

Abstract
Aims
The aims of the present study were to evaluate the risk of cardiac failure (CF) associated with 15 anticancer protein kinase inhibitors (PKIs) through a case/noncase analysis and to identify which PK(s) and pathways are involved in PKI‐induced CF.
Methods
In order to evaluate the risk of CF, adjusted reporting odds ratios (aRORs) were calculated for the 15 anticancer PKIs in the World Health Organization safety report database (VigiBase®). We realised a literature review to identify 21 protein kinases (PKs) that were possibly involved in CF caused by PKIs. Pearson correlation coefficients (r) between aRORs and affinity data of the 15 PKIs for the 21 PKs were calculated to identify the cellular target most likely to be involved in PKI‐induced CF.
Results
A total of 141 601 individual case safety reports (ICSRs) were extracted from VigiBase® for the following PKIs: afatinib, axitinib, bosutinib, crizotinib, dasatinib, erlotinib, gefitinib, imatinib, lapatinib, nilotinib, pazopanib, ruxolitinib, sorafenib, sunitinib and vandetanib. Among them, 2594 ICSRs concerned CF. The disproportionality analysis revealed that, for dasatinib, imatinib, bosutinib, sunitinib and nilotinib, disproportionality for CF was significantly higher than for other PKIs, with aRORs of 2.52 [95% CI 2.26, 2.82], 1.79 (95% CI 1.57, 2.03), 1.73 (95% CI 1.18, 2.54), 1.67 (95% CI 1.51, 1.84) and 1.38 (95% CI 1.18, 1.61), respectively. Significant values for correlation coefficients between the product of dissociation constant (pKd) and aROR were observed for two non‐receptor protein kinases: ABL1 (non‐phosphorylated and phosphorylated forms) and ABL2 protein kinases, with values of r = 0.83 (P = 0.0001), r = 0.75 (P = 0.0014) and r = 0.78 (P = 0.0006), respectively.
Conclusion
We observed a higher disproportionality for CF with dasatinib, imatinib, bosutinib, sunitinib and nilotinib than with other PKIs. In addition, the study highlighted the role of ABL tyrosine kinases in CF caused by anticancer PKIs.
Keywords: Abl, cardiac failure, pharmacovigilance, pharmacodynamics, protein kinase inhibitor, oncology
What is Already Known about this Subject
Cardiac failure (CF) is an adverse drug reaction (ADR) shared by several protein kinase inhibitors (PKIs) used in oncology.
PKI‐induced CF in ‘real life’ conditions of use has never been assessed.
Cellular target(s) and pathway(s) leading to clinical CF with PKIs are not known.
What this Study Adds
We assessed the risk of CF associated with 15 PKIs used in oncology.
We developed a new approach, based on pharmacovigilance and pharmacodynamics data, to identify cellular pathways involved in ADRs.
We identified cellular targets likely to be involved in CF ADR: ABL1 and ABL2 tyrosine kinases (Abelson murine leukemia viral oncogene homolog).
Tables of Links
| TARGETS | |
|---|---|
| Enzymes 1 | IRE1 |
| ABL1 | JAK2 |
| ABL2 | JNK |
| AMPKA1 | KIT |
| AMPKA2 | MET |
| B‐Raf | PDGFRα |
| CSFR | PDGFRβ |
| EGFR | PDK1 |
| ERK5 | PERK |
| FGFR1 | PKCδ |
| FGFR3 | RAF1 |
| FLT3 | VEGFR‐1 |
| HER2 | VEGFR‐2 |
Introduction
Protein‐kinase inhibitors (PKIs) were introduced in 1999 for use in various clinical areas. Nowadays, 38 PKIs are available worldwide and hundreds are in development in clinical trials, with expected approval for usage in additional illnesses 3. The introduction of PKIs into oncology was a revolution in many domains. For example, imatinib dramatically increased the 5‐year overall survival rate from 11% to 90% among Philadelphia chromosome‐positive patients with chronic myelogenous leukaemia (CML) 4.
Despite this success, PKIs are also associated with serious adverse drug reactions (ADRs). As a consequence, in addition to cancer treatment, active follow‐up of patients is essential, especially because PKIs are used for significantly long‐term treatments in this at‐risk population. Cardiotoxicity [e.g. cardiac failure (CF), cardiomyopathy, conduction abnormalities, QT prolongation, acute coronary syndromes, myocardial injury, arterial thrombosis and hypertension] represents a common class of ADRs caused by several PKIs 5.
The human protein kinase (PK) gene family targeted by PKIs consists of 518 members 6. The activity of kinases involves the transfer of a phosphate group from a nucleoside triphosphate, which is covalently attached to specific amino acids with a free hydroxyl group. Although PKIs share a common mechanism of action when acting on protein kinases, there are different classes and subclasses. First, the PK targeted by a PKI could be a receptor or a non‐receptor PK. Secondly, the targeted PK could belong to one of three large families: the tyrosine kinase family (<20% of the whole kinome), the serine/threonine kinase family (>70%) and the atypical protein kinase family (<10%). Thirdly, PKIs are specific for the inhibitory binding site: type I PKIs bind to the ATP binding site of the PK, which is a very conservative site that leads to poor drug selectivity; type II PKIs bind to the hydrophobic pocket adjacent to the ATP‐binding site, and are more selective; type III PKIs bind to a cysteine residue that can be variably located in the kinase domain, and is very selective for a single PK 7. Selectivity is a crucial but relative issue in developing PKIs. Ideally, a drug should be able to bind only to the on‐target site with high affinity, to avoid extra toxicity. ADRs can be caused by two separate mechanisms: on‐target toxicity, which leads to an ADR being induced by inhibition of the PKI's target of interest; and off‐target toxicity, leading to an ADR being induced by the inhibition of a secondary or unexpected target(s) of the drug 8.
Cardiological ADRs are often serious, sometimes irreversible and can lead to life‐threatening interventions or the need for a new therapy, which can increase the number of multidrug interactions and the risk of toxicity. ADRs may also lead to discontinuation of the anticancer treatment, to underdosing and, finally, to a poor response to treatment, thus decreasing the survival rate of patients and increasing the medical costs of hospitalization.
Identification of the target(s) and pathway(s) involved in PKI‐induced cardiac outcomes could improve the management and resolution of ADRs, as well as identifying at‐risk conditions 9. Moreover, a better understanding of the mechanisms involved in PKI‐induced cardiotoxicity could be crucial to anticipate the potential cardiotoxicity of new in‐development PKIs and influence the design of future cancer treatment strategies 10.
PKI‐induced CF is one of the most serious cardiac ADRs experienced by PKI‐exposed patients. It can lead to a decreased left‐ventricular ejection fraction, contractile dysfunction, hypertensive cardiac remodelling and cardiac ischaemia 11. At present, the mechanism involved in PKI‐induced CF is unclear.
