

Abstract
The Gram-negative pathogen Pseudomonas aeruginosa encodes multiple protein export systems, the substrates of which contain export signals such as N-terminal signal peptides. Here we report the first genome-wide computational and laboratory screen for N-terminal signal peptides in this important opportunistic pathogen. The computational identification of signal peptides was based on a consensus between multiple predictive tools and showed that 38% of the P. aeruginosa PAO1 proteome was predicted to encode exported proteins, most of which utilize cleavable type I signal peptides or uncleavable transmembrane helices. In addition, known and novel lipoproteins (type II), twin arginine transporter (TAT), and prepilin peptidase substrates (type IV) were also identified. A laboratory-based screen using the alkaline phosphatase (PhoA) fusion method was then used to test our predictions. In total, 310 nonredundant PhoA fusions were successfully identified, 296 of which possess a predicted export signal. Analysis of the PhoA fusion proteins lacking an export signal revealed that three proteins have alternate translation start sites that encode signal peptides, two proteins may use an unknown export signal, and the remaining nine proteins are likely cytoplasmic proteins and represent false positives associated with the PhoA screen. Our approach to identify exported proteins illustrates how computational and laboratory-based methods are complementary, where computational analyses provide a large number of accurate predictions while laboratory methods both confirm predictions and reveal unique cases meriting further analysis.
The completion of the Pseudomonas aeruginosa PAO1 genome sequence has provided many insights into the biology and pathogenesis of this organism and serves as the starting point for genome-wide studies of this important opportunistic pathogen (Stover et al. 2000). P. aeruginosa is the primary cause of chronic lung infections and mortality in patients with cystic fibrosis and is also the third most frequently isolated nosocomial pathogen, causing 10% of all hospital-acquired infections (Govan and Deretic 1996; Fridkin and Gaynes 1999; Hancock and Speert 2000). Pseudomonas infections are difficult to treat due to the high intrinsic antibiotic resistance of this organism, which is attributed to low outer membrane permeability coupled with additional resistance mechanisms such as active drug efflux and antibiotic modification (Hancock and Speert 2000).
A major subset of the P. aeruginosa proteome is dedicated to proteins that are exported out of the cytoplasm to the cell envelope (the cytoplasmic membrane, the periplasm, and the outer membrane) or that are secreted out of the cell to the extracellular environment (Nouwens et al. 2000; Guina et al. 2003). This subset of proteins is involved in essential cellular processes that include cell wall assembly, nutrient uptake, virulence, antibiotic resistance, pili and flagella biogenesis, immunogenicity, adherence, energy generation, and environmental sensing. The importance of these proteins is illustrated by the fact that many cell envelope proteins are the targets of current antimicrobials (Drew et al. 2003) or vaccines, and thus identifying novel envelope proteins may provide new targets for drug discovery and immunoprophylaxis (Cachia and Hodges 2003).
P. aeruginosa proteins destined to noncytoplasmic subcellular localizations utilize various protein export systems, as recently reviewed in Ma et al. (2003). The Sec machinery facilitates the majority of protein transport across the cytoplasmic membrane (Pugsley 1993; Filloux et al. 1998). Proteins may be recognized by the SecB chaperone after translation, maintaining their appropriate conformation to permit recognition by the SecYEG translocation machinery (Pugsley 1993; de Gier and Luirink 2001; Drew et al. 2003), or they may be recognized by the signal recognition particle (SRP). SRP is a nucleoprotein complex of the Ffh protein and a 4.5S ribosomal RNA that directs proteins to the FtsY receptor and ultimately to the SecYEG translocase (Bernstein 2000; de Gier and Luirink 2001; Drew et al. 2003). Export systems that are Sec-independent are also found in P. aeruginosa, including the twin-arginine translocation (TAT) pathway, which is responsible for the translocation of certain prefolded proteins (Voulhoux et al. 2001; Ochsner et al. 2002), as well as type I and type III secretion systems that translocate proteins across the cytoplasmic and outer membranes in a single step (Ma et al. 2003).
Proteins utilizing the Sec or TAT machinery are recognized by their N-terminal signal peptides. These are typically short sequences with a tripartite positive-hydrophobic-polar character suitable for partitioning into lipid bilayers. The N region is positively charged, the hydrophobic H region is a minimum of eight hydrophobic residues in length and forms a membrane-spanning helix, and the C region often contains a signal peptidase recognition site (von Heijne 1985). The majority of tripartite signal peptides are cleaved by signal peptidase I (SPase I), and are known as type I signal peptides. In some cases however, such signal peptides may remain uncleaved, serving as signal anchors for inner membrane proteins (von Heijne 1988). Other types of tripartite signal peptides exhibit unique characteristics in their structure and are cleaved by different types of signal peptidases. For example, TAT substrates contain signal peptides similar to those processed by SPase I; however, they also contain the twin arginine motif RRXFL[KR] upstream of their hydrophobic region (Chaddock et al. 1995). Signal peptidase II cleaves type II signal peptides, which are associated with lipoproteins. Although these are similar to type I signal peptides, cleavage occurs immediately upstream of a Cys residue, which is part of the N-terminal lipobox motif that characterizes these signal peptides (von Heijne 1989). Prepilin peptidase cleaves type IV signal peptides, which differ from traditional N-terminal signal peptides in that they are short (approximately six residues) with no tripartite structure. Instead cleavage occurs downstream of a glycine residue that precedes a long N-terminal stretch of hydrophobic amino acids in the mature protein (LaPointe and Taylor 2000). A GFTLIE motif is often found in prepilin peptidase substrate signal peptides (Lory 1994).
The availability of the P. aeruginosa PAO1 genome sequence, combined with knowledge of the defined structures and motifs found in most N-terminal signal peptides, permitted us to perform a genome-wide computational survey of proteins that use N-terminal signal peptides for export out of the cytoplasm. Our definition of “exported protein” includes all proteins exported out of the cytoplasm, including those incorporated into the cytoplasmic membrane through the presence of transmembrane helices.
Laboratory-based surveys of signal peptide-encoding genes are possible through the use of the alkaline phosphatase (PhoA) fusion technique (Bina et al. 1997; Wiker et al. 2000). In this approach, signal peptide-containing genes are fused in frame with a truncated ′phoA gene lacking its native signal peptide. The signal peptide in the fused genomic fragment targets the PhoA moiety across the inner membrane to the periplasm, where PhoA folds and becomes enzymatically active.
In this report, we describe a combined computational and laboratory survey of P. aeruginosa signal peptide-encoding genes. We first screened the P. aeruginosa PAO1 genome for potential export candidates using computational techniques, and then performed a random cloning PhoA fusion screen to test our predictions. This study represents the most comprehensive analysis of exported proteins in P. aeruginosa to date, and illustrates the utility of a combined approach for genome-scale studies.
