

Abstract
Although PKC-mediated phosphorylation of protein kinase D1 (PKD1) has been extensively characterized, little is known about PKD1 regulation by other upstream kinases. Here we report that stimulation of epithelial or fibroblastic cells with G protein-coupled receptor agonists, including angiotensin II or bombesin, induced rapid and persistent PKD1 phosphorylation at Ser203, a highly conserved residue located within the PKD1 N-terminal domain. Exposure to PKD or PKC family inhibitors did not prevent PKD1 phosphorylation at Ser203, indicating that it is not mediated by autophosphorylation. In contrast, several lines of evidence indicated that the phosphorylation of PKD1 at Ser203 is mediated by kinases of the class I PAK subfamily, specifically 1) exposing cells to four structurally unrelated PAK inhibitors (PF-3758309, FRAX486, FRAX597, and IPA-3) that act via different mechanisms abrogated PKD1 phosphorylation at Ser203, 2) siRNA-mediated knockdown of PAK1 and PAK2 in IEC-18 and Swiss 3T3 cells blunted PKD1 phosphorylation at Ser203, 3) phosphorylation of Ser203 markedly increased in vitro when recombinant PKD1 was incubated with either PAK1 or PAK2 in the presence of ATP. PAK inhibitors did not interfere with G protein-coupled receptor activation-induced rapid translocation of PKD1 to the plasma membrane but strikingly prevented the dissociation of PKD1 from the plasma membrane and blunted the phosphorylation of nuclear targets, including class IIa histone deacetylases. We conclude that PAK-mediated phosphorylation of PKD1 at Ser203 triggers its membrane dissociation and subsequent entry into the nucleus, thereby regulating the phosphorylation of PKD1 nuclear targets, including class IIa histone deacetylases.
Keywords: angiotensin II, G protein-coupled receptor (GPCR), protein kinase C (PKC), protein kinase D (PKD), serine/threonine-protein kinase PAK 1 (PAK1)
Introduction
Protein kinase D (PKD)5 is an evolutionarily conserved protein kinase family with structural, enzymological, and regulatory properties different from the PKC family members. Accumulating evidence implicates PKD1, the founding and most studied member of the family, in the regulation of multiple biological responses, including signal transduction (1–6), chromatin organization (7), Golgi function, gene expression (5, 6, 8–10) and cell survival, polarity, adhesion, motility, differentiation, DNA synthesis, and proliferation, (for review see Refs. 11 and 12). In fibroblasts, PKD1 overexpression potently enhances ERK activation, DNA synthesis, and cell proliferation induced by Gq-coupled receptor agonists (1, 13, 14). In intestinal epithelial cells, PKD1 promotes proliferative and migratory responses in vitro and in crypt intestinal epithelial cells in vivo (3, 15). Furthermore, PKD family members are increasingly implicated in inflammation, T cell development, angiogenesis, cardiac hypertrophy, and cancer (11, 12, 16–18). Recently, PRKD1 hotspot mutations have been identified in adenocarcinomas of the salivary gland tumors (19). The involvement of PKD1 in mediating such a diverse array of normal and abnormal biological functions depends on dynamic changes in its spatial localization combined with its distinct substrate specificity. Consequently, the mechanisms that coordinate and modulate PKD multisite phosphorylation with its subcellular localization are important and attract intense interest.
We proposed a model of PKD1 activation that integrates the spatial and temporal changes in PKD1 localization with its multisite phosphorylation (11). In the framework of this model, PKD1 is kept in an inactive state in unstimulated cells through N-terminal domain repression of its catalytic domain activity (11). PKD1 can be activated within intact cells by a remarkable array of stimuli acting through receptor-mediated pathways. Our own studies demonstrated rapid, protein kinase C (PKC)-dependent, PKD1 activation in response to phorbol esters (13, 20, 21), G protein-coupled receptor (GPCR) agonists (1, 10, 13, 22–29) that act through Gq, G12, Gi, and Rho (24, 28–32), growth factors that signal via tyrosine-kinase receptors (22, 33), cross-linking of B-cell receptor and T-cell receptor in B and T lymphocytes, respectively (34–36), and oxidative stress (37, 38). The phosphorylation of Ser744 and Ser748 in the PKD1 activation loop, also referred as activation segment or T-loop, is critical for PKD1 activation (11, 27, 30, 39, 40). Rapid PKC-dependent PKD1 activation is followed by a late, PKC-independent phase of activation induced by Gq-coupled receptor agonists (3, 14, 41). PKD1 catalytic activation within cells leads to its autophosphorylation at Ser916 and Ser748 (1, 3, 14, 36, 41).