In the present study, we investigated which PK(s) and pathway(s) are involved in PKI‐induced CF. For this purpose: (i) we used the pharmacovigilance (PV) data for 15 PKIs to perform a disproportionality analysis and to assess the risk for these drugs to induce CF; (ii) from a literature review, we identified 21 PKs involved in cardiovascular and/or cardiac function; and (iii) we identified the cellular targets/pathways most likely to be involved in PKI‐induced CF using PV data and pharmacodynamic (PD) data for the 21 selected PKs.
Methods
Data sources
Pharmacovigilance database
VigiBase® is the World Health Organization's (WHO) safety report database, using data collected from 121 countries 12. It contains approximatively 13 million individual case safety reports (ICSRs), which contain information on drugs suspected to cause ADRs. Drugs are coded using the WHO drug dictionary, covering 150 000 medicines and vaccines. VigiBase® gathers ICSRs from national centres which contain information on patient age, gender, medical history, country, drugs taken, drug initiation and stop dates, indications for the drug, ADRs and their seriousness, and outcomes.
Cardiac failure
ADRs are coded according to the Medical Dictionary for Regulatory Activities (MedDRA). There are five levels in the MedDRA hierarchy, arranged from very specific to very general. The most general level of MedDRA gathers codes grouped into 27 system organ classes (SOCs) according to aetiology, manifestation site and purpose. We identified CF in VigiBase® using the standardized MedDRA query ‘cardiac failure’.
PKIs
To be included in our analysis, a PKI had to have at least 100 ICSRs reported in VigiBase® between January 2001 and December 2015. In addition, their affinity for several PKs should have been screened by Davis et al. 13. Thus, we selected the 15 following PKIs: afatinib, axitinib, bosutinib, crizotinib, dasatinib, erlotinib, gefitinib, imatinib, lapatinib, nilotinib, pazopanib, ruxolitinib, sorafenib, sunitinib and vandetanib. All ICSRs reported for any of these drugs were extracted from VigiBase® for analyses.
Concomitant drugs
At‐risk drugs for CF, known to induce CF independently of exposure to a PKI, were identified from reported comedication in ICSRs 14. Drugs interacting with PKIs are likely to increase plasma concentrations of PKI through pharmacokinetic interactions. Drug–drug interactions (DDIs) reported in the summary of the product characteristics (SPC) were identified between PKIs and comedications.
Affinity data
Data on the binding affinity of PKIs for a PK involved in cardiovascular function and cardiac homeostasis (see below) were extracted from the study by Davis et al. 13. Dissociation constants [expressed in Kd (nM)] for the 15 PKIs selected were retrieved for analyses and convert into product of dissociation constants (pKd) (Table S1).
Identification of PKs involved in cardiovascular function and/or cardiac homeostasis
Cellular targets of PKIs were selected according to our literature search. We searched in the MEDLINE database for titles or abstracts that mentioned a PK and its role in cardiovascular homeostasis, and for a title or abstract that mentioned the mechanisms of PKI cardiotoxicity. Key words used for the search were: ‘protein kinase inhibitors’, ‘protein kinases’, ‘cardiac failure’, ‘cardiotoxicity’ and ‘adverse drug reactions’. This search was completed by including references cited in the selected articles.
We collected the following data from the PK studies: type of study (in vivo or in vitro study, review), the use of a PKI or derivative, the model used (animal, animal cells or human biopsies), effect of the inhibition and/or inactivation of the PK, and the effect of the PKI on cardiomyocytes (CMs) or heart functions. We selected studies according to the presence of (i) the identification of a PK as a potential mediator for a cardiac pathological condition and/or (ii) discussion or assessment of a cardiotoxic effect from exposure to a PKI at the cellular level.
Analyses
Descriptive analyses
Information contained in the ICSRs and used in our analyses were as follows: age, gender and comedications used by the patient at the time of the ADR (at‐risk drugs for CF CF or DDI). Missing data were categorized as ‘unknown’ in the analyses. The SOCs of ADRs were analysed.
Disproportionality analyses
Disproportionality analyses (also known as a case/non‐case analysis) were performed for the 15 selected PKIs. This method compares the proportion of specific a ADR reported for a single drug with the proportion of the same ADR for all other drugs, or for a selected panel of control drugs. Disproportionality was estimated by calculating the reporting odds ratio (ROR). This indicator is easily reproducible and could be adjusted for potential confounders using logistic regression. Calculation of the ROR has been described elsewhere 15, 16. Briefly, if the proportion of ‘Y’ ADRs in patients exposed to drug X (cases) is greater than the proportion of ‘Y’ ADRs in patients not exposed to drug X (non‐cases), this suggests an association between the specific drug and the reaction, and is a potential signal for safety. In the present study, disproportionality was calculated using the adjusted RORs (aRORs). For each PKI, to compare the proportion of CF ADRs (cases) with the proportion of CF ADRs for the 14 other PKIs (noncases), a logistic regression model was used to consider major confounders – that is, the gender and age of the patient at the time of the reaction, comedications potentially related to the CF, DDIs responsible for an increase in PKI concentration, and time since market approval. All aRORs were calculated with their 95% confidence intervals (CIs).
Relationship between disproportionality and PKI affinity
To determine which suspected cellular targets were involved in PKI‐induced CF, we calculated the Pearson correlation coefficients (r) between the affinities of the PKIs for each selected target, and their disproportionality for CF. We assumed that the higher the affinity of the PKI for the cellular target involved in the drug‐induced CF (expressed as pKd), the higher the ‘risk’ of drug‐induced CF (expressed as the aROR for CF). From multiple comparisons, the threshold P‐value of the test was adapted using a Bonferroni correction (File S1). This correction reduced the risk of finding a spurious association with an increased type I error.
To assess the strength of the correlation, we calculated the determination coefficient (r 2) and the adjusted correlation coefficient (r adj). r 2 indicated the proportion of variance predicted by the model and its goodness of fit, and radj allowed for the adjustment of r according to the number of observations in the analysis. Details about the calculation of r and r adj are presented in File S1.
Linear regression lines for each PK tested were constructed to analyse the characteristics of the linear relationship (File S1). Sensitivity analyses were carried out to evaluate the weighting of the outlier data points in the linear relationship. All analyses were performed using the statistical software package SAS®, version 9.4 (SAS institute Inc., Cary, NC, USA). The drug and molecular target nomenclature used here conforms to the British Journal of Pharmacology's Concise Guide to PHARMACOLOGY 2015/16 2.
Results
Pharmacovigilance data
Baseline information from ICSRs
A total of 141 601 ICSRs (including 730 718 different ADRs) were reported in VigiBase® for the 15 selected PKIs (Table 1). Among them, 2594 ICSRs (including 35 481 different ADRs) contained at least one MedDRA preferred term for a CF.
