

Abstract
Objective
Psychiatric symptoms are a significant aspect of Huntington disease (HD), an inherited neurodegenerative illness. The presentation of these symptoms is highly variable in patients, and their course does not fully correlate with motor or cognitive disease progression. We sought to better understand the development and longitudinal course of psychiatric manifestations in patients who carry the HD mutation starting from the prodromal period prior to motor diagnosis.
Method
Longitudinal measures for up to 10 years of psychiatric symptoms from the Symptom Checklist-90-Revised were obtained from 1305 participants (1007 carrying the HD mutation) and 1235 companions enrolled in the Neurobiological Predictors of Huntington’s Disease (PREDICT-HD) study. Participants with the HD mutation were stratified into three groups according to probability of motor diagnosis within five years. Using linear mixed effects regression models, differences in psychiatric symptoms at baseline and over time between HD mutation positive groups and controls were compared as well as between HD mutation participants and their companions.
Results
19 of 24 psychiatric measures showed significant increases at baseline and longitudinally in HD mutation carrying individuals or their companions versus controls. The differences were greatest when comparing symptom reports from companions (versus self-report), especially in participants who were closest to motor diagnosis.
Conclusions
Results indicate psychiatric manifestations develop more often than previously thought in the HD prodrome. Symptoms also increase with progression of disease severity. Companions of HD mutation carriers also report greater psychiatric symptoms over time compared to affected individuals, consistent with decreasing awareness.
Introduction
Huntington disease is an autosomal dominant neurodegenerative disease that typically manifests in adulthood with motor, cognitive and psychiatric symptoms. The disease occurs due to an expanded number of cytosine-adenine-guanine (CAG) trinucleotide repeats in the protein-coding DNA sequence of the Huntingtin gene on chromosome 4. A formal diagnosis of Huntington disease is usually given based on the presence of significant motor symptoms, including chorea, rigidity, and bradykinesia. Huntington disease is progressive over several years, leading to functional decline and premature death (1, 2). Cognitive changes include progressive deficits in learning, executive and sensory functions and attention, resulting in dementia (3–6). Psychiatric manifestations in Huntington disease include depression, irritability, apathy, perseverations, obsessions, and occasionally psychosis (7–14). With a few exceptions (7, 15), psychiatric changes are not typically related to indices of progression.
The neurodegeneration of striatal and cortical structures underlying the symptomatic presentation of Huntington disease has been known for some time, and while there is an association between the number of CAG repeats and age of onset, there is significant unexplained variability in the presentation and severity of symptoms, in particular its psychiatric manifestations. The ability to test for the Huntington disease mutation prior to onset of symptoms, known as the illness prodrome, has provided the opportunity to study the development of disease manifestations over time. The longitudinal Neurobiological Predictors of Huntington’s Disease study (PREDICT-HD) has identified motor, cognitive, psychiatric, and brain imaging changes that occur in individuals with the Huntington disease mutation (16–19). We reported an increase of psychiatric syndromes for individuals in the prodrome of Huntington disease at the time of study entry, and we have shown increases in symptoms cross-sectionally as estimated proximity to motor diagnosis nears (4, 19–21).
Here we report longitudinal psychiatric ratings from up to ten years of PREDICT-Huntington disease follow-up in order to better characterize psychiatric symptoms that occur in the Huntington disease prodrome up to and through motor conversion. We have also developed statistical models to evaluate whether there are differences in the trajectories of psychiatric manifestations as reported by participants versus companions, which has implications for psychiatric assessment in Huntington disease and other disorders where changes in awareness are prominent (4, 22).
Method
Participants
Participants were from the PREDICT-HD study at 33 sites in six countries (USA, Canada, Germany, Australia, Spain, and UK). Eligible participants had to have independently underwent testing for the Huntington disease gene mutation and knew their gene status prior to study participation. That is, independent from study enrollment, every participant was aware of their family history and gene mutation status for HD. The study did not enroll participants who were at-risk but had not undergone predictive testing for the HD gene mutation. Recruitment efforts included the following: talks at regional, national and international Huntington disease lay meetings, flyer dissemination to genetic counseling and Huntington disease clinics, development of a PREDICT-HD website and links to/from other Huntington disease sites. Individuals with the CAG repeat expansion (CAG≥36) (23, 24) served as cases while those who tested negative for the gene expansion (CAG<36) served as controls. All were 18 years of age or older and not diagnosed with manifest Huntington disease at study entry. Exclusion criteria included unstable medical or psychiatric illness, active substance abuse, history of a significant developmental cognitive disorder, significant history of head trauma or other central nervous system disease, the presence of a pacemaker or other metallic implants, use of antipsychotic medication in the six months before enrollment, or the use of phenothiazine antiemetic medication in the three months before enrollment. Study protocol was approved by each site’s respective Institutional Review Board. After complete description of the study to the participants, written informed consent was obtained.
All participants were seen at study sites annually. The analysis used data from 1305 participants with 6112 observations and 1235 companions with 5365 observations. Companions were predominately spouse/partner (74%) followed by friend/neighbor (8%), parent (7%), and sibling (5%), and approximately 75% of companions reported living with the participants. The mean number of years companions reported knowing the participants was 20.77 (SD=13.59) years. The median number of follow-up visits was 5 (range=1–10).
Progression Groups
Individuals entered PREDICT-HD with different genetic exposure according to their CAG repeat lengths and current ages. To yield valid inferences, gene-expanded participants were classified into three groups based on their CAG-Age Product (CAP) score (25) computed as CAP=(Age at baseline)×(CAG–33.66). The CAG-Age Product formula was derived from a parametric accelerated failure time model predicting motor diagnosis from age at entry, CAG length, and their interaction. CAG-Age Product is similar to the “disease burden” score of Penny et al. (26) and purports to index the cumulative toxicity of mutant huntingtin at the time of study entry. Cutoffs were derived for the best fitting subgroups based on an optimization algorithm using an earlier sample of PREDICT-HD participants (25). Based on gene status and the CAG-Age Product distribution, four groups were defined in this analysis: Control (gene non-expanded), Low, Medium, and High probability of motor diagnosis within five years. The estimated time to motor diagnosis for each CAG-Age Product group is >12.8 years for the Low group, 7.6–12.8 years for the Medium group, and <7.6 years for the High group.
Measures
The Symptom Checklist-90-Revised (SCL-90-R) is a 90-item self-report scale of psychiatric symptomatology with each item rated on a scale of 0=not at all to 4=extremely, based on the degree of distress over the previous seven days (27). Three global measures of psychiatric symptoms and scores for nine specific symptom domains are produced from the scale: GSI=Global Severity Index; PST=Positive Symptom Total; PSDI=Positive Symptom Distress Index; SOM=Somatization; O-C=Obsessive–Compulsive; I-S=Interpersonal Sensitivity; DEP=Depression; ANX=Anxiety; HOS=Hostility; PHOB=Phobic Anxiety; PAR=Paranoid Ideation; and PSY=Psychoticism. For all participants, the SCL-90-R was administered by a trained staff member. The PREDICT-HD protocol requested annual exams including standardized cognitive, motor, functional and psychiatric ratings as well as brain scans.
Statistical Methods
Baseline characteristics were compared by group using an analysis of variance (ANOVA) for continuous variables and a chi-square test for categorical variables. All tests were two-tailed tests.
