novel set of TFs and DNA motifs.
Maximum weighted matching algorithm
A maximum weighted matching algorithm (MWM) has been used to predict RNA secondary structures (Cary and Stormo 1995; Tabaska et al. 1998). To apply MWM to our problem, we convert the posterior probability table (containing the probabilities for all possible TF-DNA-motif pairs) into a bipartite graph, only allowing edges between each TF and each DNA motif. The edge weight between TFj and motif Mi is calculated as follows:
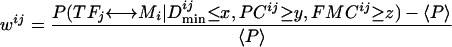 |
where 〈P〉 is the average of the probabilities of associating TFj with each of the DNA motifs.
To find the best set of TF-DNA-motif pairs, the algorithm finds the match with the highest total edge weight. MWM is guaranteed to reach the optimal solution (Gabow 1976).
Calculation of P-value for functional category enrichment
Genes having a binding site for a DNA motif, scored above the cutoff, are regarded as members of the regulon. The hypergeometric distribution is used to calculate the P-value of observing the number of genes from a particular COG functional category within a regulon. Specifically, the probability of having at least k genes from a functional category within a regulon of size n is given by:
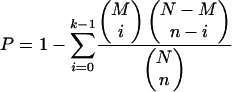 |
where M is the total number of genes in a functional category, and N is the total number of genes in the E. coli K-12 genome (4279). Since we tested 21 COG functional categories for each regulon, a significance value of 2.4 × 10-3 is used for each functional category to give an overall significance value of 0.05 for each regulon.
Calculation of P-value for binding sites
Scores of all possible l-mers (l is the length of the DNA motif) in the genome is calculated using the program PATSER. The scores conform to a normal distribution. The tail probability of finding a site having the observed score is calculated by converting the score distribution into a standard normal distribution.
Acknowledgments
We thank Charles Lawrence for suggestions on probability calculations and comments on the manuscript, Gabriel Moreno-Hagelsieb for help with transcription unit prediction, and Yongmei Ji for the collection of known regulatory RNA motifs. We also thank members of the Stormo lab for their comments on the manuscript. T.K. and G.D.S. are supported by National Institute of Health (NIH) grant HG 00249. L.A.M. is supported by Department of Energy (DOE) grants DE-FG02-01ER63204 and DE-FG02-04ER63942.
Footnotes
Article and publication are at http://www.genome.org/cgi/doi/10.1101/gr.3069205. Article published online ahead of print in January 2005.
[Supplemental material is available online at www.genome.org.]
References
- Babu, M.M. and Teichmann, S.A. 2003. Evolution of transcription factors and the gene regulatory network in Escherichia coli. Nucleic Acids Res. 31: 1234-1244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bar-Joseph, Z., Gerber, G.K., Lee, T.I., Rinaldi, N.J., Yoo, J.Y., Robert, F., Gordon, D.B., Fraenkel, E., Jaakkola, T.S., Young, R.A., et al. 2003. Computational discovery of gene modules and regulatory networks. Nat. Biotechnol. 21: 1337-1342. [DOI] [PubMed] [Google Scholar]
- Blanchette, M. and Tompa, M. 2002. Discovery of regulatory elements by a computational method for phylogenetic footprinting. Genome Res. 12: 739-748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blattner, F.R., Plunkett III, G., Bloch, C.A., Perna, N.T., Burland, V., Riley, M., Collado-Vides, J., Glasner, J.D., Rode, C.K., Mayhew, G.F., et al. 1997. The complete genome sequence of Escherichia coli K-12. Science 277: 1453-1474. [DOI] [PubMed] [Google Scholar]
- Cary, R.B. and Stormo, G.D. 1995. Graph-theoretic approach to RNA modeling using comparative data. Proc. Int. Conf. Intell. Syst. Mol. Biol. 3: 75-80. [PubMed] [Google Scholar]
- De Wulf, P., McGuire, A.M., Liu, X., and Lin, E.C. 2002. Genome-wide profiling of promoter recognition by the two-component response regulator CpxR-P in Escherichia coli. J. Biol. Chem. 277: 26652-26661. [DOI] [PubMed] [Google Scholar]
- Dickson, R.C., Abelson, J., Barnes, W.M., and Reznikoff, W.S. 1975. Genetic regulation: The Lac control region. Science 187: 27-35. [DOI] [PubMed] [Google Scholar]
- Eichler, K., Buchet, A., Lemke, R., Kleber, H.P., and Mandrand-Berthelot, M.A. 1996. Identification and characterization of the caiF gene encoding a potential transcriptional activator of carnitine metabolism in Escherichia coli. J. Bacteriol. 178: 1248-1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabow, H. 1976. “Implementation of algorithms for maximum matching on non-bipartite graphs.” Ph.D thesis, Stanford University, Stanford, CA.