Additional studies demonstrated that PKD family members undergo rapid subcellular redistributions in response to stimulation by GPCR agonists and growth factors. Specifically, PKD1 translocates from the cytosol to the plasma membrane followed by its reverse translocation from the plasma membrane to the cytosol and Golgi followed by subsequent accumulation in the nucleus after activation (3, 26, 38, 42–44). Despite the importance of the N-terminal region of PKD1 in mediating autoinhibition, membrane translocation, nuclear import, interaction with other proteins and Golgi localization, surprisingly little is known about its regulation by post-translational modifications. In this context, the highly conserved Ser203 in the N-terminal region of PKD1 (equivalent to Ser205 in the human PKD1) is of interest because it is highly represented in phosphoproteomic databases (45), but neither its signal-dependent regulation nor the kinase responsible for its phosphorylation has been identified.
The p21-activated kinase (PAK) family, which are effectors of Rac and/or Cdc42 in their GTP-bound state, regulate fundamental cellular processes, including motility, proliferation, apoptosis, and gene transcription (46). PAKs are subdivided into two groups: type I PAKs (PAK1, PAK2, and PAK3) and type II PAKs (PAK4, PAK5, and PAK6), which have distinct modes of catalytic activation and both unique and common substrates (47). The PAKs are overexpressed or mutated in many cancer cells (47), including cancers of the gastrointestinal tract (48–51), and promote pro-oncogenic signaling in these cells (52). Although several pathways, including Raf/MEK/ERK and Wnt/β-catenin, have been implicated in PAK signaling, it is recognized that downstream targets in PAK-initiated cascades remain to be identified (53). It is not known whether the PAKs can regulate the phosphorylation and/or the dynamic subcellular distribution of the PKDs during cell activation.
Here, we demonstrate that agonist-mediated activation of GPCRs in multiple cellular model systems, including epithelial and fibroblastic cells, induces rapid and striking phosphorylation of PKD1 on Ser203, revealing novel input in PKD1 regulation. Based on pharmacological, biochemical, and genetic evidence, we identify the PAK family I as the upstream protein kinase that phosphorylates PKD1 on Ser203 in response to GPCR agonists. The phosphorylation of this residue regulates the time of residence in the membrane of activated PKD1 via accelerating its reverse translocation from the plasma membrane to the cytosol. The inhibition of PAK1/2 retains PKD1 in the membrane and prevents PKD-mediated phosphorylation and nuclear extrusion of histone deacetylase 5 (HDAC5), a member of the class IIa HDACs characterized by nuclear/cytoplasmic shuttling. Thus, our results uncover a new regulatory cross-talk between PKD1 and PAK1/2 that stimulates PKD1 phosphorylation at a site that regulates its subcellular localization and identify a new PAK/PKD1/HDAC-signaling pathway in intact cells.
Results
Rapid phosphorylation of PKD1 on Ser203 in response to GPCR agonists in multiple cell model systems
To examine regulation of PKD1 phosphorylation in its N-terminal region, we used intestinal epithelial IEC-18 cells (54, 55), which endogenously express Gαq/11-coupled receptors for angiotensin II (ANG II) and vasopressin (24, 25, 56–60). These cells have been extensively used as a model system to examine signal transduction pathways in response to GPCR activation (3, 15, 24, 25, 56, 58, 59, 61). Lysates of IEC-18 cells stimulated with ANG II for various times (0.5–300 min) were analyzed by Western blotting with an antibody that detects the phosphorylated state of PKD1 at Ser203, a highly conserved residue located between the cysteine-rich motifs (cys1 and cys2), which comprise the cysteine-rich domain (CRD) in the N-terminal region of PKD1 (Fig. 1A). ANG II induced a rapid (detectable within 30 s), sustained (persisted for at least 5 h), and dramatic increase in PKD1 phosphorylation at Ser203 in intestinal epithelial IEC-18 cells (Fig. 1B). A similar increase in PKD1 phosphorylation at Ser203 was induced when these cells were challenged with vasopressin (50 nm) instead of ANG II (data not shown).