Group comparison of baseline and longitudinal change
To examine potential differences between the controls and each of the gene expanded groups at baseline and over time, participant and companion ratings were analyzed separately using linear mixed-effects regression (28). The time metric was duration, defined as the time since study entry expressed in years. All models included sex, years of education, and age at entry as covariates. Three linear mixed-effects regression models were fitted for each outcome variable: no effect, baseline differences only, and baseline and longitudinal differences among groups. The models were evaluated using Akaike’s information criterion (AIC), corrected for small-sample bias (AICc). The effect sizes were the t-values of either the intercept (baseline difference) or slope difference (longitudinal change) among groups, each computed as the difference of the sample estimates divided by its standard error. Detail of all statistical analyses are provided in the supplemental data.
Participant and companion comparison
To test whether there were longitudinal differences between participant and companion ratings in each group, participant and companion ratings were modeled simultaneously using multiresponse linear mixed-effects regression. All models included sex, years of education, and age at entry as covariates. The models were assessed by AICc values, as described above, to examine which groups had statistically reliable slope differences. The relative importance of the longitudinal change discrepancy of each group was assessed by the sum of the weights (wAICc) across all models with unequal group slopes, as described by Burnham and Anderson (29). A sum closer to 1 indicates higher importance. Model parameters were averaged over all the candidate models after multiplying the weight of the model and the estimated parameters for the given model. Using these model-averaged parameters over all models, fitted curves for participant and companion ratings were drawn as described by Burnham and Anderson (29).
Results
Baseline and Longitudinal Change in Self and Companion-Reported SCL-90-R
Participant characteristics at study entry are presented in Table 1. As expected there were significant age differences between the prodromal Huntington disease groups with increasing age associated with increasing proximity to estimated motor diagnosis. Of the 24 psychiatric outcome variables examined, 19 showed significant baseline and longitudinal differences between prodromal Huntington disease and the healthy controls (eight participant ratings: Global Severity Index, Positive Symptom Total, Positive Symptom Distress Index, Obsessive–Compulsive, Interpersonal Sensitivity, Depression, Hostility, and Phobic Anxiety; 11 companion ratings: Global Severity Index, Positive Symptom Total, Positive Symptom Distress Index, Somatization, Obsessive–Compulsive, Interpersonal Sensitivity, Depression, Anxiety, Hostility, Phobic Anxiety, and Psychoticism). Three participant ratings (Anxiety, Paranoid Ideation, Psychoticism) and one companion rating (Paranoid Ideation) showed only baseline differences between the prodromal Huntington disease cases and the healthy controls (i.e., no longitudinal differences). Please see Table 2.
TABLE 1.
Participant characteristics at study entry
| Group | Group Comparison | |||||
|---|---|---|---|---|---|---|
| Control | Low | Medium | High | |||
| N of participants | 298 | 280 | 356 | 371 | ||
| N of companions | 282 | 262 | 335 | 356 | ||
| Sex (% female) | 64.43 | 67.5 | 65.45 | 58.76 | X2(3)=6.21 (p=0.10) | |
| Age (years) | Mean | 44.11 | 34.16 | 40.45 | 44.42 | F(3,1301)=66.45 (p<0.001) |
| SD | 11.76 | 8.47 | 9.6 | 10.34 | ||
| CAG repeat | Median | 19 | 41 | 42 | 43 | F(3,1291)=4985.84 (p<0.001) |
| Range | 12–35 | 36–47 | 38–49 | 39–61 | ||
| Education (years) | Mean | 14.85 | 14.51 | 14.54 | 14.18 | F(3,1301)=3.58 (p=0.014) |
| SD | 2.57 | 2.47 | 2.62 | 2.79 | ||
Low, Med, High indicate low, medium, or high probability of diagnosis within five years as estimated by CAG-Age Product (CAP) score.
TABLE 2.
Model comparison for participant and companion SCL-90-R measures
| Variable | Model | Group Effect | Participant AICc |
Participant dAICc |
Participant wAICc |
Companion AIC |
Companion dAICc |
Companion wAICc |
|---|---|---|---|---|---|---|---|---|
| GSI | 1 | Null | 44820.28 | 31.77 | 0.00 | 40001.38 | 54.79 | 0.00 |
| 2 | Baseline | 44792.62 | 4.11 | 0.11 | 39968.83 | 22.24 | 0.00 | |
| 3 | Baseline + Longitudinal | 44788.51 | 0.00 | 0.89 | 39946.59 | 0.00 | 1.00 | |
|
| ||||||||
| PST | 1 | Null | 42831.61 | 33.29 | 0.00 | 38466.49 | 68.84 | 0.00 |
| 2 | Baseline | 42805.20 | 6.88 | 0.03 | 38430.43 | 32.78 | 0.00 | |
| 3 | Baseline + Longitudinal | 42798.31 | 0.00 | 0.97 | 38397.65 | 0.00 | 1.00 | |
|
| ||||||||
| PSDI | 1 | Null | 45463.96 | 23.74 | 0.00 | 40298.05 | 41.08 | 0.00 |
| 2 | Baseline | 45445.56 | 5.35 | 0.07 | 40263.78 | 6.82 | 0.03 | |
| 3 | Baseline + Longitudinal | 45440.22 | 0.00 | 0.94 | 40256.97 | 0.00 | 1.00 | |
|
| ||||||||
| SOM | 1 | Null | 42803.06 | 0.00 | 0.46 | 37634.13 | 10.62 | 0.01 |
| 2 | Baseline | 42803.70 | 0.64 | 0.34 | 37633.26 | 9.75 | 0.01 | |
| 3 | Baseline + Longitudinal | 42804.76 | 1.70 | 0.20 | 37623.51 | 0.00 | 0.99 | |
|
| ||||||||
| O-C | 1 | Null | 46037.23 | 66.08 | 0.00 | 40225.99 | 90.69 | 0.00 |
| 2 | Baseline | 45983.99 | 12.84 | 0.00 | 40165.93 | 30.63 | 0.00 | |
| 3 | Baseline + Longitudinal | 45971.15 | 0.00 | 1.00 | 40135.30 | 0.00 | 1.00 | |
|
| ||||||||
| I-S | 1 | Null | 45460.40 | 21.75 | 0.00 | 40387.99 | 32.74 | 0.00 |
| 2 | Baseline | 45439.80 | 1.16 | 0.36 | 40364.90 | 9.65 | 0.01 | |
| 3 | Baseline + Longitudinal | 45438.64 | 0.00 | 0.64 | 40355.25 | 0.00 | 0.99 | |
|
| ||||||||
| DEP | 1 | Null | 46069.77 | 30.01 | 0.00 | 40702.42 | 55.57 | 0.00 |
| 2 | Baseline | 46040.05 | 0.29 | 0.46 | 40663.07 | 16.22 | 0.00 | |
| 3 | Baseline + Longitudinal | 46039.76 | 0.00 | 0.54 | 40646.85 | 0.00 | 1.00 | |
|
| ||||||||
| ANX | 1 | Null | 44524.35 | 24.61 | 0.00 | 39287.96 | 43.24 | 0.00 |
| 2 | Baseline | 44499.73 | 0.00 | 0.51 | 39260.88 | 16.16 | 0.00 | |
| 3 | Baseline + Longitudinal | 44499.82 | 0.09 | 0.49 | 39244.72 | 0.00 | 1.00 | |
|
| ||||||||
| HOS | 1 | Null | 45763.65 | 33.04 | 0.00 | 41923.67 | 36.12 | 0.00 |
| 2 | Baseline | 45731.09 | 0.48 | 0.44 | 41889.31 | 1.77 | 0.29 | |
| 3 | Baseline + Longitudinal | 45730.61 | 0.00 | 0.56 | 41887.55 | 0.00 | 0.71 | |
|
| ||||||||
| PHOB | 1 | Null | 44385.08 | 21.60 | 0.00 | 38414.95 | 25.84 | 0.00 |
| 2 | Baseline | 44368.30 | 4.82 | 0.08 | 38405.41 | 16.30 | 0.00 | |
| 3 | Baseline + Longitudinal | 44363.48 | 0.00 | 0.92 | 38389.11 | 0.00 | 1.00 | |
|
| ||||||||
| PAR | 1 | Null | 41461.59 | 6.78 | 0.02 | 39364.64 | 13.05 | 0.00 |
| 2 | Baseline | 41454.81 | 0.00 | 0.64 | 39351.59 | 0.00 | 1.00 | |
| 3 | Baseline + Longitudinal | 41456.09 | 1.28 | 0.34 | 39391.12 | 39.53 | 0.00 | |
|
| ||||||||
| PSY | 1 | Null | 45879.04 | 29.96 | 0.00 | 40685.39 | 35.15 | 0.00 |
| 2 | Baseline | 45849.08 | 0.00 | 0.66 | 40653.05 | 2.81 | 0.20 | |
| 3 | Baseline + Longitudinal | 45850.41 | 1.33 | 0.34 | 40650.24 | 0.00 | 0.80 | |
The models with smallest AICc values are the best models and are displayed in boldface type. Models with smaller dAICc and wAICc closest to 1 indicate better fit. GSI=Global Severity Index; PST=Positive Symptom Total; PSDI=Positive Symptom Distress Index; SOM=Somatization; O-C=Obsessive–Compulsive; I-S=Interpersonal Sensitivity; DEP=Depression; ANX=Anxiety; HOS=Hostility; PHOB=Phobic Anxiety; PAR=Paranoid Ideation; PSY=Psychoticism.