- Gelfand, M.S., Koonin, E.V., and Mironov, A.A. 2000. Prediction of transcription regulatory sites in archaea by a comparative genomic approach. Nucleic Acids Res. 28: 695-705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hertz, G.Z. and Stormo, G.D. 1999. Identifying DNA and protein patterns with statistically significant alignments of multiple sequences. Bioinformatics 15: 563-577. [DOI] [PubMed] [Google Scholar]
- Heumann, J.M., Lapedes, A.S., and Stormo, G.D. 1994. Neural networks for determining protein specificity and multiple alignment of binding sites. Proc. Int. Conf. Intell. Syst. Mol. Biol. 2: 188-194. [PubMed] [Google Scholar]
- Huynen, M.A. and Bork, P. 1998. Measuring genome evolution. Proc. Natl. Acad. Sci. 95: 5849-5856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji, Y., Xu, X., and Stormo, G.D. 2004. A graph theoretical approach for predicting common RNA secondary structure motifs including pseudoknots in unaligned sequences. Bioinformatics 20: 1591-1602. [DOI] [PubMed] [Google Scholar]
- Kao, K.C., Yang, Y.-L., Boscolo, R., Sabatti, C., Roychowdhury, V., and Liao, J.C. 2004. Transcriptome-based determination of multiple transcription regulator activities in Escherichia coli by using network component analysis. Proc. Natl. Acad. Sci. 101: 641-646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korf, I., Flicek, P., Duan, D., and Brent, M.R. 2001. Integrating genomic homology into gene structure prediction. Bioinformatics 17 Suppl 1: S140-S148. [DOI] [PubMed] [Google Scholar]
- Lawrence, J.G. 1999. Selfish operons: The evolutionary impact of gene clustering in prokaryotes and eukaryotes. Curr. Opin. Genet. Dev. 9: 642-648. [DOI] [PubMed] [Google Scholar]
- Liao, J.C., Boscolo, R., Yang, Y.-L., Tran, L.M., Sabatti, C., and Roychowdhury, V.P. 2003. Network component analysis: Reconstruction of regulatory signals in biological systems. Proc. Natl. Acad. Sci. 100: 15522-15527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luscombe, N.M., Austin, S.E., Berman, H.M., and Thornton, J.M. 2000. An overview of the structures of protein-DNA complexes. Genome Biol. 1: REVIEWS001. [DOI] [PMC free article] [PubMed]
- Madera, M., Vogel, C., Kummerfeld, S.K., Chothia, C., and Gough, J. 2004. The SUPERFAMILY database in 2004: Additions and improvements. Nucleic Acids Res. 32: D235-D239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcotte, E.M., Pellegrini, M., Ng, H.L., Rice, D.W., Yeates, T.O., and Eisenberg, D. 1999. Detecting protein function and protein-protein interactions from genome sequences. Science 285: 751-753. [DOI] [PubMed] [Google Scholar]
- Martinez-Antonio, A. and Collado-Vides, J. 2003. Identifying global regulators in transcriptional regulatory networks in bacteria. Curr. Opin. Microbiol. 6: 482-489. [DOI] [PubMed] [Google Scholar]
- McCue, L.A., Thompson, W., Carmack, C.S., Ryan, M.P., Liu, J.S., Derbyshire, V., and Lawrence, C.E. 2001. Phylogenetic footprinting of transcription factor binding sites in proteobacterial genomes. Nucleic Acids Res. 29: 774-782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCue, L.A., Thompson, W., Carmack, C.S., and Lawrence, C.E. 2002. Factors influencing the identification of transcription factor binding sites by cross-species comparison. Genome Res. 12: 1523-1532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mironov, A.A., Koonin, E.V., Roytberg, M.A., and Gelfand, M.S. 1999. Computer analysis of transcription regulatory patterns in completely sequenced bacterial genomes. Nucleic Acids Res. 27: 2981-2989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagakubo, S., Nishino, K., Hirata, T., and Yamaguchi, A. 2002. The putative response regulator BaeR stimulates multidrug resistance of Escherichia coli via a novel multidrug exporter system, MdtABC. J. Bacteriol. 184: 4161-4167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Overbeek, R., Fonstein, M., D'Souza, M., Pusch, G.D., and Maltsev, N. 1999. The use of gene clusters to infer functional coupling. Proc. Natl. Acad. Sci. 96: 2896-2901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palacios, S. and Escalante-Semerena, J.C. 2000. prpR, ntrA, and ihf functions are required for expression of the prpBCDE operon, encoding enzymes that catabolize propionate in Salmonella enterica Serovar Typhimurium LT2. J. Bacteriol. 