Table 1.
Pharmacodynamic properties of 15 protein kinase inhibitors and adverse drug reactions contained in VigiBase®
| Drug name (year of approval) | Indications | Main targets |
ICSRs reported in VigiBase® N (%) N = 141 601 |
ICSRs of CF reported in VigiBase® n (%) n = 2594 |
|---|---|---|---|---|
| Afatinib (2013) | Advanced/metastatic NSCLC (EGFR+) | EGFR, HER2, HER3, HER4 | 1442 (1.0) | 15 (0.6) |
| Axitinib (2012) | Advanced RCC | VEGFR‐1, VEGFR‐2, VEGFR‐3 | 3000 (2.1) | 39 (1.5) |
| Bosutinib (2012) | CML (ph+) | Bcr‐abl, CSK, Lyn, Hck, PDGFRs, KIT | 987 (0.7) | 28 (1.1) |
| Crizotinib (2011) | Advanced NSCLC (ALK+) | ALK, MET | 3141 (2.2) | 34 (1.3) |
| Dasatinib (2006) | CML (ph+) | Bcr‐abl, KIT, PDGFRβ, Eph | 9861 (7.0) | 375 (14.5) |
| ALL (ph+) | ||||
| Erlotinib (2004) | Advanced/metastatic NSCLC (EGFR+) | EGFR | 24 891 (17.6) | 240 (9.3) |
| Gefitinib (2003) | Advanced/metastatic NSCLC (EGFR+) | EGFR | 4812 (3.4) | 46 (1.8) |
| Imatinib (2001) | CML (ph+) | Bcr‐abl, KIT, DDR1, DDR2, CSFR, PDGFRα, PDGFRβ | 25 503 (18.0) | 577 (22.2) |
| ALL (ph+) | ||||
| MD/MPD (PDGFR+) | ||||
| HES/CEL (FIP1L1+/PDGFRα+) | ||||
| GIST | ||||
| DFSP | ||||
| Lapatinib (2007) | BC (HER2+) | EGFR, HER2 | 12 030 (8.5) | 164 (6.3) |
| Nilotinib (2007) | CML (ph+) | Bcr‐abl, PDGFRα, PDGFRβ, KIT, EPH | 7589 (5.4) | 175 (6.8) |
| Pazopanib (2009) | Advanced RCC | VEGFR‐1, VEGFR‐2, VEGFR‐3, PDGFRα, PDGFRβ, KIT | 8378 (5.9) | 117 (4.5) |
| Soft‐tissue sarcoma | ||||
| Ruxolitinib (2011) | Myelofibrosis | JAK1, JAK2 | 5513 (3.9) | 79 (3.1) |
| Polycythaemia vera | ||||
| Sorafenib (2005) | Hepatocellular carcinoma | B‐Raf, B‐RafV600E, KIT, FLT3, c‐Raf, VEGFR‐2, VEGFR‐3, PDGFRβ | 15 482 (10.9) | 188 (7.3) |
| Advanced RCC | ||||
| Differentiated thyroid carcinoma | ||||
| Sunitinib (2006) | GIST | PDGFRα, PDGFRβ, VEGFR‐1, VEGFR‐2, VEGFR‐3, KIT, FLT3, Ret, CSFR | 18 303 (12.9) | 508 (19.56) |
| Metastatic RCC | ||||
| Pancreatic neuroendocrine tumours | ||||
| Vandetanib (2011) | Medullary thyroid cancer | VEGFR‐2, EGFR | 669 (0.5) | 9 (0.4) |
| Total | 141 601 (100) | 2594 (100) |
ALK, anaplastic lymphoma receptor tyrosine kinase; ALL, acute lymphoblastic leukaemia; BC, breast cancer; Bcr‐abl, Bcr‐abl fusion protein; B‐Raf, B‐Raf proto‐oncogene, serine/threonine kinase; CEL, chronic eosinophilic leukaemia; CF, cardiac failure; CML, chronic myeloid leukaemia; c‐Raf, Raf‐1 proto‐oncogene, serine/threonine kinase; CSFR, colony‐stimulating factor 1 receptor; CSK, c‐src tyrosine kinase; DDR1, discoidin domain receptor tyrosine kinase 1/2; DFSP, dermofibro sarcoma protuberans; EGFR, epidermal growth factor receptor; EPH, ephrin receptor; FI1L1: FIP1‐like 1 protein; FLT3, receptor‐type tyrosine protein kinase FLT3; GIST, gastrointestinal stromal tumour; Hck, HCK proto‐oncogene, Src family tyrosine kinase; HER2/3/4, erb‐b2 receptor tyrosine kinase 2/3/4; HES, hypereosinophilic syndrome; ICRS, individual case safety report; JAK1/2, tyrosine protein kinase JAK1/2; KIT, KIT proto‐oncogene receptor tyrosine kinase; Lyn: LYN proto‐oncogene, Src family tyrosine kinase; MD/MPD, myelodysplastic/myeloproliferative syndrome; MET, MET proto‐oncogene, receptor tyrosine kinase; NSCLC, nonsmall cell lung cancer; PDGFRα/β, platelet‐derived growth factor receptor alpha/beta; Ph, Philadelphia chromosome; RCC, renal cell carcinoma; Ret, ret proto‐oncogene; VEGFR‐1/2/3, vascular endothelial growth factor 1/2/3
Imatinib, erlotinib and sunitinib were the three drugs most frequently reported in all ICSRs (18.0%, 17.6% and 12.9%, respectively). Imatinib, sunitinib and dasatinib represented the drugs most frequently reported with a CF (22.2%, 19.6% and 14.5%, respectively). For dasatinib, bosutinib and sunitinib, CF represented 3.8%, 2.8% and 2.8%, respectively, of the total number of ICSRs for each drug.
The baseline information contained in the ICSRs is presented in Table 2. Among the 141 601 ICSRs for PKIs, ADRs concerned patients of both genders equally. They had a reported mean age of 60.9 years (standard deviation 14.81; missing in 39.7% of ICSRs). In the ICSRs, 30 257 patients (21.37%) died. Among the 2594 ICSRs with CF, ADRs affected equal numbers of male and female patients; these had a mean reported age of 66.3 years. The number of reactions related to an outcome of death was slightly increased in CF ICSRs compared with global ICSR data (25.3% vs. 21.4%). DDIs between CF‐induced PKIs and comedications were identified in 0.7% of ICSRs (Table S4). Comedications associated with a risk of CF were identified in 26.0% of ICSRs.
Table 2.