Results
Type I signal peptides are the most common N-terminal export signal
A combination of four signal peptide and two transmembrane helix prediction methods, as well as manual motif searching, was used to identify potential export signals in the P. aeruginosa PAO1 genome. Type I or type II signal peptides and their cleavage sites were predicted using SignalP v.3.0's neural network and hidden Markov model tools (Bendtsen et al. 2004), LipoP v.1.0 (Juncker et al. 2003), and Phobius (Kall et al. 2004). Phobius and TMHMM (Krogh et al. 2001) were used to identify membrane-targeting transmembrane segments. Type IV and TAT signal peptides were identified manually. The predictions for each of the 5570 ORFs in the P. aeruginosa PAO1 genome are presented in Supplemental Table 1.
The majority of proteins using an N-terminal signal peptide for export are substrates of the Sec pathway and are cleaved by SPase I. In the P. aeruginosa genome, 801 (14.4%) proteins were predicted by at least three of the four methods to contain a cleavable type I signal peptide (Table 1; Supplemental Table 2). The programs typically agree in their predictions, with 518 out of 801 signal peptides having four identically predicted cleavage sites and an additional 56 signal peptides with three identically predicted cleavage sites.
Table 1.
Summary of predicted export signals in the P. aeruginosa PAO1 proteome
| Type of export signal | Number | % of genome | % of export signals |
|---|---|---|---|
| Type I | 801 | 14.4 | 37.7 |
| Possible type I | 57 | 1.0 | 2.7 |
| Type II (lipoprotein) | 185 | 3.3 | 8.7 |
| Type IV (prepilin) | 23 | 0.4 | 1.1 |
| TAT | 15 | 0.3 | 0.7 |
| Transmembrane helix | 1042 | 18.7 | 49.1 |
| Total with export signals | 2123 | 38.1 | — |
| No export signal | 3447 | 61.9 | — |
To depict representative type I signal peptides, amino acid sequence logos were constructed for P. aeruginosa type I signal peptides (Fig. 1A). The logo illustrates that predicted P. aeruginosa type I signal peptides are similar to those of other Gram-negative bacteria with respect to the N region charge and length and the H region length and hydrophobicity (von Heijne 1985, 1989). The A-X-A motif for the SPase I cut site in the C region is also conserved, although this reflects the biases in data used to train the predictive methods.
Figure 1.
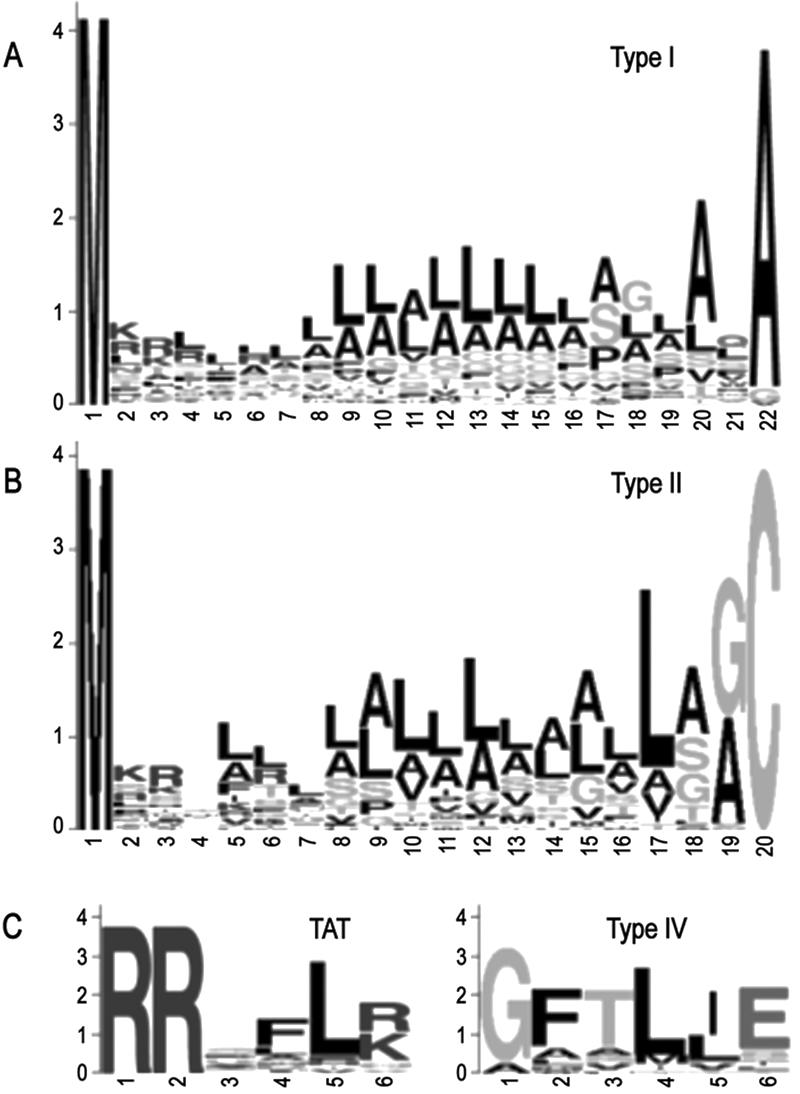
Amino acid sequence logos of predicted P. aeruginosa PAO1 type I signal peptides (A), type II signal peptides (B), as well as the TAT and type IV motifs (C). For type I, all signal sequences where the cleavage site predictions were in agreement and that were 22 amino acids in length (mode) were used (n = 77). For type II, all lipoproteins 19 amino acids in length (mode) plus the Cys residue were used (n = 31). All predicted TAT motifs (n = 27) and type IV motifs (n = 23) were used.
In addition to the 801 proteins with strongly predicted signal peptides, 57 proteins yielded inconclusive results, with only two out of four methods making a signal peptide prediction. These proteins were therefore classified as possible type I signal peptides (Table 1; Supplemental Table 3). This list includes the known TAT substrate phospholipase PlcH (PA0844) (Voulhoux et al. 2001), a lectin protein thought to be anchored in the outer membrane (PA3361), and the probable outer membrane protein OprC (PA3790). Interestingly, this list also contains three regulatory proteins that are predicted to be cytoplasmic (PA1949, PA1998, PA2267) by PSORTb v.2.0 (Gardy et al. 2004). These three proteins likely represent the small number of false positives inherent in any predictive technique.
The PAO1 genome is predicted to encode a high proportion of lipoproteins
Signal peptidase II processes lipoproteins, which are tethered to the cytoplasmic or outer membrane via a lipid anchor. LipoP v.1.0 (Juncker et al. 2003) was used to identify potential lipoproteins in the P. aeruginosa genome. There were 185 proteins, or 3.3% of the genome, predicted to contain a type II signal peptide (Table 1; Supplemental Table 4). This number is considerably higher than the 76 PSORT I-predicted lipoproteins (Nakai and Horton 1999) annotated at http://www.pseudomonas.com, likely due to the improved identification algorithm of LipoP. LipoP also reports the +2 residue of each predicted lipoprotein, as this residue is thought to act in targeting of the lipoprotein to one or the other membrane. The majority of P. aeruginosa lipoproteins contain Ser (54), Ala (49), or Gly (32) residues at this position, indicating that they are likely localized to the outer membrane (Yamaguchi et al. 1988). The L[ASG][GA]C lipobox motif of P. aeruginosa type II signal peptides is very similar to that of other Gram-negative bacteria (Fig. 1B) (Juncker et al. 2003).