Table 3 shows the comparison of each prodromal group (Low, Medium and High probability of motor diagnosis within five years) compared with the healthy controls on each baseline and longitudinal psychiatric outcome. As shown in the top of the table, 11 of the 12 participant psychiatric variables and 12 of the 12 companion psychiatric variables showed significant differences between the healthy controls and the prodromal Huntington disease groups with Medium and High probabilities of diagnosis. In the prodromal Huntington disease group with a lower probability of motor diagnosis, nine of the 12 participant-rated psychiatric variables and only one of the companion-rated psychiatric outcomes were significantly different than the healthy controls. When the groups were compared longitudinally, findings showed companion-rated psychiatric outcomes showed significant change for 11 of the 12 outcomes for the High group, six of the 12 outcomes for the Medium group, and two of the 12 variables for the Low group. Prodromal Huntington disease participant-reported psychiatric symptoms showed significant change over time on seven of the 12 variables for the High group. None of the participant-rated variables showed significant change over time for the Medium or Low prodromal Huntington disease groups.
TABLE 3.
Baseline and Longitudinal prodromal HD group differences relative to controls for participant (P) and companion (C) SCL-90-R
| Measure | Baseline Group Difference Relative to Controls | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Low | Medium | High | ||||||||||
| Est | SE | tval | pval | Est | SE | tval | pval | Est | SE | tval | pval | |
| P GSI | 2.54 | 1.14 | 2.23 | 0.026* | 4.89 | 1.03 | 4.74 | <0.001*** | 4.32 | 1.02 | 4.24 | <0.001*** |
| P PST | 2.03 | 0.97 | 2.10 | 0.036* | 4.24 | 0.88 | 4.84 | <0.001*** | 3.31 | 0.86 | 3.83 | <0.001*** |
| P PSDI | 3.15 | 0.92 | 3.43 | <0.001*** | 3.55 | 0.83 | 4.26 | <0.001*** | 2.76 | 0.82 | 3.36 | <0.001*** |
| P O-C | 2.81 | 1.19 | 2.36 | 0.018* | 5.81 | 1.08 | 5.38 | <0.001*** | 6.02 | 1.07 | 5.65 | <0.001*** |
| P I-S | 2.24 | 1.12 | 2.01 | 0.045* | 3.46 | 1.01 | 3.42 | <0.001*** | 3.94 | 1.00 | 3.95 | <0.001*** |
| P DEP | 2.78 | 1.12 | 2.49 | 0.013* | 4.89 | 1.01 | 4.84 | <0.001*** | 4.39 | 1.00 | 4.40 | <0.001*** |
| P ANX | 2.08 | 0.94 | 2.20 | 0.028* | 4.14 | 0.85 | 4.87 | <0.001*** | 3.94 | 0.84 | 4.71 | <0.001*** |
| P HOS | 2.98 | 1.06 | 2.81 | 0.005** | 5.21 | 0.96 | 5.42 | <0.001*** | 4.44 | 0.95 | 4.67 | <0.001*** |
| P PHOB | 1.46 | 0.96 | 1.52 | 0.13 | 2.92 | 0.87 | 3.36 | <0.001*** | 3.12 | 0.86 | 3.64 | <0.001*** |
| P PAR | 1.42 | 0.75 | 1.89 | 0.059 | 1.70 | 0.68 | 2.50 | 0.013* | 2.35 | 0.67 | 3.52 | <0.001*** |
| P PSY | 2.86 | 1.09 | 2.61 | 0.009** | 4.52 | 0.99 | 4.59 | <0.001*** | 5.55 | 0.97 | 5.72 | <0.001*** |
| C GSI | 0.87 | 1.07 | 0.81 | 0.417 | 3.59 | 0.97 | 3.71 | <0.001*** | 3.21 | 0.95 | 3.38 | <0.001*** |
| C PST | 0.94 | 0.98 | 0.96 | 0.338 | 3.46 | 0.89 | 3.91 | <0.001*** | 2.96 | 0.87 | 3.39 | <0.001*** |
| C PSDI | 0.64 | 0.95 | 0.67 | 0.504 | 3.09 | 0.87 | 3.56 | <0.001*** | 2.80 | 0.85 | 3.29 | 0.001** |
| C SOM | −0.35 | 0.84 | −0.42 | 0.674 | 1.14 | 0.76 | 1.50 | 0.134 | −0.18 | 0.74 | −0.25 | 0.804 |
| C O-C | 1.56 | 1.08 | 1.45 | 0.148 | 4.09 | 0.98 | 4.18 | <0.001*** | 5.18 | 0.96 | 5.40 | <0.001*** |
| C I-S | 1.00 | 1.10 | 0.91 | 0.363 | 2.86 | 1.00 | 2.86 | 0.004** | 2.94 | 0.98 | 3.00 | 0.003** |
| C DEP | 0.80 | 1.08 | 0.74 | 0.460 | 3.51 | 0.98 | 3.56 | <0.001*** | 3.53 | 0.97 | 3.65 | <0.001*** |
| C ANX | 0.42 | 0.91 | 0.47 | 0.639 | 2.76 | 0.82 | 3.36 | <0.001*** | 2.34 | 0.81 | 2.90 | 0.004** |
| C HOS | 2.36 | 1.25 | 1.88 | 0.059 | 4.56 | 1.14 | 4.00 | <0.001*** | 4.41 | 1.12 | 3.94 | <0.001*** |
| C PHOB | 0.69 | 0.77 | 0.90 | 0.370 | 1.36 | 0.69 | 1.97 | 0.049* | 1.75 | 0.68 | 2.57 | 0.010* |
| C PAR | 0.76 | 0.86 | 0.88 | 0.378 | 2.81 | 0.78 | 3.61 | <0.001*** | 2.66 | 0.76 | 3.49 | <0.001*** |
| C PSY | 2.60 | 1.02 | 2.55 | 0.011* | 4.03 | 0.93 | 4.35 | <0.001*** | 3.37 | 0.91 | 3.70 | <0.001*** |
| Longitudinal Group Difference Relative to Controls | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Low | Medium | High | ||||||||||
| Est | SE | tval | pval | Est | SE | tval | pval | Est | SE | tval | pval | |
| P GSI | 0.21 | 0.21 | 1.02 | 0.308 | 0.05 | 0.19 | 0.26 | 0.796 | 0.51 | 0.19 | 2.74 | 0.006** |
| P PST | 0.27 | 0.17 | 1.54 | 0.124 | 0.06 | 0.16 | 0.35 | 0.73 | 0.49 | 0.16 | 3.10 | 0.002** |
| P PSDI | −0.18 | 0.20 | −0.93 | 0.354 | −0.05 | 0.19 | −0.29 | 0.772 | 0.37 | 0.18 | 2.04 | 0.041* |