Baseline information reported in the 141 160 individual case safety reports (ICSRs) concerning the 15 PKIs in VigiBase®
| Total ICSRs (N = 141 601) | ICSRs with CF (n = 2594) | |
|---|---|---|
| Gender, n (%) | ||
| Female | 64 115 (45.3) | 1166 (45.0) |
| Male | 68 215 (48.2) | 1266 (48.8) |
| Unknown | 9271 (6.6) | 162 (6.3) |
| Age | ||
| mean (SD) | 60.90 (14.81) | 66.26 (13.69) |
| n (%) | ||
| 0–39 | 8835 (5.1) | 63 (2.4) |
| 40–59 | 34 409 (19.9) | 377 (14.5) |
| 60–69 | 29 866 (17.3) | 479 (18.5) |
| 70–80 | 23 076 (13.3) | 530 (20.4) |
| ≥80 | 8066 (4.7) | 260 (10.0) |
| Unknown | 68 724 (39.7) | 885 (34.1) |
| Coadministration of drugs interacting with a PKI, n (%) | 583 (0.4) | 18 (0.7) |
| Coadministration of a drug associated with a risk for CF, n (%) | 26 758 (18.9) | 674 (26.0) |
| Deaths, n (%) | 30 257 (21.4) | 656 (25.3) |
CF, cardiac failure; ICSR, individual case safety report; PKI, protein kinase inhibitor; SD, standard deviation
Disproportionality analysis
The aRORs, with their 95% CIs, are presented in Figure 1. For dasatinib, imatinib, bosutinib, sunitinib and nilotinib, disproportionality for CF was significantly higher than for other PKIs, with aRORs of 2.52 (CI 2.26, 2.82), 1.79 (CI 1.57, 2.03), 1.73 (CI 1.18, 2.54), 1.67 (CI 1.51, 1.84]) and 1.38 (CI 1.18, 1.61), respectively.
Figure 1.
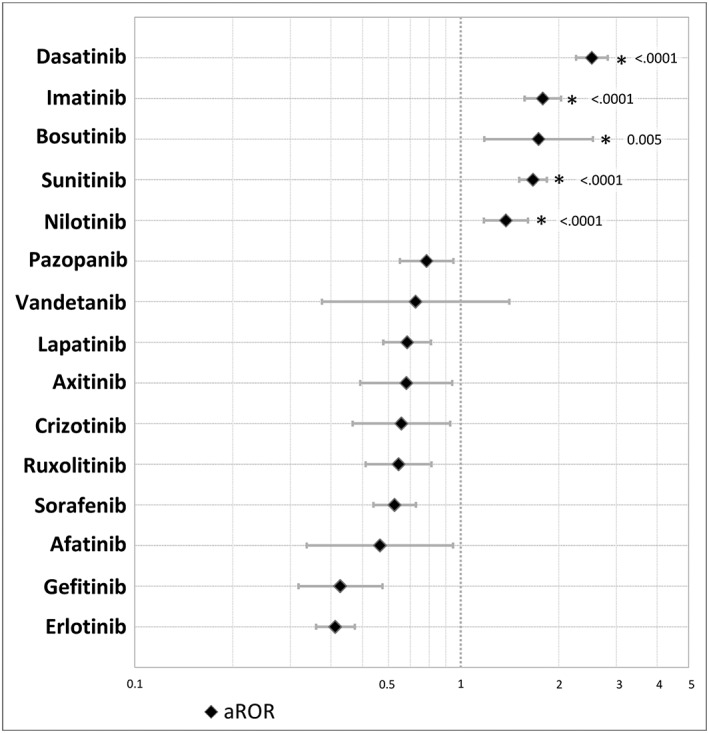
Disproportionality analysis for cardiac failure with 15 protein kinase inhibitors, showing the adjusted reporting odds ratios (aRORs) with confidence intervals and P‐values. aRORs were calculated in multivariate logistic regression, with adjustment for age, gender, drug–drug interactions, at‐risk comedications for cardiac failure, time since approval
Selection of PKs involved in cardiovascular and cardiac homeostasis
We identified 21 PKs involved in cardiovascular functions and/or cardiac homeostasis in the literature review. The results are depicted in File S1 and Table S3.
Relationship between disproportionality for CF and PKI affinity
Figure 2 shows Pearson correlation coefficients (r) between aRORs and pKd values. A significant correlation coefficient between pKd and aROR was observed for two non‐receptor protein kinases: ABL1 (non‐phosphorylated and phosphorylated forms) and ABL2 protein kinases, with values of r = 0.83, r = 0.75 and r = 0.78, respectively. Colony‐stimulating factor 1 receptor (CSF1R) was also found to have a high positive r of 0.70, but this did not reach statistical significance. r, r 2 and r adj data are presented in Table S2. The proportions of variance (r 2) explained by the model were 0.70, 0.56 and 0.61, respectively, for non‐phosphorylated ABL1, phosphorylated ABL1 and ABL2. Values of r adj were similar for ABL1 (non‐phosphorylated and phosphorylated forms) and ABL2 protein kinases (0.82, 0.72 and 0.76, respectively).
Figure 2.

Results of Pearson correlation coefficients (r) (with P‐values) between adjusted reporting odds ratios and product of dissociation constants (pKd) (threshold P‐value adjusted with Bonferroni's correction = 0.0022). ABL1/2, ABL proto‐oncogene 1, nonreceptor tyrosine kinase 1/2; AMPKA1/2, protein kinase AMP‐activated catalytic subunit alpha 1/2; B‐Raf, B‐Raf proto‐oncogene, serine/threonine kinase; CSFR, colony‐stimulating factor 1 receptor; EGFR, epidermal growth factor receptor; ERK5, mitogen‐activated protein kinase 7; FGFR1/3, fibroblast growth factor receptor 1/3; FLT3, receptor‐type tyrosine protein kinase FLT3; JAK2, Janus kinase 2; KIT, mast/stem cell growth factor receptor Kit; MET, MET proto‐oncogene, receptor tyrosine kinase; PDGFR α/β, platelet‐derived growth factor receptor α/β; PDK1, 3‐phosphoinositide‐dependent protein kinase 1; r, correlation coefficient; RAF1, RAF proto‐oncogene serine/threonine‐protein kinase; VEGFR1/2, vascular endothelial growth factor receptor 1/2
The linear regression lines for phosphorylated ABL1, non‐phosphorylated ABL1 and ABL2 are presented in Figure 3. The linear regression lines for phosphorylated ABL1, non‐phosphorylated ABL1 and ABL2 were similar. For non‐phosphorylated ABL1, phosphorylated ABL1 and ABL2, the slopes of the regression lines were positive, indicating a positive association between aRORs and pKd values. The scatters had a relatively balanced spread. The relationship between aRORs and pKd values seemed to be linear, even though some of the outlier points may have influenced the shape of the line (dasatinib, sunitinib). Sensitivity analysis (data not shown), with the exclusion of dasatinib, showed a slight decrease in the correlation coefficient for non‐phosphorylated ABL1, phosphorylated ABL1 and ABL2 (rABL1 non‐pho = 0.73, rABL1 pho = 0.60 and rABL2 = 0.64, respectively). However, excluding sunitinib data from the analyses increased the correlation coefficient for the three PKs (rABL1 nonpho = 0.90, rABL1 pho = 0.80 and rABL2 = 0.86, respectively).