Manual scanning reveals novel putative prepilin peptidase substrates
Type IV signal peptides are typically associated with prepilins and prepilin-like proteins and are cleaved by prepilin peptidase (Nunn and Lory 1991). Because type IV signal peptides are extremely short and do not exhibit a tripartite structure, this class of signal sequence is not well recognized by signal peptide prediction methods. Sequences must be manually scanned for the occurrence of a GFTLIE-like motif (Fig. 1C) preceding a stretch of hydrophobic residues. By using this screen, we initially identified 13 candidate type IV signal peptides, all of which were present in proteins annotated as pseudopilins, type II secretion proteins, general secretion pathway proteins, pilins, or fimbrial subunits—classes of proteins known or suspected to be processed by prepilin peptidase.
The 13 predicted prepilin peptidase substrates occurred in clusters along the genome. Reasoning that neighboring proteins might exhibit type IV-like signal sequences missed in the initial scan, we manually inspected the 10 proteins both upstream and downstream of the clusters. A further 10 sequences representing possible prepilin peptidase substrates were identified in this fashion. In total, 23 proteins were predicted to represent prepilin peptidase substrates, six of which have not been previously described (Table 2).
Table 2.
Proteins predicted to contain a type IV signal peptide
| PAID | Gene | Protein | Sequencei |
|---|---|---|---|
| PA0677a | HxcW putative pseudopilin | MSGYAERATTRRRQAGFTLIE | |
| PA0678a | HxcU putative pseudopilin | MRRPRQSGFTLIE | |
| PA0680a | HscV putative pseudopilin | MSTRLSSRGFTLIE | |
| PA0681a | HxcT pseudopilin | MDVVQFSSSPKGHRGQRGFTLIE | |
| PA0682a | HxcX atypical pseudopilin | MNGHRQRGMAIIS | |
| PA2671 | hypothetical protein | MISDGARRHAASARSSGFVLVG | |
| PA2672 | probable type II secretion system protein | MIRSRRKQGAFTLL | |
| PA2673 | probable type II secretion system protein | MPARQEGFTLLE | |
| PA2674 | probable type II secretion system protein | MPMSVTRPNVDQAAFTLLE | |
| PA2675 | probable type II secretion system protein | MNRAAQQGFTLLE | |
| PA3097b | xcpX | general secretion pathway protein K | MRRGQNGVALIT |
| PA3098c | xcpW | general secretion pathway protein J | MRLQRGFTLLE |
| PA3099c | xcpV | general secretion pathway protein I | MKRARGFTLLE |
| PA3100c | xcpU | general secretion pathway protein H | MRASRGFTLIE |
| PA3101c | xcpT | general secretion pathway protein G | MNQSRPLQRRQQSGFTLIE |
| PA4525d | pilA | type 4 fimbrial precursor PilA | MKAQKGFTLIE |
| PA4549e | fimT | type 4 fimbrial biogenesis protein FimT | MVERSQRALTLTE |
| PA4550e | fimU | type 4 fimbrial biogenesis protein FimU | MSYRSNSTGFTLIE |
| PA4551f | pilV | type 4 fimbrial biogenesis protein PilV | MLLKSRHRSLHQSGFSMIE |
| PA4552g | pilW | type 4 fimbrial biogenesis protein PilW | MSMNNRSRRQSGLSMIE |
| PA4553g | pilX | type 4 fimbrial biogenesis protein PilX | MNNFPAQQRGATLVI |
| PA4554 | pilY1 | type 4 fimbrial biogenesis protein PilY1 | MKSVLHQIGKTSLAAALSGAVLLS |
| PA4556h | pilE | type 4 fimbrial biogenesis protein PilE | MRTRQKGFTLLE |
Reported in Ball et al. (2002).
Reported in Bleves et al. (1998).
Reported in Bally et al. (1992).
Reported in Strom and Lory (1987).
Reported in Alm and Mattick (1996).
Reported in Alm and Mattick (1995).
Reported in Alm et al. (1996).
Reported in Russell and Darzins (1994).
Bolding in the sequence identifies the type IV motif as described in Figure 1D.
TAT motifs are found in 10 novel putative substrates
Proteins exported via the TAT machinery display an RRXFL[KR]-like motif at their N terminus, which otherwise contains a leader sequence that resembles a tripartite signal peptide (Berks 1996). The P. aeruginosa genome has been scanned for the presence of TAT substrates in two previous studies. Ochsner et al. (2002) identified 18 putative substrates through a manual inspection approach. Their criteria included the presence of twin arginines, a match to at least one of the remaining residues in the motif, a hydrophobic tract following the motif, and an AXA cleavage site. Dilks et al. (2003) implemented their TATFIND v.1.2 program to identify 57 putative substrates, using the criteria of the presence of an XRRXXX motif within residues 1-34 as well as an uncharged region of ≥13 residues downstream of the twin arginines.
In the present analysis, we first scanned proteins with predicted type I or II signal peptides for the presence of the TAT motif RRXFL[KR] immediately N-terminal to a stretch of hydrophobic residues. We predicted 14 proteins with type I signal peptides that also possess TAT motifs, while one protein with a predicted type II signal peptide contains the TAT motif (PA4712) (Table 3A). However, several putative TAT substrates reported in the earlier two studies were not identified in this analysis. We attribute this to the fact that many of these proteins do not contain traditional type I or type II signal peptides. Thus a second analysis was performed in which we eliminated the requirement for a predicted signal peptide. When the entire genome was searched without the signal peptide filtering step, an additional 12 putative TAT substrates were found (Table 3B).
Table 3.