| P O-C | 0.32 | 0.23 | 1.41 | 0.157 | 0.16 | 0.21 | 0.76 | 0.448 | 0.81 | 0.21 | 3.93 | <0.001*** |
| P I-S | 0.26 | 0.21 | 1.22 | 0.222 | 0.20 | 0.20 | 1.02 | 0.306 | 0.51 | 0.19 | 2.61 | 0.009** |
| P DEP | 0.11 | 0.21 | 0.51 | 0.613 | 0.07 | 0.20 | 0.37 | 0.714 | 0.42 | 0.19 | 2.19 | 0.028* |
| P HOS | −0.05 | 0.21 | −0.24 | 0.809 | −0.27 | 0.20 | −1.36 | 0.175 | 0.19 | 0.20 | 0.95 | 0.340 |
| P PHOB | 0.13 | 0.19 | 0.67 | 0.506 | 0.26 | 0.18 | 1.43 | 0.153 | 0.55 | 0.18 | 3.08 | 0.002** |
| C GSI | 0.40 | 0.22 | 1.82 | 0.069 | 0.51 | 0.20 | 2.50 | 0.012* | 1.04 | 0.20 | 5.21 | <0.001*** |
| C PST | 0.45 | 0.19 | 2.35 | 0.019* | 0.48 | 0.18 | 2.69 | 0.007** | 1.08 | 0.18 | 6.14 | <0.001*** |
| C PSDI | 0.31 | 0.22 | 1.44 | 0.150 | 0.27 | 0.20 | 1.31 | 0.191 | 0.69 | 0.20 | 3.46 | <0.001*** |
| C SOM | 0.39 | 0.16 | 2.38 | 0.017* | 0.38 | 0.15 | 2.46 | 0.014* | 0.60 | 0.15 | 3.98 | <0.001*** |
| C O-C | 0.44 | 0.24 | 1.84 | 0.065 | 0.65 | 0.22 | 2.90 | 0.004** | 1.30 | 0.22 | 5.90 | <0.001*** |
| C I-S | 0.09 | 0.22 | 0.42 | 0.672 | 0.23 | 0.20 | 1.16 | 0.246 | 0.70 | 0.20 | 3.50 | <0.001*** |
| C DEP | 0.43 | 0.23 | 1.87 | 0.062 | 0.46 | 0.22 | 2.14 | 0.032* | 0.99 | 0.21 | 4.67 | <0.001*** |
| C ANX | 0.27 | 0.19 | 1.42 | 0.155 | 0.36 | 0.18 | 1.99 | 0.047* | 0.81 | 0.18 | 4.54 | <0.001*** |
| C HOS | 0.08 | 0.26 | 0.32 | 0.749 | 0.29 | 0.24 | 1.20 | 0.230 | 0.59 | 0.24 | 2.49 | 0.013* |
| C PHOB | 0.16 | 0.20 | 0.79 | 0.430 | 0.29 | 0.19 | 1.56 | 0.118 | 0.79 | 0.18 | 4.31 | <0.001*** |
| C PSY | −0.12 | 0.23 | −0.50 | 0.619 | 0.17 | 0.22 | 0.78 | 0.435 | 0.47 | 0.22 | 2.18 | 0.029* |
p<0.001,
p<0.01,
p<0.05,
P=participant; C=companion; GSI=Global Severity Index; PST=Positive Symptom Total; PSDI=Positive Symptom Distress Index; O-C=Obsessive–Compulsive; I-S=Interpersonal Sensitivity; DEP=Depression; ANX=Anxiety; HOS=Hostility; PHOB=Phobic Anxiety; PAR=Paranoid Ideation; PSY=Psychoticism; SOM=Somatization. Low, Med, High indicate low, medium, or high probability of diagnosis within five years as estimated by CAG-Age Product (CAP) score.
Comparisons Between Participant and Companion Ratings
A multi-response linear mixed-effects regression analysis was performed to examine whether the differences in longitudinal change among participants and companions were statistically reliable. When the importance of the longitudinal change discrepancy between participants and companions was compared across CAG-Age Product groups for each measure as shown in the Table 4, differences (weight>0.7) were observed for seven measures: Global Severity Index, Positive Symptom Total, Obsessive–Compulsive, Depression, Anxiety, Phobic Anxiety, and Paranoid Ideation. For all the seven measures, the High group had relatively high importance with weights of 0.75 and above. The Medium group had relatively high importance only for Positive Symptom Total and Obsessive–Compulsive. The Low group did not have relatively high importance for any measure. For these seven measures – Global Severity Index, Positive Symptom Total, Obsessive–Compulsive, Depression, Anxiety, Phobic Anxiety, and Paranoid Ideation – fitted curves are displayed in Figure 1 using model-averaged coefficients across all possible models. For the other five measures which did not have groups with longitudinal change discrepancies – Positive Symptom Distress Index, Somatization, Interpersonal Sensitivity, Hostility and Psychoticism – fitted curves are displayed in the supplemental data (Figure SF1).
TABLE 4.
Slope discrepancy between participants and companions by group as measured by the sum of weights over all models with unequal group slopes
| Measure | Control | Low | Medium | High |
|---|---|---|---|---|
| GSI | 0.43 | 0.27 | 0.55 | 0.80 |
| PST | 0.39 | 0.28 | 0.73 | 0.99 |
| PSDI | 0.45 | 0.54 | 0.28 | 0.28 |
| SOM | 0.67 | 0.33 | 0.51 | 0.40 |
| O-C | 0.27 | 0.28 | 0.89 | 0.90 |
| I-S | 0.27 | 0.33 | 0.28 | 0.44 |
| DEP | 0.50 | 0.28 | 0.30 | 0.75 |
| ANX | 0.34 | 0.31 | 0.49 | 0.86 |
| HOS | 0.41 | 0.28 | 0.58 | 0.36 |
| PHOB | 0.27 | 0.29 | 0.33 | 0.81 |
| PAR | 0.66 | 0.28 | 0.42 | 0.80 |
| PSY | 0.29 | 0.43 | 0.34 | 0.29 |
Values close to 1 indicate higher importance. Weights > 0.7 are displayed in boldface type. Low, Med, High indicate low, medium, or high probability of diagnosis within five years as estimated by CAG-Age Product (CAP) score. GSI=Global Severity Index; PST=Positive Symptom Total; PSDI=Positive Symptom Distress Index; SOM=Somatization; O-C=Obsessive–Compulsive; I-S=Interpersonal Sensitivity; DEP=Depression; ANX=Anxiety; HOS=Hostility; PHOB=Phobic Anxiety; PAR=Paranoid Ideation; PSY=Psychoticism.