Figure 3.
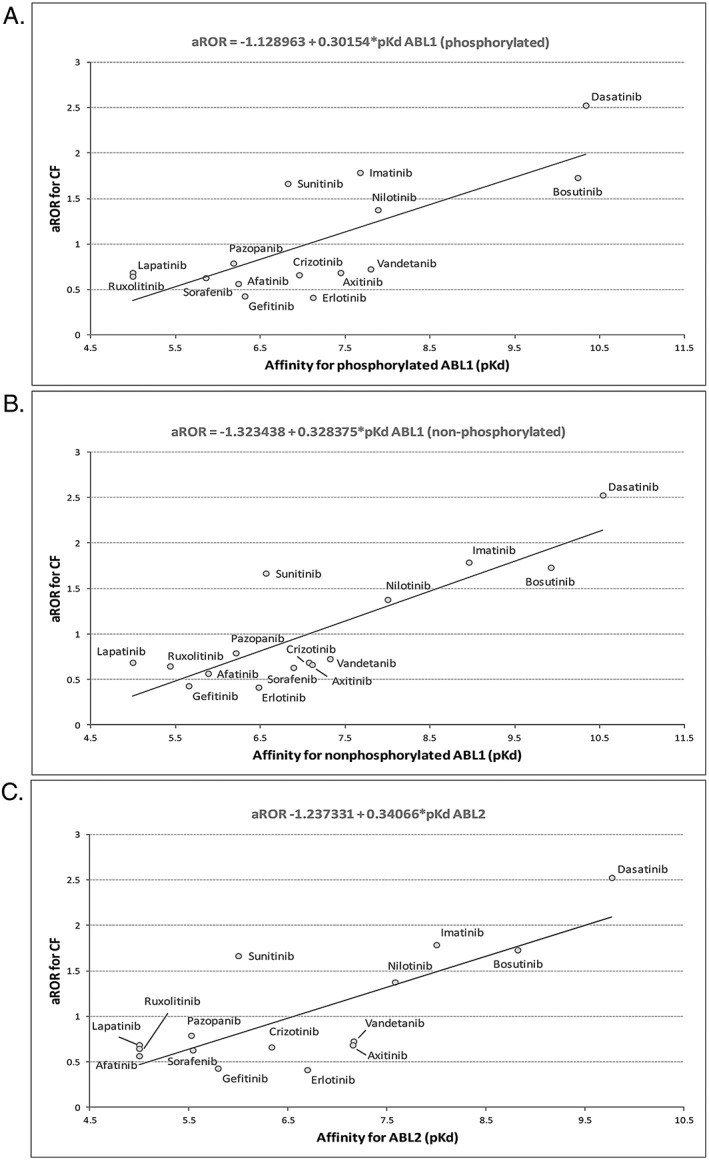
Linear regression lines between aRORs of cardiac failure and affinity (pKd) for (A) non‐phosphorylated ABL1, (B) phosphorylated ABL1 and (C) ABL2. ABL1/2, ABL proto‐oncogene 1, non‐receptor tyrosine kinase 1/2; aROR, adjusted reporting odds ratio; CF, cardiac failure; pKd, product of the dissociation constant
Discussion
Main results
In VigiBase®, 141 601 ICSRs were reported for the selected PKIs. Among them, 1.8% concerned CF. Disproportionality analyses showed that dasatinib, imatinib, bosutinib, sunitinib and nilotinib had significantly higher aRORs than the other PKIs. As far as we know, this was the first time that a method combining PV and PD knowledge had been used to identify the cellular target most probably involved in CF induced by a PKI. This original approach, consisting of comparisons between PV data and the binding properties of PKIs used in oncology, highlighted two cellular targets: ABL1 and ABL2 non‐receptor tyrosine kinases.
The disproportionality results were mostly in accordance with the literature data regarding the potential for the induction of CF for each PKI tested. Indeed, PKIs with the lowest disproportionality (erlotinib, gefitinib, afatinib, ruxolitinib) were also those without any information on CF ADRs in the SPC, whereas other PKIs presented information on CF ADRs in the SPC. Only two drugs were incorrectly ranked in the disproportionality analysis, according to the information on CF ADRs in the SPC: sorafenib ranked with the low‐disproportionality drugs (but with CF ADRs in the SPC), and bosutinib ranked with the high‐disproportionality drugs (but without CF ADRs in the SPC). However, the results in the literature are more heterogeneous. Hasinoff compared the toxicity of seven PKIs in CMs (gefitinib, lapatinib, dasatinib, sorafenib, erlotinib, sunitinib and imatinib), and the damaging effect of these drugs on CMs (lactate dehydrogenase release) was in line with our disproportionality results 17. Erlotinib and gefitinib were found to have low toxicity, whereas imatinib, dasatinib, sorafenib and sunitinib were found to be associated with CM damage. Only two drugs were ranked differently, compared with our results: sorafenib was associated with little CM damage, and lapatinib with none. Regarding the identification of the PK suspected to be involved in CF, Hasinoff et al. also calculated a correlation between the cardiotoxic potential of each PKI and their binding affinity for 242 PKs, but the binding affinity for ABL was not found to have a high correlation with toxicity, and CSF1R PK was found to have the highest correlation. In another study carried out in mice, imatinib was re‐engineered with a lower binding affinity for Abl 18. The compound derived from imatinib was less cardiotoxic in mice, suggesting a role for Abl tyrosine kinase in the PKI‐induced cardiotoxicity, in accordance with our results.
ABL: suspect number one
ABL1 is a non‐receptor tyrosine kinase that plays a role in many key processes linked to cell growth and survival, the response to DNA damage and apoptosis, cell motility, cell adhesion, receptor endocytosis and autophagy. Phosphorylated ABL1 is the active form of the PK, whereas non‐phosphorylated ABL1 is the inactive form. Among selected PKIs, several are known to inhibit ABL1 – that is, bosutinib, dasatinib, imatinib and nilotinib. Bosutinib and dasatinib show type I inhibition, directly on the ATP binding site of the target, whereas nilotinib and imatinib show type II inhibition, using both the ATP binding site and the hydrophobic pocket of the PK. In accordance with the results of Davis et al. 13 (72 PKIs vs. 442 PKs), we observed that more PKIs than expected in clinic had a relative affinity [dissociation constant (Kd) ≤300 nm; threshold from Davis et al. 13) for ABL, although this is not related to the anticancer activity of these drugs – i.e. axitinib (KdABL1 pho = 36 nm), crizotinib (KdABL1 pho = 110 nm), erlotinib (KdABL1 pho = 76 nm), sorafenib (KdABL1nonpho = 130 nm), sunitinib (KdABL1 pho = 150 nm) and vandetanib (KdABL1 pho = 16 nm). This relative affinity for ABL may not be strong enough to induce anticancer activity via inhibition of the breakpoint cluster region Bcr‐abl, but could be sufficient to induce cardiac failure in frail or comorbid patients.