Proteins predicted to have potential TAT signal peptides
| PAID | Gene | Protein | Sequencec |
|---|---|---|---|
| a. Proteins predicted to contain a TAT signal peptide | |||
| PA0144a | hypothetical protein | MSRSNGSSSRRTFLR | |
| PA1601a | probable aldehyde dehydrogenase | MSLANPSRRGFLK | |
| PA2065a | pcoA | copper resistance protein A precursor | MHRTSRRTFVK |
| PA2328b | hypothetical protein | MCLDDPTHSRRDILK | |
| PA2394a | pvdN | PvdN | MNDRRTFLK |
| PA3319a | plcN | non-hemolytic phospholipase C precursor | MISKSRRSFIR |
| PA3493b | conserved hypothetical protein | MDAATRRSMLR | |
| PA3713b | hypothetical protein | MTISRRDFLN | |
| PA4160 | fepD | ferric enterobactin transport protein FepD | MQASPMRRRRLR |
| PA4431b | probable iron-sulfur protein | MSNDGVNAGRRRFLV | |
| PA4621a | probable oxidoreductase | MSNRDISRRAFLO | |
| PA4650 | hypothetical protein | MPMSSRRLFLS | |
| PA4712 | hypothetical protein | MRRFSLR | |
| PA4812a | fdnG | formate dehydrogenase-O, major subunit | MDMNRROFFK |
| PA4858b | conserved hypothetical protein | MKRRSLLK | |
| b. Additional putative TAT substrates | |||
| PA0205b | probable permease of ABC transporter | MHPIPSPGAALQRROALR | |
| PA1880a | probable oxidoreductase | MNSKIDLSNALPGSRRGFLK | |
| PA2195 | hcnC | hydrogen cyanide synthase HcnC | MNRTYDIVIAGGGVIGASCAYQLSRRGNLR |
| PA2378a | probable aldehyde dehydrogenase | MKRSYPDDLVIGNLSRRGFLK | |
| PA3377 | conserved hypothetical protein | MSLSPQASAAPEQGYNFAYLDEQTKRMIRRGLLK | |
| PA3392a | nosZ | nitrous-oxide reductase precursor | MSDDTKSPHEETHGLNRRGFLG |
| PA3750 | hypothetical protein | MRRLATLAWWAAALPLLPLAVPLAIRTRRRALR | |
| PA3825b | hypothetical protein | MPIPVKRARRKALR | |
| PA4608 | hypothetical protein | MSDQHDERRRFHR | |
| PA4882 | hypothetical protein | MKGPEKKRAKIAIDPSSERQMVDLQRRLLLR | |
| PA5280 | sss | site-specific recombinase Sss | MRADLDAFLEHLRSERQVSAHTLDGYRRDLLK |
| PA5354 | glcE | glycolate oxidase subunit GlcE | MSQPYFDPDAGADHAGALLERVRDALARRAPLR |
In total, 27 potential TAT substrates were identified, 10 of which were not described in either of the two previous studies. The sequence logo derived from the twin arginine motif from these 27 proteins is shown in Figure 1C. The 10 novel substrates we predicted include FepD, HcnC, Sss, GlcE, and six hypothetical or conserved hypothetical proteins. Sss, however, is annotated as a site-specific recombinase and likely represents a false positive associated with the scanning procedure. We believe that these proteins may have been missed in the previous two analyses due to the requirement for an AXA cleavage site in the Ochsner et al. (2002) study, and the minimum of 13 uncharged residues downstream of the twin arginines required by TATFIND.
Eight proteins identified by Ochnser et al. (2002) were not found in the present study. Inspection of these revealed that the proteins either exhibit weakly hydrophobic regions downstream of their motifs or have less than two residues in common with the FL[KR] portion of the TAT motif. We also did not identify 41 proteins reported in Dilks et al. (2003), which we attribute to the fact that our RRXFL[KR] motif requirement is significantly more stringent than is the XRRXXX used in the TATFIND program.
Transmembrane helices for membrane targeting
Many cytoplasmic membrane proteins do not require a cleavable signal sequence in order to insert into the membrane. Instead, the presence of one or more transmembrane α helices and recognition by the SRP is sufficient for membrane-targeting (de Gier et al. 1998; Bernstein 2000; de Gier and Luirink 2001). Of the proteins without a predicted cleavable N-terminal signal peptide, 1042 (18.7%) were predicted by Phobius and TMHMM (Krogh et al. 2001) to contain at least one transmembrane helix (Table 1; Supplemental Table 5). Some of these likely represent N-terminal uncleaved signal anchors, while in most cases it appears that internal helices may be sufficient for cytoplasmic membrane targeting.
PhoA fusion screening to identify exported proteins
In an attempt to test our predictions regarding exported P. aeruginosa proteins, we performed an PhoA fusion screen to identify membrane-localized proteins. Genomic DNA fragments were cloned upstream of an N-terminally truncated PhoA gene (′phoA) to create a library of plasmid-encoded ′phoA fusions. This plasmid library was introduced into Escherichia coli, and the transformants were screened for PhoA activity. In total, we isolated 1035 PhoA positive colonies. After growth in liquid media for plasmid DNA isolation, cultures were re-inoculated onto PhoA indicator agar to examine the stability of the PhoA phenotype. In contrast to colonies transferred multiple times on solid media, cultures grown in liquid media had highly unstable PhoA phenotypes. We reasoned that growth in liquid media strongly selects for mutations that limit the amount of PhoA fusion protein expressed, due to the toxicity of membrane-localized PhoA fusion proteins (Manoil and Traxler 1995) or due to high expression levels. The plasmid used has a high copy number and contains the lac promoter upstream of the ′phoA gene (Mdluli et al. 1995). Cloned P. aeruginosa fragments that produce successful PhoA fusions may also contain strong Pseudomonas promoters; thus, it is possible that the PhoA fusions may be expressed from either the lac promoter, the Pseudomonas promoter, or from both.
Plasmids were purified from all PhoA positive colonies regardless of the stability of the PhoA phenotype. Only plasmids with high and intermediate yields were used as templates in sequencing reactions. In some cases, we observed extremely low plasmid yields, suggesting that plasmid loss had occurred. A total of 646 plasmids were sequenced and mapped to the P. aeruginosa genome to identify the gene randomly cloned upstream of the ′phoA gene. This analysis yielded a total of 474 proteins cloned in the correct orientation to produce a PhoA fusion protein, while the majority of remaining sequences were of poor quality and did not produce high-scoring BLAST hits to the PAO1 genome. Eliminating the redundant BLAST hits reduced the list to 310 unique P. aeruginosa-PhoA fusion proteins (Supplemental Table 6).
The ability of these proteins to direct PhoA to the cytoplasmic membrane is likely due to the presence of an export signal. Of the 310 proteins identified in the PhoA screen, 296 displayed a predicted cleavable N-terminal signal peptide or contained one or more predicted transmembrane helices (Table 4). These data indicate that our consensus computational prediction strategy displayed high recall—in other words, a low number of false-negative results was encountered. However, we are unable to comment on the precision, or false-positive rate, of the strategy, as the PhoA fusion simply indicates export and does not provide information on the nature of the export signal itself.
Table 4.
Export signals predicted in 310 PhoA fusion proteins
| Type of export signal | Number | % of export signals |
|---|---|---|
| Type I | 169 | 57.1 |
| Possible type I | 1 | 0.3 |
| Type II (lipoprotein) | 31 | 10.5 |
| Type IV (prepilin) | 5 | 1.7 |
| TAT | 1 | 0.3 |
| Transmembrane helix | 89 | 30.1 |
| Total with export signals | 296 | — |
| No export signal | 14 | — |
Export signals were annotated as type I, probable type I, type II, type IV, TAT, or transmembrane helix based on the predictions generated in the initial computational screen. Similar proportions of type II and type IV signal peptides were observed in both the whole genome predictions and among the PhoA fusion proteins, indicating that the prediction of these two types of signals might be relatively straightforward, particularly compared with prediction of type I signal peptides. The proportion of type I signal peptides identified in the PhoA screen was ∼20% higher than the proportion predicted genome-wide, indicating that the predictive methods may be missing some noncanonical N-terminal signal peptides or that the PhoA fusion method preferentially identifies type I signal peptides.