Figure 1.
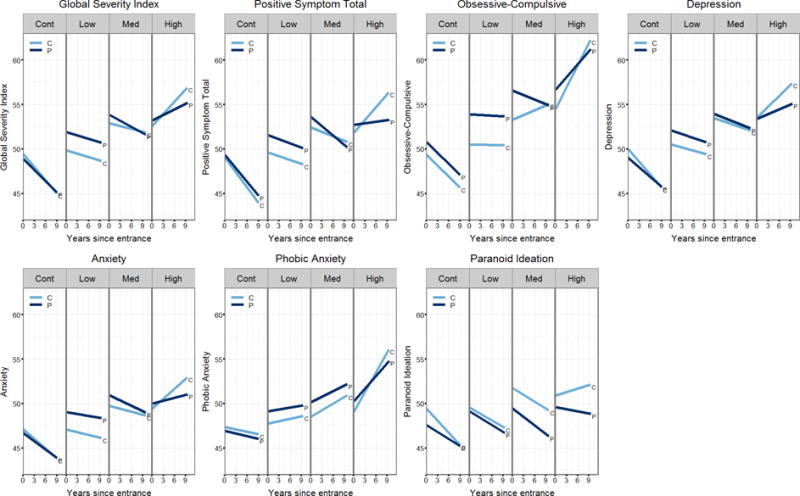
Fitted Linear Mixed-Effects Regression (LMER) curves by group for participant and companion Symptom Checklist-90-Revised (SCL-90-R) ratings. All model coefficients were estimated adjusting for gender, years of education, and age at entry. The plots show the SCL-90-R score as a function of duration, person (participant or companion) and group. Low, Med, High indicate low, medium, or high probability of diagnosis within five years as estimated by CAG-Age Product (CAP) score.
Discussion
To our knowledge, this is the largest longitudinal study of psychiatric manifestations in prodromal Huntington disease published. Some previous research has suggested that severity of psychiatric and/or behavioral manifestations of HD are not associated with progression of the disease (2, 30). Contrary to this popularly held belief, 19 of 24 measures examined showed cross-sectional and longitudinal differences between prodromal Huntington disease and gene-mutation negative controls. Of the remaining five measures analyzed, three also showed elevated baseline scores although change over time did not differ. Although previous research has consistently emphasized the clinical importance of the psychiatric symptoms associated with Huntington disease, and a wealth of studies have noted the higher prevalence of psychiatric symptoms and increased psychiatric distress in Huntington disease, few studies have documented longitudinal progression of the psychiatric disturbances in Huntington disease. These findings are consistent with Craufurd and colleagues (7) who provide longitudinal data for 111 persons with a diagnosis of Huntington disease, finding that all three subscales of the Problem Behaviors Assessment for Huntington’s Disease (PBA-HD) showed significant change over time. Further, Craufurd (7) showed whereas all three psychiatric subscales (Apathy, Irritability, Depression) worsened over time in the earliest stages of Huntington disease (Stage I and II), only Apathy continued to progress with disease progression into Stages II–IV. Comparisons with other research in prodromal Huntington disease, however, are more central to the current findings. Kirkwood and colleagues (31) followed 45 prodromal gene mutation carriers and reported a greater increase in psychiatric abnormalities over an interval of 3.7 years. The greatest changes were noted in Irritability and Hostility. More recently, Tabrizi and colleagues (32) followed 120 prodromal Huntington disease gene mutation carriers and reported worsening apathy over 36 months. The current findings replicate and extend these reports and provide confidence that the psychiatric symptoms seen in Huntington disease progress with disease severity and are likely secondary to the neurodegenerative disease process.
The detection of neurodegenerative diseases has progressed considerably as reflected in the fifth edition of the Diagnostic and Statistical Manual of Mental Disorders. In this latest edition the field describes the emergence of mild cognitive impairment (MCI) as a precursor, or prodromal stage, evident in progressive brain disease. MCI in Huntington disease has been documented in at least 40% of persons carrying the gene mutation (33, 34), and numerous publications demonstrate that cognitive decline can be detected a decade prior to motor onset (35). Given the plethora of publications demonstrating early cognitive decline in Huntington disease in concert with the findings of our study showing psychiatric symptom progression prior to motor onset, the mono-symptomatic diagnostic criteria seems out of date. Since mental health professionals may see prodromal Huntington disease in clinical practice a decade before movement disorder specialists, an earlier diagnosis may be prudent for many individuals.
The current findings suggest the source of measurement can significantly alter psychiatric outcomes in prodromal and diagnosed Huntington disease. All findings from this research were more robust when psychiatric symptoms were rated by companions (rather than self-reported by gene-expanded participants) for prodromal Huntington disease. For instance, companion ratings worsened over time for 11/12 symptoms in the prodromal Huntington disease group with a high probability of imminent motor diagnosis, 6/12 symptoms in those with a medium probability of diagnosis and 2/12 symptoms in persons considered to be far from their estimated motor onset. Only 7/12 participant ratings showed significant change over time and only in the group in closest proximity to motor diagnosis. When analyses were conducted to examine the importance of discrepant ratings between the companion and the prodromal Huntington disease participants, findings showed that ratings on 7/12 psychiatric symptom scales were inconsistent between the raters. In participants closer to estimated motor onset, the companion ratings appear to be more valid and self-reported psychiatric symptoms may be more appropriate in gene-mutation carriers who are furthest from estimated motor onset.
These findings are consistent with prior cross-sectional observations that individuals with Huntington disease have decreased awareness of symptoms (36–39). Previous studies on awareness described above were cross-sectional (although Ho et al. had two time points, they were only about six weeks apart) and involved individuals diagnosed with Huntington disease. It is important to note that, even in Huntington disease mutation carriers who do not yet have motor diagnosis, decreased awareness of both motor (22) and psychiatric (4) symptoms have been documented. The current findings are the first to provide longitudinal data of reduced awareness in a prodromal Huntington disease cohort over such a long follow-up time period. Our findings indicate unawareness increases in concert with Huntington disease progression during the prodromal Huntington disease stages as indicated by participant-companion differences arising mostly in the Medium and High CAG-Age Product groups.