Under pathological conditions, bosutinib, dasatinib, imatinib and nilotinib target the Bcr‐abl fusion protein, an abnormal protein mostly found in patients with CML. The catalytic domain of PKs is highly conserved from ABL1 to mutant Bcr‐abl (SH1 or SHT site, or ATP binding site). Thus, PKIs with on‐target Bcr‐abl (bosutinib, dasatinib, ponatinib, imatinib and nilotinib) share a common off‐target – namely, ABL1.
ABL1 and ABL2 have unique physiological roles but share some functions and have highly conserved sequences and structural domains: SH1 (catalytic kinase activity), SH2 and SH3 domains. Specifically, they share amino‐terminal regulatory and catalytic domains, which are over 90% identical 19. Not surprisingly, PKIs that target the SH1 domain and the hydrophobic pocket in Bcr‐abl also have off‐target activity on ABL2 and ABL1 nonreceptor tyrosine kinases 19.
The findings from our literature review indicated that ABL has a role in cardiac homeostasis and responds to CF/hypertrophy (Table S3) 18, 20, 21, 22, 23. The present study, carried out in ‘real‐life’ conditions, in a worldwide population and outside clinical trials, supports this hypothesis.
CMs require enormous amounts of ATP for contractility, and the hypothesis that certain PKIs lead to apoptosis by inhibiting the prosurvival pathway and impairing mitochondrial/endoplasmic reticulum (ER) function is of interest. Bcr‐abl has direct effects on the prosurvival cellular pathways located in tumour cells but the mechanism involved in cardiac failure is most probably caused by ABL. Figure 4 presents the cellular pathway that is likely to be involved in cardiac failure induced by PKIs. In CM, ABL (located next to the plasma membrane or ER) plays a role in ER homeostasis. Inhibition of ABL by PKIs induces ER stress, which leads to activation of the PKR‐like ER kinase (PERK) and inositol‐requiring enzyme 1 (IRE1) pathways, and to overexpression of protein kinase Cdelta (PKCδ). PERK activates protein translation as part of a protective response. IRE1, which upregulates factors involved in the degradation of misfolded proteins in the ER to restore homeostasis, activates Jun N‐terminal kinases, which leads to mitochondrial depolarization, ATP depletion, cytochrome C release and cell death 24. In addition, direct activation of PKCδ is highly probable, although the exact mechanism is not yet known.
Figure 4.
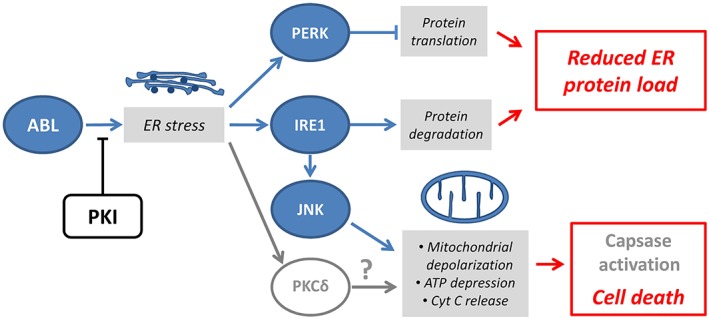
Possible mechanism of PKI‐induced cardiac failure. ABL plays a role in ER homeostasis. Inhibition of ABL by PKI induces ER stress, which leads to activation of the PERK and IRE1, and to overexpression of PKCδ. PERK activates protein translation as part of a protective response. IRE1, which upregulates factors involved in the degradation of misfolded proteins in the ER to restore homeostasis, activates JNKs. This leads to mitochondrial depolarization, ATP depletion, Cyt C release and cell death. Direct activation of PKCδ is also highly probable. ABL, Abelson tyrosine protein kinase; Cyt C, cytochrome C; ER, endoplasmic reticulum; IRE1, serine/threonine protein kinase/endoribonuclease IRE1; JNK, Jun N‐terminal kinases; PERK, PKR‐like ER kinase; PKCδ, protein kinase C delta; PKI, protein kinase inhibitor
Finally, ABL is highly present and active in heart muscle compared with other tissues. Although the activity of PKIs is focused at the location of a cancer, the distribution of the drug is not limited to the target tissue and its distribution in the body is a function of several parameters. Vascularization of the organs is one of the parameters governing the distribution of drugs in the body. As the vascularization of the heart is highly developed, this organ is critical in ADRs and is highly sensitive to toxicity. Furthermore, the volume of distribution is a theoretical volume that is directly correlated with the amount of drug distributed into the tissues, compared with the concentration of the drug in the plasma. Most of the PKIs tested in the present study had a large volume of distribution, suggesting important exposure to these agents in organs other than the plasma.
PV/PD: identification of the suspects
Identifying the pharmacological mechanisms involved in an ADR is a major current challenge in drug safety. Tagging the cellular target as well as the cellular pathways involved in the toxic mechanism is crucial to elucidate the full range of activities of a drug in humans. It is necessary to understand better the roles and interactions between cellular components and pathways. In addition, an understanding of the mechanism of toxicity could help to resolve an ADR. This would enable the establishment of a tailored safety programme to detect early ADRs, help to choose the most appropriate treatment to resolve an ADR and enable an appropriate switch to other drugs with a different mechanism, when and if possible.
The PD properties of drugs in development are known long before their first administration to patients as these suggest its possible efficacy and future indication. If the mechanisms involved in ADRs caused by marketed drugs were to be identified, we could estimate the toxicity of unknown compounds and anticipate appropriate risk management plans for new drugs, even before the first administration to humans. The PV/PD method could also be complementary to the long PV process (phase IV trials) following the marketing of a new drug under ‘real‐life’ conditions.
A safety assessment is essential before the full approval of a new drug; however, clinical trials are mainly designed with the objective of efficacy, with safety often considered a secondary objective. During the last decade, rapid innovation has led to a shortened duration of drug development, from 10 years before 2010 to 6 years or less today 25. This trend has led to a reduced duration of exposure to new drugs and a decreased number of exposed subjects in clinical trials. Furthermore, little is known about the effects of these drugs on comorbid or frail patients, who are usually excluded from clinical trials. Big data collection in healthcare and analyses on pharmacoepidemiology and PV databases may be the only way to assess rapidly the safety of marketed drugs after approval in ‘real‐life’ conditions of use 26.