PhoA fusions reveal misannotated start sites and confirm expression of 150 hypothetical proteins
There were 14 proteins identified in the PhoA screen that lacked a predicted export signal. These proteins may possess noncanonical export signals not identified by the methods used, or else incorrect assignment of start sites may have caused their true export signals to be missed. Alternatively, they may represent false positives associated with the PhoA screen. To explore these possibilities, these proteins were selected for further examination.
The protein sequences and upstream regions were examined for alternative start sites by manual scanning and GeneMark 2.4 analysis (Lukashin and Borodovsky 1998). Ten proteins displayed potential alternative translation products, which were reanalyzed by the four signal peptide prediction methods. The alternative translation products associated with three proteins (PA0667, PA0259, PA3348) were predicted by all four methods to be exported by a cleavable type I signal peptide.
Next, the remaining proteins were compared to the PSORTdb database of proteins of experimentally verified localization (http://db.psort.org) using a BLASTp search and an Evalue cutoff of 1e - 10. None of the proteins were homologous to exported proteins, but four proteins (PA2451, PA3919, PA4091, PA2744) showed similarity to cytoplasmic proteins. Lastly, the protein sequences were similarly compared to the PSORTdb database of proteins with computationally predicted subcellular localizations. The predictions contained in the database were generated with PSORTb v.2.0, the most precise bacterial subcellular localization predictor available (Gardy et al. 2004). Five proteins showed similarity to predicted cytoplasmic proteins (PA1389, PA3357, PA3673, PA4124, PA4577), and none showed similarity to PSORBdb predicted exported proteins. Of the two remaining proteins, PA1531 is similar to the periplasmic protein of ABC transporters and PA5044 (PilM) is similar to the actin-like protein MreB from E. coli and Bacillus (Mattick 2002). PilM is involved in type IV pili biosynthesis and is necessary for twitching motility (Mattick 2002). These two proteins have no predicted export signal but may contain a unique export signal not identified by our methods. Thus, of the 14 PhoA fusions with no apparent export signal, only five appear to be candidates for export while the remaining nine proteins are probable false positives.
Among the proteins that produced active PhoA fusions, 150 are annotated as hypothetical or conserved hypothetical proteins. This finding suggests that these proteins are likely localized to the membrane and thus provides some preliminary information regarding the function of these proteins that have not as yet been characterized.
Discussion
In the first part of our analysis, a genome-wide computational screen for exported proteins was performed. Multiple predictive methods, including machine learning methods and manual pattern matching, were used to identify P. aeruginosa PAO1 proteins containing possible export signals. In large-scale genome studies, it is critical to employ a consensus approach in order to reduce the number of false positives and to increase the confidence of the prediction. Our study reports ∼100% agreement between the genome-wide predictions and the experimental PhoA fusion data.
The consensus prediction method used here indicates that 38% of the genome encodes proteins exported via five types of export signal: type I signal peptides, type II (lipoprotein) signal peptides, type IV (prepilin) signal peptides, TAT (twin arginine transporter) signal peptides, and membrane-targeting transmembrane helices. Approximately 40% of these predicted exported proteins appear to utilize cleavable type I signal peptides, according to three or more of the predictive methods. A small number of false positives were observed that include four of the 719 proteins with strongly predicted signal peptides (PA2003, PA2554, PA3883, PA5389) that show significant similarity to cytoplasmic proteins and three proteins with weakly predicted signal peptides (PA1949, PA1998, PA2267). Furthermore, predictions may also reflect biases in the programs' training data, such that certain noncanonical type I signal peptides might be missed. This is likely the case with many of the 57 proteins with weakly predicted type I signal peptides since many of them appear to be candidates for export based on their annotated functions; however, at least two of the four methods failed to predict a signal peptide.
The methods used to identify the other classes of signal peptide are more specialized and appear to result in better predictions compared with type I signal peptide methods. LipoP v.1.0 predicted 185 potential lipoproteins in the genome, 109 more than are presently annotated in the pseudomonas.com database. The Pseudomonas Genome Database annotations were calculated using PSORT I, and the increase in predicted lipoproteins reported here illustrates the importance of using up to date computational methods.
We predicted 23 putative prepilin peptidase substrates in the PAO1 genome, of which six represent novel candidates. These include five proteins occurring in a cluster, four of which are annotated as probable type II secretion system proteins and may represent novel type II secretion proteins, similar to the Xcp and Hxc machinery. There are likely more prepilin peptidase substrates within the P. aeruginosa genome, which could be identified through searching with a more degenerate motif. For example, the supposedly invariant Gly residue preceding the cleavage site appears to be replaceable by an Ala residue, as seen in the previously identified FimT protein, as well as in PA2672 and PA2674 reported here.
Fifteen putative TAT substrates were initially identified in the present analysis, which utilized stringent criteria, including the presence of a predicted N-terminal signal peptide and a match of at least five of the six residues in the RRXFL[KR] motif. An expanded analysis searching for potential TAT substrates across the whole genome—not just within the subset of proteins with predicted signal peptides—identified a further 12 possible TAT substrates. This indicates that it is important not to overlook proteins without a predicted signal peptide, as they may contain functioning TAT-directing motifs. In fact, of the 18 previously identified TAT substrates reported by Ochsner et al. (2002), only 11 contain predicted type I signal peptides. Of the 57 putative substrates reported by Dilks et al. (2003), just 28 are predicted to contain a type I signal peptide. Overall, 27 potential substrates were identified, 10 of which have not been previously described. However, there are likely many more TAT substrates within the genome with more degenerate motifs, since our prediction strategy was unable to identify the known TAT substrates phospholipase PlcH and the ferripyoverdine receptor FpvA (Ochsner et al. 2002).
Our computational analysis showed that the majority of exported proteins are likely cytoplasmic membrane proteins that lack cleavable signal peptides but possess one or more transmembrane helices as an export signal. This estimate of proteins possessing transmembrane helices, 18.7% of the P. aeruginosa proteome, is similar to the 18.5% of proteins predicted by PSORTb to be localized to the cytoplasmic membrane. This may reflect the fact that computational prediction of transmembrane helices is generally regarded to be more accurate than the prediction of targeting signals, due to the sequence constraints associated with crossing a lipid bilayer. As signal peptide prediction methods improve, we are likely to see an increase in the number of predicted exported proteins.