Difficulties with awareness or insight occur across the spectrum of neuropsychiatric disorders, including stroke, brain injury, dementia, psychosis, and mood disorders (40). The etiologies of these disparate conditions vary, and it is not known whether there are common mechanisms that underlie the development of unawareness between disorders. Several studies in different disorders have noted associations between deceased awareness of symptoms and pathology in the right hemisphere, dorsolateral frontal, and less often the parieto-temporal region. However, the findings are not consistent enough to indicate shared mechanisms with certainty. The results presented here and in other studies on Huntington disease symptoms and brain pathology are consistent with these anatomic findings, but more research is needed to support this connection. As described previously by others (41), it is likely that neural networks connecting several regions are responsible for the phenomenon of decreased awareness. The presentation of lack of awareness also varies among disorders. In fronto-temporal dementia, for example, insight is reduced early in the course of the illness, while this is a later manifestation in Alzheimer dementia (42). Unawareness is also irreversible in dementias, while it is often transient in other disorders such as stroke, traumatic brain injury, mood disorders, or schizophrenia. The progressive brain degeneration that occurs in Huntington disease may indicate reduced insight is permanent but additional study is needed to understand its course and causes in the illness. There are some limitations to consider in the interpretation of these results. Some issues relate to the nature of the assessments. It is important to note that individuals are asked to provide ratings over the previous seven days for the SCL-90-R. As these measures are obtained annually, they may miss episodes of psychopathology that occur between assessments. While these findings support changes in participant-companion reporting over time, more detailed assessments at more time points would be beneficial. Another issue relates to potential biases of the raters. Both participants and companions are aware of the Huntington disease genetic status of the participants, which may affect their perception of how they rate their symptoms. Selection bias of the sample may also impact the generalizability of these results. Only a fraction of those at risk for Huntington disease obtain genetic testing, which is required prior to enrollment in the PREDICT-HD study. There may be factors unique to those who obtain genetic testing and participate in research studies not present in the Huntington disease population. It is interesting to observe, however, that a recent study of premanifest Huntington disease not requiring known genetic status, and thus no predictive genetic testing, showed highly similar demographics, including education (43). Differences in awareness may also be present in our sample compared to the overall population. However, as previously described by Duff and colleagues (4), it is equally likely that individuals with lower or higher awareness would participate in the study. Despite these limitations, the results here provide initial information regarding psychiatric symptoms that occur in individuals who will develop Huntington disease and the importance of obtaining assessments from companions. This is critical to consider in future studies that assess behavioral manifestations and psychiatric symptoms, including those that investigate their underlying pathophysiology in Huntington disease and in therapeutic trials, as well as clinical assessment and management of persons who will develop Huntington disease.
Supplementary Material
Acknowledgments
We thank the PREDICT-HD sites, the study participants, the National Research Roster for Huntington Disease Patients and Families, the Huntington’s Disease Society of America and the Huntington Study Group. This publication was supported by the National Center for Advancing Translational Sciences, and the National Institutes of Health (NIH), through Grant 2 UL1 TR000442-06. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
This research is supported by the National Institutes of Health, National Institute of Neurological Disorders and Stroke (5R01NS040068) awarded to Jane Paulsen, CHDI Foundation, Inc (A6266; A2015) awarded to Jane Paulsen, Cognitive and Functional Brain Changes in Preclinical Huntington’s Disease (HD) (5R01NS054893) awarded to Jane Paulsen.
Location of Work
Department of Psychiatry, Carver College of Medicine, The University of Iowa, Iowa City, IA, USA
PREDICT-HD Investigators, Coordinators, Motor Raters, Cognitive Raters
Isabella De Soriano, Courtney Shadrick, and Amanda Miller (University of Iowa, Iowa City, Iowa, USA);
Edmond Chiu, Joy Preston, Anita Goh, Stephanie Antonopoulos, and Samantha Loi (St. Vincent’s Hospital, The University of Melbourne, Kew, Victoria, Australia);
Phyllis Chua and Angela Komiti (The University of Melbourne, Royal Melbourne Hospital, Melbourne, Victoria, Australia);
Lynn Raymond, Joji Decolongon, Mannie Fan, and Allison Coleman (University of British Columbia, Vancouver, British Columbia, Canada);
Christopher A. Ross, Mark Varvaris, Maryjane Ong, and Nadine Yoritomo (Johns Hopkins University, Baltimore, Maryland, USA);
William M. Mallonee and Greg Suter (Hereditary Neurological Disease Centre, Wichita, Kansas, USA);
Ali Samii, Emily P. Freney, and Alma Macaraeg (University of Washington and VA Puget Sound Health Care System, Seattle, Washington, USA);
Randi Jones, Cathy Wood-Siverio, and Stewart A. Factor (Emory University School of Medicine, Atlanta, Georgia, USA);
Roger A. Barker, Sarah Mason, and Natalie Valle Guzman (John van Geest Centre for Brain Repair, Cambridge, UK);
Elizabeth McCusker, Jane Griffith, Clement Loy, Jillian McMillan, and David Gunn (Westmead Hospital, Sydney, New South Wales, Australia);
Michael Orth, Sigurd Süβmuth, Katrin Barth, Sonja Trautmann, Daniela Schwenk, and Carolin Eschenbach (University of Ulm, Ulm, Germany);
Kimberly Quaid, Melissa Wesson, and Joanne Wojcieszek (Indiana University School of Medicine, Indianapolis, Indiana, USA);
Mark Guttman, Alanna Sheinberg, Albie Law, and Irita Karmalkar (Centre for Addiction and Mental Health, University of Toronto, Markham, Ontario, Canada);
Susan Perlman and Brian Clemente (UCLA Medical Center, Los Angeles, California, USA);
Michael D. Geschwind, Sharon Sha, Joseph Winer, and Gabriela Satris (University of California, San Francisco, San Francisco, California, USA);
Tom Warner and Maggie Burrows (National Hospital for Neurology and Neurosurgery, London, UK);
Anne Rosser, Kathy Price, and Sarah Hunt (Cardiff University, Cardiff, Wales, UK);
Frederick Marshall, Amy Chesire, Mary Wodarski, and Charlyne Hickey (University of Rochester, Rochester, New York, USA);
Peter Panegyres, Joseph Lee, Maria Tedesco, and Brenton Maxwell (Neurosciences Unit, Graylands, Selby-Lemnos & Special Care Health Services, Perth, Western Australia, Australia);
Joel Perlmutter, Stacey Barton, and Shineeka Smith (Washington University, St. Louis, Missouri, USA);
Zosia Miedzybrodzka, Daniela Rae, Vivien Vaughan, and Mariella D’Alessandro (Clinical Genetics Centre, Aberdeen, Scotland, UK);
David Craufurd, Judith Bek, and Elizabeth Howard (University of Manchester, Manchester, UK);
Pietro Mazzoni, Karen Marder, and Paula Wasserman (Columbia University Medical Center, New York, New York, USA);
Rajeev Kumar, Diane Erickson, Christina Reeves, and Breanna Nickels (Colorado Neurological Institute, Englewood, Colorado, USA);
Vicki Wheelock, Lisa Kjer, Amanda Martin, and Sarah Farias (University of California, Davis, Sacramento, California, USA);
Wayne Martin, Oksana Suchowersky, Pamela King, Marguerite Wieler, and Satwinder Sran (University of Alberta, Edmonton, Alberta, Canada);
Anwar Ahmed, Stephen Rao, Christine Reece, Alex Bura, and Lyla Mourany (Cleveland Clinic Foundation, Cleveland, Ohio, USA);
Executive Committee
Principal Investigator Jane S. Paulsen, Jeffrey D. Long, Hans J. Johnson, Thomas Brashers-Krug, Phil Danzer, Amanda Miller, H. Jeremy Bockholt, and Kelsey Montross.
Scientific Consultants
Deborah Harrington (University of California, San Diego); Holly Westervelt (Rhode Island Hospital/Alpert Medical School of Brown University); Elizabeth Aylward (Seattle Children’s Research Institute); Stephen Rao (Cleveland Clinic); David J. Moser, Janet Williams, Nancy Downing, Vincent A. Magnotta, Hans J. Johnson, Thomas Brashers-Krug, Jatin Vaidya, Daniel O’Leary, and Eun Young Kim (University of Iowa).
Core Sections
Biostatistics
Jeffrey D. Long, Ji-In Kim, Spencer Lourens (University of Iowa); Ying Zhang and Wenjing Lu (University of Indiana).
Ethics
Cheryl Erwin (Texas Tech University Health Sciences Center); Thomas Brashers-Krug, Janet Williams (University of Iowa); and Martha Nance (University of Minnesota).