To the best of our knowledge, no methods have demonstrated predictive value in predicting the targets and mechanisms involved in ADRs. In vivo and in vitro studies are the standard methods used to evaluate a potential target in the development of an ADR. The PV/PD method allows screening of several targets in a one‐shot analysis. This could be a complementary method used in in vivo and in vitro studies. Application of the PV/PD method is expected for other safety issues 27.
Strengths of the study
The RORs were calculated using VigiBase® data, the most important PV database worldwide. The ROR is a reproducible tool to evaluate disproportionality in PV.
The binding properties of a PKI have been screened in in vitro competitive binding assays 13. For each PKI, Kd values were determined using 11 serial threefold dilutions and a dimethyl sulfoxide control 13. This method, used to measure affinity, is accurate and reproducible. We chose to focus our analysis on PKIs tested using this specific method, to avoid bias and errors caused by a change in the method of measurement. Other interaction maps for affinity binding between PKIs and PKs exist and have been screened 28, 29. We gave priority to the largest interaction map for PKs.
We performed a thorough literature review to select the most probable PKs involved in drug‐induced CF. This first step in the analysis was crucial, to avoid spurious associations caused by a large number of analyses, which are a source of increased type I errors.
Limitations of the study
PV programmes are mostly based on spontaneous reporting systems. As a consequence, accuracy and the amount of information reported in cases may not be optimal. This could have led to misclassification of CF cases. PV data also suffer from other bias, such as underreporting, halo bias, a lack of information on the exposed population, and the sales data for the drugs. In addition, in the present study, disproportionality was used as a very early proxy of the relative risk, although this relationship is not clear 30.
The model used in the analyses to link PD data and CF was based on two hypotheses. First, the cardiac effect of a PKI was caused by a single PK. The model was not able to detect co‐inhibition of multiple pKs or inhibition/activation of non‐PK cellular targets (e.g. proteasomes, G protein‐coupled receptors, voltage‐gated ion channels or ligand‐gated ion channels). Secondly, we assumed that the mechanism was similar for all the selected PKIs, even though there is heterogeneity in this family. However, some similarities were observed during drug‐induced CF, such as a loss of CMs in apoptosis, caused by energy‐impaired homeostasis with ER and mitochondrial abnormalities 8, 31.
Accordingly, we observed some heterogeneity within the linear regression lines between disproportionality and the affinity of PKIs for non‐phosphorylated ABL1, phosphorylated ABL1 and ABL2. Even though some scatter points seemed to follow the regression line correctly (imatinib, dasatinib, bosutinib, sunitinib and nilotinib), it should be emphasized that the rest of the scatter did not. This difference might have been caused by the uncertainty of measuring two tested variables (especially for drugs with a very weak affinity for ABL), but also the inadequacy of the model for these drugs. Other cellular mechanisms for CF associated with some PKIs cannot be excluded.
Conclusion
The present study is the first to investigate the disproportionality of CF induced by PKIs in the WHO global safety database. We observed a significantly higher aROR for CF with dasatinib, imatinib, bosutinib, sunitinib and nilotinib than with other PKIs. In addition, we developed the first approach to combining our PV and PD knowledge to identify cellular targets related to ADRs. The study highlights the role of ABL tyrosine kinases in CF caused by anticancer PKIs. This method could be used as a complementary method in in vitro and in vivo assays to determine cellular pathways involved in ADRs. This method is reproducible and could be applied to other drugs and other types of ADR. Identification of cellular targets with PV and PD knowledge of approved drugs could allow very early prediction of the toxicity of new drugs, according to the affinity data measured during the preclinical development stage.
Competing Interests
Both authors declare: no support from any organization for the submitted work; no financial relationships with any organizations that might have an interest in the submitted work in the previous 3 years; no other relationships or activities that could appear to have influenced the submitted work.
Contributors
E.P.D.C., F.D. and M.L.M. were responsible for formulating the research question, designing and carrying out the study and analysing the data. E.P.D.C. wrote the article with F.D., and M.L.M., E.B.G., F.M., G.L. and J.L.M. revised it.
The opinions and conclusions in this study are not necessarily those of the various centres or of the WHO.
Supporting information
File S1 Additional details on methods and results
Table S1 Dissociation constants (Kd) and the product of dissociation constants (pKd) of the 15 protein kinase inhibitors for the 21 protein kinases involved in cardiovascular functions and/or cardiac homeostasis
Table S2 Pearson correlation coefficients (r), determination coefficients (r 2) and adjusted correlation coefficients (r adj) between adjusted reporting odds ratios and values of product of dissociation constants (pKd)
Table S3 Selected protein kinases involved in cardiovascular function and cardiac homeostasis
Table S4Drug–drug interactions between protein kinase inhibitors and concomitantly administered drugs
Patras de Campaigno, E. , Bondon‐Guitton, E. , Laurent, G. , Montastruc, F. , Montastruc, J.‐L. , Lapeyre‐Mestre, M. , and Despas, F. (2017) Identification of cellular targets involved in cardiac failure caused by PKI in oncology: an approach combining pharmacovigilance and pharmacodynamics. Br J Clin Pharmacol, 83: 1544–1555. doi: 10.1111/bcp.13238.