PhoA fusion methods are a versatile genetic tool to identify proteins that are translocated across the cytoplasmic membrane. By using a plasmid-based ′phoA screen, 310 unique P. aeruginosa fusion proteins were successfully identified. This approach to identify membrane-localized proteins is as efficient as reported in previous P. aeruginosa membrane proteomic studies (Nouwens et al. 2000; Guina et al. 2003). A significant disadvantage of this approach is the apparent toxicity of PhoA fusion proteins, which often selects for mutations that lead to a loss of PhoA activity or even the loss of plasmid. Furthermore, PhoA fusion analysis is of limited utility in the identification of proteins using certain export systems. Proteins secreted to the extracellular space by the type I or type III systems typically lack N-terminal signal peptides and, in the case of type I ABC transporter substrates, instead rely on a C-terminal secretion signal (Duong et al. 1996). Such proteins will not be identified through PhoA fusions. Proteins using the TAT system represent a more complex case. TAT substrates are folded in the cytoplasm prior to entering the TAT transporter. PhoA, however, is folded in the periplasm, where the necessary disulfide bonds are formed. Interestingly, one protein with a predicted TAT motif did produce an active PhoA fusion. While this could represent a false-positive prediction of a TAT export signal, it may also indicate that the TAT transporter is capable of translocating an unfolded substrate, or that an active PhoA fusion can be formed in the cytoplasm.
Although there is a possibility that certain P. aeruginosa-specific export signals may not be recognized as PhoA fusions expressed in a recombinant E. coli host, the strong conservation of the inner membrane targeting and translocation machinery should not affect the export of most PhoA fusion proteins. In addition, we have used this approach previously to identify secreted proteins in Helicobacter pylori (Bina et al. 1997).
The 310 proteins identified in this PhoA screen reflect many of the known functions associated with the cell envelope. The outer membrane proteins identified included those that function as porins, iron uptake receptors, and efflux channels, the three largest families of outer membrane proteins, and proteins involved in secretion and adhesion. Periplasmic proteins identified included the binding components of ABC transporters, cell wall biosynthesis enzymes, stress response proteases, and chaperones. Inner membrane proteins included transport proteins, chemotaxis transducers, two-component sensors, efflux pumps, cell wall biosynthesis enzymes, and proteins involved in secretion.
The PhoA fusion data provided confirmation that 14% of our predicted exported proteins are indeed exported, although the export signals themselves cannot be identified. The laboratory analysis also identified 14 proteins with no predicted export signal. Three of these 14 contained mispredicted start sites, and the new translation products displayed type I signal peptides. Nine of the remaining proteins showed significant similarity to known and predicted cytoplasmic proteins and likely represent false positives (3%) associated with the PhoA fusion technique. A second class of false positives, not counted in the 310 successful fusions, occurred at a similar rate and included fusions to genes in the opposite orientation of the ′phoA gene. The false positives found in the PhoA screen do not overlap with the false positives found in the computational screen and, had the computational screen not been performed, would have gone unnoticed. The remaining proteins without a predicted signal peptide exhibited significant similarity to an exported protein and a bacterial cytoskeletal protein, however their export signals remain unclear.
In summary, we used a combined computational and experimental approach to identify exported proteins in P. aeruginosa. Our approach illustrates the effectiveness of using two complementary methods for genome-wide analyses. Computational techniques have the advantage of yielding a large number of predictions—ideal for genome-wide studies—and when a consensus method is employed the number of false-positive results is reduced. Laboratory methods, although they generally provide fewer results, can both confirm predictions and reveal interesting cases meriting closer inspection, including erroneous annotations and potentially unusual sequence features. We believe that this combined analytical approach is readily adaptable to other bacteria—the increase of the breadth of training data available means that current export signal predictive methods can be applied to a diverse range of organisms with accurate results, and the PhoA fusion is commonly used to study exported proteins.
In addition to creating the P. aeruginosa signal peptide data set described in this report (Supplemental Table 1), we have provided laboratory-based experimental evidence to confirm the export of 14% of the predicted export candidates, as well as the existence of 150 proteins annotated as hypothetical or conserved hypothetical. The genome-wide identification of exported proteins will help define this important subset of the P. aeruginosa genome and may assist in the discovery of novel drug targets.
Methods
Data set
The version of the P. aeruginosa PAO1 genome used in the present analysis was downloaded from http://www.pseudomonas.com, updated June 10, 2004. This version of the genome contains 5570 proteins.
Prediction of N-terminal signal sequences
Type I signal peptides were predicted by a consensus approach utilizing four signal peptide prediction methods: SignalP v.3.0's neural network and hidden Markov model implementations (Bendtsen et al. 2004), LipoP v.1.0 (Juncker et al. 2003), and Phobius (Kall et al. 2004). A protein was noted as having a type I signal peptide if three or more methods predicted one, and as not having a signal peptide if three or more methods did not predict one. Fifty-seven proteins were noted as having possible type I signal peptides, representing cases where two methods predicted a signal peptide while two methods did not. Type II signal peptides were predicted exclusively by the program LipoP.
Sequences of type IV prepilin precursors and related proteins (Lory 1994) were used to construct the motif G[FIMLSY] [TS][LT][ILVP]E. The motif was then used to scan the P. aeruginosa PA01 genome for possible prepilin peptidase substrates. Downstream hydrophobic tracts were identified using the Kyte and Doolittle (1982) hydrophobicity scale.
Possible TAT substrates were identified by searching for occurrences of the RRXFL[KR] motif (Chaddock et al. 1995), where a protein exhibited the dual arginines as well as matches to at least two of the FL[KR] residues. This set of proteins was filtered to remove proteins with little to no hydrophobic character in the region immediately C-terminal to the TAT motif, again using the Kyte-Doolittle hydrophobicity index.
Proteins utilizing a transmembrane helix for targeting were identified by both Phobius and TMHMM (Krogh et al. 2001). In the absence of a strongly predicted signal peptide (three or more predictions) and given a prediction of two or more transmembrane helices by either Phobius or TMHMM, a protein was annotated as using a TMH for export.
Construction of a P. aeruginosa-PhoA fusion library
E. coli DH5α was used as the recombinant host for a P. aeruginosa-PhoA fusion library. Genomic DNA from P. aeruginosa H103 was isolated, partially digested with Sau3AI, and size fractionated on 1% agarose-Tris-acetate-EDTA (TAE) gels. Digested DNA in the size range 1-3 kb was excised, gel-purified, and ligated into BamHI digested, phosphatase-treated plasmids pJDT1, pJDT2, and pJDT3 that contain single base additions to permit the coding fragment to fuse in the correct reading frame to ′phoA (Mdluli et al. 1995). Ligation products were transformed into electrocompetent E. coli DH5α. Transformants were recovered and screened for PhoA activity on PhoA indicator LB agar containing 75 mM Na2HPO4 to repress endogenous PhoA activity, 100 μg/mL ampicillin, and 90 μg/mL of the chromogenic PhoA substrate BCIP (5-bromo-4-chloro-3-indolyl phosphate) as previously described (Bina et al. 1997). All blue colonies were picked to 96-well microtitre plates and subcultured in LB broth containing 50 μg/mL ampicillin. Plasmids from PhoA positive clones were purified in 96-well format (Qiagen), visualized for yield on 96-lane 1% agarose-TAE gels and sequenced using Big Dye Terminator chemistry (Applied Biosystems) on a Basestation 51 Fragment Analyzer (MJ Research) and a ′phoA-specific sequencing primer directed toward the upstream cloned region as previously described (Bina et al. 1997). The P. aeruginosa genes upstream of the truncated ′phoA gene were mapped to the P. aeruginosa PAO1 genome sequence by BLASTN and BLASTX analyses (Altschul et al. 1990). Routine genetic manipulations were carried out according to Sambrook et al. (Maniatis et al. 1989).