Biomedical Informatics.
H. Jeremy Bockholt, Jason Evans, and Roland Zschiegner (University of Iowa).
Footnotes
Disclosures: Dr. Eric Epping has served as a consultant for Lundbeck, Inc. Dr. Ji-In Kim reports no financial relationships with commercial interests. Dr. David Craufurd has received payments for advisory panel membership or honoraria for speaking at meetings from F. Hoffmann-La Roche AG and AOP Orphan Pharmaceuticals AG. He does not have any conflicts to declare in relation to this paper. Dr. Thomas Brashers-Krug reports no financial relationships with commercial interests. Dr. Karen Anderson reports no financial relationships with commercial interests. Dr. Elizabeth McCusker reports no financial relationships with commercial interests. Ms. Jolene Luther reports no financial relationships with commercial interests. Dr. Jeffrey D. Long has a consulting agreement with NeuroPhage, LLC. Dr. Jane S. Paulsen has served on an advisory board for Lundbeck, LLC and has a consulting agreement with ProPhase, LLC.
Clinical trial registration
The study associated with this manuscript, PREDICT-HD, is registered with www.clinicaltrials.gov (identifier NCT00051324)
References
- 1.Walker FO. Huntington’s Disease. Semin Neurol. 2007;27:143–150. doi: 10.1055/s-2007-971176. [DOI] [PubMed] [Google Scholar]
- 2.Ross CA, Tabrizi SJ. Huntington’s disease: from molecular pathogenesis to clinical treatment. Lancet Neurol. 2011;10:83–98. doi: 10.1016/S1474-4422(10)70245-3. [DOI] [PubMed] [Google Scholar]
- 3.Beglinger LJ, O’Rourke JJ, Wang C, Langbehn DR, Duff K, Paulsen JS, Huntington Study Group Investigators Earliest functional declines in Huntington disease. Psychiatry Res. 2010;178:414–418. doi: 10.1016/j.psychres.2010.04.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Duff K, Paulsen JS, Beglinger LJ, Langbehn DR, Wang C, Stout JC, Ross CA, Aylward E, Carlozzi NE, Queller S, Predict-HD Investigators of the Huntington Study Group “Frontal” behaviors before the diagnosis of Huntington’s disease and their relationship to markers of disease progression: evidence of early lack of awareness. J Neuropsychiatry Clin Neurosci. 2010;22:196–207. doi: 10.1176/appi.neuropsych.22.2.196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Paulsen JS. Cognitive impairment in Huntington disease: diagnosis and treatment. Curr Neurol Neurosci Rep. 2011;11:474–483. doi: 10.1007/s11910-011-0215-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Paulsen JS, Smith MM, Long JD, PREDICT-HD investigators and coordinators of the Huntington Study Group Cognitive decline in prodromal Huntington Disease: implications for clinical trials. J Neurol Neurosurg Psychiatry. 2013;84:1233–1239. doi: 10.1136/jnnp-2013-305114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Craufurd D, Thompson JC, Snowden JS. Behavioral changes in Huntington Disease. Neuropsychiatry Neuropsychol Behav Neurol. 2001;14:219–226. [PubMed] [Google Scholar]
- 8.Paulsen JS, Zhao H, Stout JC, Brinkman RR, Guttman M, Ross CA, Como P, Manning C, Hayden MR, Shoulson I, Huntington Study Group Clinical markers of early disease in persons near onset of Huntington’s disease. Neurology. 2001;57:658–662. doi: 10.1212/wnl.57.4.658. [DOI] [PubMed] [Google Scholar]
- 9.Paulsen JS, Conybeare RA. Cognitive changes in Huntington’s disease. Adv Neurol. 2005;96:209–225. [PubMed] [Google Scholar]
- 10.van Duijn E, Kingma EM, van der Mast RC. Psychopathology in verified Huntington’s disease gene carriers. J Neuropsychiatry Clin Neurosci. 2007;19:441–448. doi: 10.1176/jnp.2007.19.4.441. [DOI] [PubMed] [Google Scholar]
- 11.Julien CL, Thompson JC, Wild S, Yardumian P, Snowden JS, Turner G, Craufurd D. Psychiatric disorders in preclinical Huntington’s disease. J Neurol Neurosurg Psychiatry. 2007;78:939–943. doi: 10.1136/jnnp.2006.103309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kingma EM, van Duijn E, Timman R, van der Mast RC, Roos RA. Behavioural problems in Huntington’s disease using the Problem Behaviours Assessment. Gen Hosp Psychiatry. 2008;30:155–161. doi: 10.1016/j.genhosppsych.2007.11.005. [DOI] [PubMed] [Google Scholar]
- 13.Tabrizi SJ, Langbehn DR, Leavitt BR, Roos RA, Durr A, Craufurd D, Kennard C, Hicks SL, Fox NC, Scahill RI, Borowsky B, Tobin AJ, Rosas HD, Johnson H, Reilmann R, Landwehrmeyer B, Stout JC, TRACK-HD investigators Biological and clinical manifestations of Huntington’s disease in the longitudinal TRACK-HD study: cross-sectional analysis of baseline data. Lancet Neurol. 2009;8:791–801. doi: 10.1016/S1474-4422(09)70170-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.van Duijn E, Kingma EM, Timman R, Zitman FG, Tibben A, Roos RA, van der Mast RC. Cross-sectional study on prevalences of psychiatric disorders in mutation carriers of Huntington’s disease compared with mutation-negative first-degree relatives. J Clin Psychiatry. 2008;69:1804–1810. doi: 10.4088/jcp.v69n1116. [DOI] [PubMed] [Google Scholar]
- 15.van Duijn E, Reedeker N, Giltay EJ, Roos RA, van der Mast RC. Correlates of apathy in Huntington’s disease. J Neuropsychiatry Clin Neurosci. 2010;22:287–294. doi: 10.1176/jnp.2010.22.3.287. [DOI] [PubMed] [Google Scholar]
- 16.Paulsen JS, Hayden M, Stout JC, Langbehn DR, Aylward E, Ross CA, Guttman M, Nance M, Kieburtz K, Oakes D, Shoulson I, Kayson E, Johnson S, Penziner E, Predict-HD Investigators of the Huntington Study Group Preparing for preventive clinical trials: the Predict-HD study. Arch Neurol. 2006;63:883–890. doi: 10.1001/archneur.63.6.883. [DOI] [PubMed] [Google Scholar]
- 17.Paulsen JS, Langbehn DR, Stout JC, Aylward E, Ross CA, Nance M, Guttman M, Johnson S, MacDonald M, Beglinger LJ, Duff K, Kayson E, Biglan K, Shoulson I, Oakes D, Hayden M, PREDICT-HD Investigators and Coordinators of the Huntington Study Group Detection of Huntington’s disease decades before diagnosis: the Predict-HD study. J Neurol Neurosurg Psychiatry. 2008;79:874–880. doi: 10.1136/jnnp.2007.128728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Paulsen JS, Long JD, Johnson HJ, Aylward EH, Ross CA, Williams JK, Nance MA, Erwin CJ, Westervelt HJ, Harrington DL, Bockholt HJ, Zhang Y, McCusker EA, Chiu EM, Panegyres PK, PREDICT-HD Investigators Coordinators of the Huntington Study Group Clinical and Biomarker Changes in Premanifest Huntington Disease Show Trial Feasibility: A Decade of the PREDICT-HD Study. Front Aging Neurosci. 2014;6:78. doi: 10.3389/fnagi.2014.00078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Duff K, Paulsen JS, Beglinger LJ, Langbehn DR, Stout JC, Predict-HD Investigators of the Huntington Study Group Psychiatric symptoms in Huntington’s disease before diagnosis: the Predict-HD study. Biol Psychiatry. 2007;62:1341–1346. doi: 10.1016/j.biopsych.2006.11.034. [DOI] [PubMed] [Google Scholar]