References
- 1. Southan C, Sharman JL, Benson HE, Faccenda E, Pawson AJ, Alexander SP, et al. The IUPHAR/BPS Guide to PHARMACOLOGY in 2016: towards curated quantitative interactions between 1300 protein targets and 6000 ligands. Nucl Acids Res 2016; 44 (Database Issue): D1054–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Alexander SPH, Fabbro D, Kelly E, Marrion N, Peters JA, Benson HE, et al. The Concise Guide to PHARMACOLOGY 2015/16: Enzymes. Br J Pharmacol 2015; 172: 6024–6109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Roskoski R. A historical overview of protein kinases and their targeted small molecule inhibitors. Pharmacol Res 2015; 100: 1–23. [DOI] [PubMed] [Google Scholar]
- 4. Bower H, Björkholm M, Dickman PW, Höglund M, Lambert PC, Andersson TM‐L. Life expectancy of patients with chronic myeloid leukemia approaches the life expectancy of the general population. J Clin Oncol 2016; 34: 2851–2857. [DOI] [PubMed] [Google Scholar]
- 5. Moslehi JJ, Deininger M. Tyrosine kinase inhibitor‐associated cardiovascular toxicity in chronic myeloid leukemia. J Clin Oncol 2015; 33: 4210–4218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Manning G, Whyte DB, Martinez R, Hunter T, Sudarsanam S. The protein kinase complement of the human genome. Science 2002; 298: 1912–1934. [DOI] [PubMed] [Google Scholar]
- 7. Smyth LA, Collins I. Measuring and interpreting the selectivity of protein kinase inhibitors. J Chem Biol 2009; 2: 131–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Cheng H, Force T. Why do kinase inhibitors cause cardiotoxicity and what can be done about it? Prog Cardiovasc Dis 2010; 53: 114–120. [DOI] [PubMed] [Google Scholar]
- 9. Mellor HR, Bell AR, Valentin J‐P, Roberts RRA. Cardiotoxicity associated with targeting kinase pathways in cancer. Toxicol Sci 2011; 120: 14–32. [DOI] [PubMed] [Google Scholar]
- 10. Kerkelä R, Grazette L, Yacobi R, Iliescu C, Patten R, Beahm C, et al Cardiotoxicity of the cancer therapeutic agent imatinibmesylate. Nat Med 2006; 12: 908–9016. [DOI] [PubMed] [Google Scholar]
- 11. Chen MH, Kerkela R, Force T. Mechanisms of cardiomyopathy associated with tyrosine kinase inhibitor cancer therapeutics. Circulation 2008; 118: 84–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Strandell J, Wahlin S. Pharmacodynamic and pharmacokinetic drug interactions reported to VigiBase, the WHO global individual case safety report database. Eur J Clin Pharmacol 2011; 67: 633–641. [DOI] [PubMed] [Google Scholar]
- 13. Davis MI, Hunt JP, Herrgard S, Ciceri P, Wodicka LM, Pallares G, et al. Comprehensive analysis of kinase inhibitor selectivity. Nat Biotechnol 2011; 29: 1046–1051. [DOI] [PubMed] [Google Scholar]
- 14. Insuffisances cardiaques médicamenteuses en bref. Rev Prescrire 2014; 34 (Suppl. 374): 602. [Google Scholar]
- 15. Montastruc J‐L, Sommet A, Bagheri H, Lapeyre‐Mestre M. Benefits and strengths of the disproportionality analysis for identification of adverse drug reactions in a pharmacovigilance database. Br J Clin Pharmacol 2011; 72: 905–908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. van Puijenbroek EP, Bate A, Leufkens HGM, Lindquist M, Orre R, Egberts ACG. A comparison of measures of disproportionality for signal detection in spontaneous reporting systems for adverse drug reactions. Pharmacoepidemiol Drug Saf 2002; 11: 3–10. [DOI] [PubMed] [Google Scholar]
- 17. Hasinoff BB. The cardiotoxicity and myocyte damage caused by small molecule anticancer tyrosine kinase inhibitors is correlated with lack of target specificity. Toxicol Appl Pharmacol 2010; 244: 190–195. [DOI] [PubMed] [Google Scholar]
- 18. Fernández A, Sanguino A, Peng Z, Ozturk E, Chen J, Crespo A, et al. An anticancer C‐Kit kinase inhibitor is reengineered to make it more active and less cardiotoxic. J Clin Invest 2007; 117: 4044–4054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Greuber EK, Smith‐Pearson P, Wang J, Pendergast AM. Role of ABL family kinases in cancer: from leukaemia to solid tumours. Nat Rev Cancer 2013; 13: 559–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Van Etten RA. Cycling, stressed‐out and nervous: cellular functions of c‐Abl. Trends Cell Biol 1999; 9: 179–186. [DOI] [PubMed] [Google Scholar]
- 21. Sun X, Majumder P, Shioya H, Wu F, Kumar S, Weichselbaum R, et al. Activation of the cytoplasmic c‐Abl tyrosine kinase by reactive oxygen species. J Biol Chem 2000; 275: 17237–17240. [DOI] [PubMed] [Google Scholar]
- 22. Hu W, Lu S, McAlpine I, Jamieson JD, Lee DU, Marroquin LD, et al. Mechanistic investigation of imatinib‐induced cardiac toxicity and the involvement of c‐Abl kinase. Toxicol Sci 2012; 129: 188–199. [DOI] [PubMed] [Google Scholar]
- 23. Li B, Wang X, Rasheed N, Hu Y, Boast S, Ishii T, et al. Distinct roles of c‐Abl and Atm in oxidative stress response are mediated by protein kinase C delta. Genes Dev 2004; 18: 1824–1837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Cheng H, Kari G, Dicker AP, Rodeck U, Koch WJ, Force T. A novel preclinical strategy for identifying cardiotoxic kinase inhibitors and mechanisms of cardiotoxicity. Circ Res 2011; 109: 1401–1409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Preziosi P. Faster drug approval: challenges for safety. Expert Opin Drug Saf 2016; 1–14. [DOI] [PubMed] [Google Scholar]
- 26. Laporte J‐R. Fifty years of pharmacovigilance – medicines safety and public health. Pharmacoepidemiol Drug Saf 2016; 25: 725–732. [DOI] [PubMed] [Google Scholar]
- 27. Nguyen TTH, Pariente A, Montastruc J‐L, Lapeyre‐Mestre M, Rousseau V, Rascol O, et al. An original pharmacoepidemiologic – pharmacodynamic method: application to antipsychotic‐induced movement disorders. Br J Clin Pharmacol 2016. (In Press) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Anastassiadis T, Deacon SW, Devarajan K, Ma H, Peterson JR. Comprehensive assay of kinase catalytic activity reveals features of kinase inhibitor selectivity. Nat Biotechnol 2011; 29: 1039–1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Karaman MW, Herrgard S, Treiber DK, Gallant P, Atteridge CE, Campbell BT, et al. A quantitative analysis of kinase inhibitor selectivity. Nat Biotechnol 2008; 26: 127–132. [DOI] [PubMed] [Google Scholar]
- 30. Maciá‐Martínez M‐A, de Abajo FJ, Roberts G, Slattery J, Thakrar B, Wisniewski AFZ. An empirical approach to explore the relationship between measures of disproportionate reporting and relative risks from analytical studies. Drug Saf 2016; 39: 29–43. [DOI] [PubMed] [Google Scholar]
- 31. Force T, Krause DS, Van Etten RA. Molecular mechanisms of cardiotoxicity of tyrosine kinase inhibition. Nat Rev Cancer 2007; 7: 332–344. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
File S1 Additional details on methods and results
Table S1 Dissociation constants (Kd) and the product of dissociation constants (pKd) of the 15 protein kinase inhibitors for the 21 protein kinases involved in cardiovascular functions and/or cardiac homeostasis
Table S2 Pearson correlation coefficients (r), determination coefficients (r 2) and adjusted correlation coefficients (r adj) between adjusted reporting odds ratios and values of product of dissociation constants (pKd)
Table S3 Selected protein kinases involved in cardiovascular function and cardiac homeostasis
Table S4Drug–drug interactions between protein kinase inhibitors and concomitantly administered drugs