Acknowledgments
We thank Agnes Kwasnika, Amélie Casgrain, and Jiesong Hua for their technical assistance. S.L. is supported by a Canadian Cystic Fibrosis Foundation (CCFF) fellowship. J.L.G. and F.S.L.B. are a Michael Smith Foundation for Health Research trainee and scholar, respectively. R.E.W.H. holds a Canada Research Chair. Funding for this research was from the FPMI program supported by Genome Canada, the Canadian Institutes of Health Research (CIHR), and from CCFF.
Footnotes
Article and publication are at http://www.genome.org/cgi/doi/10.1101/gr.3257305.
[Supplemental information is available online at www.genome.org.]
References
- Alm, R.A. and Mattick, J.S. 1995. Identification of a gene, pilV, required for type 4 fimbrial biogenesis in Pseudomonas aeruginosa, whose product possesses a pre-pilin-like leader sequence. Mol. Microbiol. 16: 485-496. [DOI] [PubMed] [Google Scholar]
- ____. 1996. Identification of two genes with prepilin-like leader sequences involved in type 4 fimbrial biogenesis in Pseudomonas aeruginosa. J. Bacteriol. 178: 3809-3817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alm, R.A., Hallinan, J.P., Watson, A.A., and Mattick, J.S. 1996. Fimbrial biogenesis genes of Pseudomonas aeruginosa: pilW and pilX increase the similarity of type 4 fimbriae to the GSP protein-secretion systems and pilY1 encodes a gonococcal PilC homologue. Mol. Microbiol. 22: 161-173. [DOI] [PubMed] [Google Scholar]
- Altschul, S.F., Gish, W., Miller, W., Myers, E.W., and Lipman, D.J. 1990. Basic local alignment search tool. J. Mol. Biol. 215: 403-410. [DOI] [PubMed] [Google Scholar]
- Ball, G., Durand, E., Lazdunski, A., and Filloux, A. 2002. A novel type II secretion system in Pseudomonas aeruginosa. Mol. Microbiol. 43: 475-485. [DOI] [PubMed] [Google Scholar]
- Bally, M., Filloux, A., Akrim, M., Ball, G., Lazdunski, A., and Tommassen, J. 1992. Protein secretion in Pseudomonas aeruginosa: Characterization of seven xcp genes and processing of secretory apparatus components by prepilin peptidase. Mol. Microbiol. 6: 1121-1131. [DOI] [PubMed] [Google Scholar]
- Bendtsen, J.D., Nielsen, H., von Heijne, G., and Brunak, S. 2004. Improved prediction of signal peptides: SignalP 3.0. J. Mol. Biol. 340: 783-795. [DOI] [PubMed] [Google Scholar]
- Berks, B.C. 1996. A common export pathway for proteins binding complex redox cofactors? Mol. Microbiol. 22: 393-404. [DOI] [PubMed] [Google Scholar]
- Bernstein, H.D. 2000. The biogenesis and assembly of bacterial membrane proteins. Curr. Opin. Microbiol. 3: 203-209. [DOI] [PubMed] [Google Scholar]
- Bina, J.E., Nano, F., and Hancock, R.E.W. 1997. Utilization of alkaline phosphatase fusions to identify secreted proteins, including potential efflux proteins and virulence factors from Helicobacter pylori. FEMS Microbiol. Lett. 148: 63-68. [DOI] [PubMed] [Google Scholar]
- Bleves, S., Voulhoux, R., Michel, G., Lazdunski, A., Tommassen, J., and Filloux, A. 1998. The secretion apparatus of Pseudomonas aeruginosa: Identification of a fifth pseudopilin, XcpX (GspK family). Mol. Microbiol. 27: 31-40. [DOI] [PubMed] [Google Scholar]
- Cachia, P.J. and Hodges, R.S. 2003. Synthetic peptide vaccine and antibody therapeutic development: Prevention and treatment of Pseudomonas aeruginosa. Biopolymers 71: 141-168. [DOI] [PubMed] [Google Scholar]
- Chaddock, A.M., Mant, A., Karnauchov, I., Brink, S., Herrmann, R.G., Klosgen, R.B., and Robinson, C. 1995. A new type of signal peptide: Central role of a twin-arginine motif in transfer signals for the δ pH-dependent thylakoidal protein translocase. EMBO J. 14: 2715-2722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Gier, J.W. and Luirink, J. 2001. Biogenesis of inner membrane proteins in Escherichia coli. Mol. Microbiol. 40: 314-322. [DOI] [PubMed] [Google Scholar]
- de Gier, J.W., Scotti, P.A., Saaf, A., Valent, Q.A., Kuhn, A., Luirink, J., and von Heijne, G. 1998. Differential use of the signal recognition particle translocase targeting pathway for inner membrane protein assembly in Escherichia coli. Proc. Natl. Acad. Sci. 95: 14646-14651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dilks, K., Rose, R.W., Hartmann, E., and Pohlschröder, M. 2003. Prokaryotic utilization of the twin-arginine translocation pathway: A genomic survey. J. Bact. 185: 1478-1483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drew, D., Froderberg, L., Baars, L., and de Gier, J.W. 2003. Assembly and overexpression of membrane proteins in Escherichia coli. Biochim. Biophys. Acta 1610: 3-10. [DOI] [PubMed] [Google Scholar]
- Duong, F., Lazdunski, A., and Murgier, M. 1996. Protein secretion by heterologous bacterial ABC-transporters: The C-terminus secretion signal of the secreted protein confers high recognition specificity. Mol. Microbiol. 21: 459-470. [DOI] [PubMed] [Google Scholar]
- Filloux, A., Michel, G., and Bally, M. 1998. GSP-dependent protein secretion in gram-negative bacteria: The Xcp system of Pseudomonas aeruginosa. FEMS Microbiol. Rev. 22: 177-198. [DOI] [PubMed] [Google Scholar]
- Fridkin, S.K. and Gaynes, R.P. 1999. Antimicrobial resistance in intensive care units. Clin. Chest Med. 20: 303-316. [DOI] [PubMed] [Google Scholar]
- Gardy, J.L., Laird, M.R., Chen, F., Rey, S., Walsh, C.J., Ester, M., and Brinkman, F.S.L. 2004. PSORTb v.2.0: Expanded prediction of bacterial protein subcellular localization and insights gained from comparative proteome analysis, Bioinformatics (in press). [DOI] [PubMed]