- 20.Beglinger LJ, Paulsen JS, Watson DB, Wang C, Duff K, Langbehn DR, Moser DJ, Paulson HL, Aylward EH, Carlozzi NE, Queller S, Stout JC. Obsessive and compulsive symptoms in prediagnosed Huntington’s disease. J Clin Psychiatry. 2008;69:1758–1765. doi: 10.4088/jcp.v69n1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Epping EA, Mills JA, Beglinger LJ, Fiedorowicz JG, Craufurd D, Smith MM, Groves M, Bijanki KR, Downing N, Williams JK, Long JD, Paulsen JS, Investigators P-H, Coordinators of the Huntington Study Group Characterization of depression in prodromal Huntington disease in the neurobiological predictors of HD (PREDICT-HD) study. J Psychiatr Res. 2013;47:1423–1431. doi: 10.1016/j.jpsychires.2013.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.McCusker EA, Gunn DG, Epping EA, Loy CT, Radford K, Griffith J, Mills JA, Long JD, Paulsen JS, PREDICT-HD Investigators of the Huntington Study Group Unawareness of motor phenoconversion in Huntington disease. Neurology. 2013;81:1141–1147. doi: 10.1212/WNL.0b013e3182a55f05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Killoran A, Biglan KM, Jankovic J, Eberly S, Kayson E, Oakes D, Young AB, Shoulson I. Characterization of the Huntington intermediate CAG repeat expansion phenotype in PHAROS. Neurology. 2013;80:2022–2027. doi: 10.1212/WNL.0b013e318294b304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ha AD, Beck CA, Jankovic J. Intermediate CAG Repeats in Huntington’s Disease: Analysis of COHORT. Tremor Other Hyperkinet Mov (N Y) 2012;2 doi: 10.7916/D8FF3R2P. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhang Y, Long JD, Mills JA, Warner JH, Lu W, Paulsen JS, PREDICT-HD Investigators and Coordinators of the Huntington Study Group Indexing disease progression at study entry with individuals at-risk for Huntington disease. Am J Med Genet B Neuropsychiatr Genet. 2011;156B:751–763. doi: 10.1002/ajmg.b.31232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Penney JB, Jr, Vonsattel JP, MacDonald ME, Gusella JF, Myers RH. CAG repeat number governs the development rate of pathology in Huntington’s disease. Ann Neurol. 1997;41:689–692. doi: 10.1002/ana.410410521. [DOI] [PubMed] [Google Scholar]
- 27.Derogatis LR, Yevzeroff H, Wittelsberger B. Social class, psychological disorder, and the nature of the psychopathologic indicator. J Consult Clin Psychol. 1975;43:183–191. doi: 10.1037/h0076514. [DOI] [PubMed] [Google Scholar]
- 28.Verbeke G, Molenberghs G. Linear mixed models for longitudinal data. New York, NY: Springer-Verlag New York LLC; 2000. [Google Scholar]
- 29.Burnham KP, Anderson DR. Model selection and multimodel inference : a practical information-theoretic approach. 2nd. New York: Springer; 2002. [Google Scholar]
- 30.Roos RA. Huntington’s disease: a clinical review. Orphanet J Rare Dis. 2010;5:40. doi: 10.1186/1750-1172-5-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kirkwood SC, Siemers E, Viken R, Hodes ME, Conneally PM, Christian JC, Foroud T. Longitudinal personality changes among presymptomatic Huntington disease gene carriers. Neuropsychiatry Neuropsychol Behav Neurol. 2002;15:192–197. [PubMed] [Google Scholar]
- 32.Tabrizi SJ, Scahill RI, Owen G, Durr A, Leavitt BR, Roos RA, Borowsky B, Landwehrmeyer B, Frost C, Johnson H, Craufurd D, Reilmann R, Stout JC, Langbehn DR, TRACK-HD Investigators Predictors of phenotypic progression and disease onset in premanifest and early-stage Huntington’s disease in the TRACK-HD study: analysis of 36-month observational data. Lancet Neurol. 2013;12:637–649. doi: 10.1016/S1474-4422(13)70088-7. [DOI] [PubMed] [Google Scholar]
- 33.Paulsen JS, Duff K. Extending MCI beyond Alzheimer disease. Neurology. 2009;72:1116–1117. doi: 10.1212/01.wnl.0000345369.41620.fc. [DOI] [PubMed] [Google Scholar]
- 34.Duff K, Paulsen J, Mills J, Beglinger LJ, Moser DJ, Smith MM, Langbehn D, Stout J, Queller S, Harrington DL. Mild cognitive impairment in prediagnosed Huntington disease. Neurology. 2010;75:500–507. doi: 10.1212/WNL.0b013e3181eccfa2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Paulsen JS, Long JD. Onset of Huntington’s disease: Can it be purely cognitive? Mov Disord. 2014;29:1342–1350. doi: 10.1002/mds.25997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Deckel AW, Morrison D. Evidence of a neurologically based “denial of illness” in patients with Huntington’s disease. Arch Clin Neuropsychol. 1996;11:295–302. [PubMed] [Google Scholar]
- 37.Chatterjee A, Anderson KE, Moskowitz CB, Hauser WA, Marder KS. A comparison of self-report and caregiver assessment of depression, apathy, and irritability in Huntington’s disease. J Neuropsychiatry Clin Neurosci. 2005;17:378–383. doi: 10.1176/jnp.17.3.378. [DOI] [PubMed] [Google Scholar]
- 38.Ho AK, Robbins AO, Barker RA. Huntington’s disease patients have selective problems with insight. Mov Disord. 2006;21:385–389. doi: 10.1002/mds.20739. [DOI] [PubMed] [Google Scholar]
- 39.Hoth KF, Paulsen JS, Moser DJ, Tranel D, Clark LA, Bechara A. Patients with Huntington’s disease have impaired awareness of cognitive, emotional, and functional abilities. J Clin Exp Neuropsychol. 2007;29:365–376. doi: 10.1080/13803390600718958. [DOI] [PubMed] [Google Scholar]
- 40.Orfei MD, Robinson RG, Bria P, Caltagirone C, Spalletta G. Unawareness of illness in neuropsychiatric disorders: phenomenological certainty versus etiopathogenic vagueness. Neuroscientist. 2008;14:203–222. doi: 10.1177/1073858407309995. [DOI] [PubMed] [Google Scholar]
- 41.Flashman LA. Disorders of awareness in neuropsychiatric syndromes: an update. Curr Psychiatry Rep. 2002;4:346–353. doi: 10.1007/s11920-002-0082-x. [DOI] [PubMed] [Google Scholar]
- 42.Ecklund-Johnson E, Torres I. Unawareness of deficits in Alzheimer’s disease and other dementias: operational definitions and empirical findings. Neuropsychol Rev. 2005;15:147–166. doi: 10.1007/s11065-005-9026-7. [DOI] [PubMed] [Google Scholar]
- 43.Huntington Study Group Pharos Investigators. At risk for Huntington disease: The PHAROS (Prospective Huntington At Risk Observational Study) cohort enrolled. Arch Neurol. 2006;63:991–996. doi: 10.1001/archneur.63.7.991. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.