182: 905-910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pellegrini, M., Marcotte, E.M., Thompson, M.J., Eisenberg, D., and Yeates, T.O. 1999. Assigning protein functions by comparative genome analysis: Protein phylogenetic profiles. Proc. Natl. Acad. Sci. 96: 4285-4288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez-Rueda, E. and Collado-Vides, J. 2000. The repertoire of DNA-binding transcriptional regulators in Escherichia coli K-12. Nucleic Acids Res. 28: 1838-1847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Postma, P.W., Lengeler, J.W., and Jacobson, G.R. 1993. Phosphoenolpyruvate: carbohydrate phosphotransferase systems of bacteria. Microbiol. Rev. 57: 543-594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pruitt, K.D. and Maglott, D.R. 2001. RefSeq and LocusLink: NCBI gene-centered resources. Nucleic Acids Res. 29: 137-140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin, Z.S., McCue, L.A., Thompson, W., Mayerhofer, L., Lawrence, C.E., and Liu, J.S. 2003. Identification of co-regulated genes through Bayesian clustering of predicted regulatory binding sites. Nat. Biotechnol. 21: 435-443. [DOI] [PubMed] [Google Scholar]
- Rajewsky, N., Socci, N.D., Zapotocky, M., and Siggia, E.D. 2002. The evolution of DNA regulatory regions for proteo-γ bacteria by interspecies comparisons. Genome Res. 12: 298-308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren, B., Robert, F., Wyrick, J.J, Aparicio, O., Jennings, E.G., Simon, I., Zeitlinger, J., Schreiber, J., Hannett, N., Kanin, E., et al. 2000. Genome-wide location and function of DNA binding proteins. Science 290: 2306-2309. [DOI] [PubMed] [Google Scholar]
- Rigali, S., Derouaux, A., Giannotta, F., Dusart, J. 2002. Subdivision of the helix-turn-helix GntR family of bacterial regulators in the FadR, HutC, mocR, and YtrA subfamilies. J. Biol. Chem. 277: 12507-12515. [DOI] [PubMed] [Google Scholar]
- Rivas, E. and Eddy, S.R. 2001. Noncoding RNA gene detection using comparative sequence analysis. BMC Bioinformatics 2: 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robison, K., McGuire, A.M., and Church, G.M. 1998. A comprehensive library of DNA-binding site matrices for 55 proteins applied to the complete Escherichia coli K-12 genome. J. Mol. Biol. 284: 241-254. [DOI] [PubMed] [Google Scholar]
- Salgado, H., Moreno-Hagelsieb, G., Smith, T.F., and Collado-Vides, J. 2000. Operons in Escherichia coli: Genomic analyses and predictions. Proc. Natl. Acad. Sci. 97: 6652-6657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salgado, H., Gama-Castro, S., Martinez-Antonio, A., Diaz-Peredo, E., Sanchez-Solano, F., Peralta-Gil, M., Garcia-Alonso, D., Jimenez-Jacinto, V., Santos-Zavaleta, A., Bonavides-Martinez, C., et al. 2004. RegulonDB (version 4.0): Transcriptional regulation, operon organization and growth conditions in Escherichia coli K-12. Nucleic Acids Res. 32: D303-D306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandelin, A. and Wasserman, W.W. 2004. Constrained binding site diversity within families of transcription factors enhances pattern discovery bioinformatics. J. Mol. Biol. 338: 207-215. [DOI] [PubMed] [Google Scholar]
- Segal, E., Shapira, M., Regev, A., Pe'er, D., Botstein, D., Koller, D., and Friedman, N. 2003. Module networks: Identifying regulatory modules and their condition-specific regulators from gene expression data. Nat. Genet. 34: 166-176. [DOI] [PubMed] [Google Scholar]
- Shen-Orr, S.S., Milo, R., Mangan, S., and Alon, U. 2002. Network motifs in the transcriptional regulation network of Escherichia coli. Nat. Genet. 31: 64-68. [DOI] [PubMed] [Google Scholar]
- Tabaska, J.E., Cary, R.B., Gabow, H.N., and Stormo, G.D. 1998. An RNA folding method capable of identifying pseudoknots and base triples. Bioinformatics 14: 691-699. [DOI] [PubMed] [Google Scholar]
- Tan, K., Moreno-Hagelsieb, G., Collado-Vides, J., and Stormo, G.D. 2001. A comparative genomics approach to prediction of new members of regulons. Genome Res. 11: 566-584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tatusov, R.L., Koonin, E.V., and Lipman, D.L. 1997. A genomic perspective on protein families. Science 278: 631-637. [DOI] [PubMed] [Google Scholar]
- Torres, B., Porras, G., Garcia, J.L., and Diaz, E. 2003. Regulation of the mhp cluster responsible for 3-(3-hydroxyphenyl)propionic acid degradation in Escherichia coli. J. Biol. Chem. 278: 27575-27585. [DOI] [PubMed] [Google Scholar]
- van Nimwegen, E., Zavolan, M., Rajewsky, N., and Siggia, E.D. 2002. Probabilistic clustering of sequences: Inferring new bacterial regulons by comparative genomics. Proc. Natl. Acad. Sci. 99: 7323-7328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, T. and Stormo, G.D. 2003. Combining phylogenetic data with co-regulated genes to identify regulatory motifs. Bioinformatics 19: 2369-2380. [DOI] [PubMed] [Google Scholar]
- Workman, C.T. and Stormo, G.D. 2000. ANN-Spec: A method for discovering transcription factor binding sites with improved specificity. Pac. Symp. Biocomput. 5: 467-478. [DOI] [PubMed] [Google Scholar]
WEB SITE REFERENCES
- http://arep.med.harvard.edu/dpinteract/index.html; DPInteract database.
- http://www.wadsworth.org/resnres/bioinfo/; 113 E. coli DNA motifs.