- Govan, J.R. and Deretic, V. 1996. Microbial pathogenesis in cystic fibrosis: Mucoid Pseudomonas aeruginosa and Burkholderia cepacia. Microbiol. Rev. 60: 539-574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guina, T., Purvine, S.O., Yi, E.C., Eng, J., Goodlett, D.R., Aebersold, R., and Miller, S.I. 2003. Quantitative proteomic analysis indicates increased synthesis of a quinolone by Pseudomonas aeruginosa isolates from cystic fibrosis airways. Proc. Natl. Acad. Sci. 100: 2771-2776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hancock, R.E.W. and Speert, D.P. 2000. Antibiotic resistance in Pseudomonas aeruginosa: Mechanisms and impact on treatment. Drug Resist. Update 3: 247-255. [DOI] [PubMed] [Google Scholar]
- Juncker, A.S., Willenbrock, H., Von Heijne, G., Brunak, S., Nielsen, H., and Krogh, A. 2003. Prediction of lipoprotein signal peptides in Gram-negative bacteria. Protein Sci. 12: 1652-1662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kall, L., Krogh, A., and Sonnhammer, E.L. 2004. A combined transmembrane topology and signal peptide prediction method. J. Mol. Biol. 338: 1027-1036. [DOI] [PubMed] [Google Scholar]
- Krogh, A., Larsson, B., von Heijne, G., and Sonnhammer, E.L. 2001. Predicting transmembrane protein topology with a hidden Markov model: Application to complete genomes. J. Mol. Biol. 305: 567-580. [DOI] [PubMed] [Google Scholar]
- Kyte, J. and Doolittle, R.F. 1982. A simple method for displaying the hydropathic character of a protein. J. Mol. Biol. 157: 105-132. [DOI] [PubMed] [Google Scholar]
- LaPointe, C.F. and Taylor, R.K. 2000. The type 4 prepilin peptidases comprise a novel family of aspartic acid proteases. J. Biol. Chem. 275: 1502-1510. [DOI] [PubMed] [Google Scholar]
- Lory, S. 1994. Leader peptidases of type IV prepilins and related proteins. In Signal peptidases (ed. G. von Heijne), pp. 31-48. R.G. Landes Co., Austin, TX.
- Lukashin, A.V. and Borodovsky, M. 1998. GeneMark.hmm: New solutions for gene finding. Nucleic Acids Res. 26: 1107-1115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma, Q., Zhai, Y., Schneider, J.C., Ramseier, T.M., and Saier Jr., M.H. 2003. Protein secretion systems of Pseudomonas aeruginosa and P fluorescens. Biochim. Biophys. Acta 1611: 223-233. [DOI] [PubMed] [Google Scholar]
- Maniatis, T., Fritsch, E., and Sambrook, J. 1989. Molecular cloning: A laboratory manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, New York.
- Manoil, C. and Traxler, B. 1995. Membrane protein assembly: Genetic, evolutionary and medical perspectives. Annu. Rev. Genet. 29: 131-150. [DOI] [PubMed] [Google Scholar]
- Mattick, J.S. 2002. Type IV pili and twitching motility. Annu. Rev. Microbio. 56: 289-314. [DOI] [PubMed] [Google Scholar]
- Mdluli, K.E., Treit, J.D., Kerr, V.J., and Nano, F.E. 1995. New vectors for the in vitro generation of alkaline phosphatase fusions to proteins encoded by G+C-rich DNA. Gene 155: 133-134. [DOI] [PubMed] [Google Scholar]
- Nakai, K. and Horton, P. 1999. PSORT: A program for detecting sorting signals in proteins and predicting their subcellular localization. Trends Biochem. Sci. 24: 34-36. [DOI] [PubMed] [Google Scholar]
- Nouwens, A.S., Cordwell, S.J., Larsen, M.R., Molloy, M.P., Gillings, M., Willcox, M.D., and Walsh, B.J. 2000. Complementing genomics with proteomics: The membrane subproteome of Pseudomonas aeruginosa PAO1. Electrophoresis 21: 3797-3809. [DOI] [PubMed] [Google Scholar]
- Nunn, D.N. and Lory, S. 1991. Product of the Pseudomonas aeruginosa gene pilD is a prepilin leader peptidase. Proc. Natl. Acad. Sci. 88: 3281-3285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ochsner, U.A., Snyder, A., Vasil, A.I., and Vasil, M.L. 2002. Effects of the twin-arginine translocase on secretion of virulence factors, stress response, and pathogenesis. Proc. Natl. Acad. Sci. 99: 8312-8317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pugsley, A.P. 1993. The complete general secretory pathway in gram-negative bacteria. Microbiol. Rev. 57: 50-108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russell, M.A. and Darzins, A. 1994. The pilE gene product of Pseudomonas aeruginosa, required for pilus biogenesis, shares amino acid sequence identity with the N-termini of type 4 prepilin proteins. Mol. Microbiol. 13: 973-985. [DOI] [PubMed] [Google Scholar]
- Stover, C.K., Pham, X.Q., Erwin, A.L., Mizoguchi, S.D., Warrener, P., Hickey, M.J., Brinkman, F.S., Hufnagle, W.O., Kowalik, D.J., Lagrou, M., et al. 2000. Complete genome sequence of Pseudomonas aeruginosa PA01, an opportunistic pathogen. Nature 406: 959-964. [DOI] [PubMed] [Google Scholar]
- Strom, M.S. and Lory, S. 1987. Mapping of export signals of Pseudomonas aeruginosa pilin with alkaline phosphatase fusions. J. Bacteriol. 169: 3181-3188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Heijne, G. 1985. Signal sequences: The limits of variation. J. Mol. Biol. 184: 99-105. [DOI] [PubMed] [Google Scholar]
- ____. 1988. Transcending the impenetrable: How proteins come to terms with membranes. Biochim. Biophys. Acta 947: 307-333. [DOI] [PubMed] [Google Scholar]
- ____. 1989. The structure of signal peptides from bacterial lipoproteins. Protein Eng. 2: 531-534. [DOI] [PubMed] [Google Scholar]
- Voulhoux, R., Ball, G., Ize, B., Vasil, M.L., Lazdunski, A., Wu, L.F., and Filloux, A. 2001. Involvement of the twin-arginine translocation system in protein secretion via the type II pathway. EMBO J. 20: 6735-6741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiker, H.G., Wilson, M.A., and Schoolnik, G.K. 2000. Extracytoplasmic proteins of Mycobacterium tuberculosis: Mature secreted proteins often start with aspartic acid and proline. Microbiology 146 (Pt 7): 1525-1533. [DOI] [PubMed] [Google Scholar]
- Yamaguchi, K., Yu, F., and Inouye, M. 1988. A single amino acid determinant of the membrane localization of lipoproteins in E. coli. Cell 53: 423-432. [DOI] [PubMed] [Google Scholar]
WEB SITE REFERENCES
- http://www.pseudomonas.com; Pseudomonas Genome Database.
- http://db.psort.org; PSORTdb.