Figure 1.
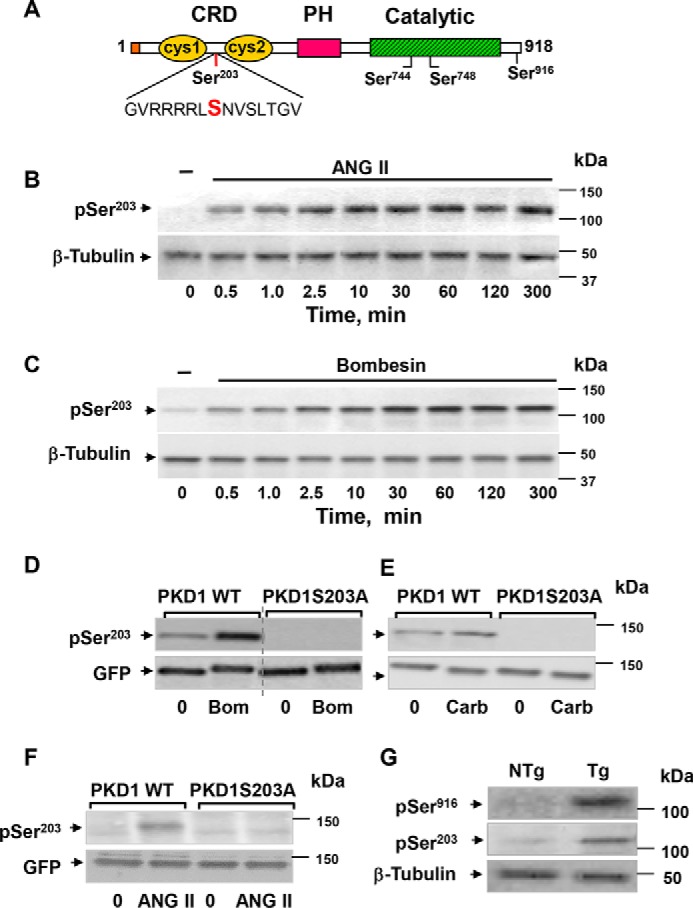
PKD1 is rapidly phosphorylated on Ser203 in response to GPCR agonists in multiple cell model systems and in intestinal cells in vivo. A, schematic representation of PKD1 domains and phosphorylation sites. Ser203 is located between the cysteine-rich motifs (cys1 and cys2), which compose the CRD in the N-terminal region. The phosphorylation sites within the activation loop of the catalytic domain (Ser744 and Ser748) as well as the autophosphorylation site (Ser916) in the C-terminal region of PKD1 are also indicated. PH, pleckstrin homology. B, confluent cultures of IEC-18 cells were incubated without (−) or with 10 nm ANG II for the indicated times. C, confluent cultures of Swiss 3T3 cells were incubated without (−) or with 10 nm bombesin for the indicated times. D, cultures of COS-7 cells transiently co-transfected with bombesin receptor and GFP-PKD1 and either wild type (PKD1 WT) or with Ser203 mutated to Ala (PKD1S203A) were stimulated without (−) or with 10 nm bombesin (Bom) for 10 min. E, cultures of HEK 293 cells were transiently transfected with either PKD1WT or with PKD1S203A and stimulated with 100 μm carbachol (Carb) for 10 min. F, cultures of IEC-18 cells were transiently transfected with either PKD1WT or PKD1S203A and stimulated without (−) or with 10 nm ANG II for 10 min. In panels B–F, the cultures were lysed with 2×SDS-PAGE sample buffer, and the extracts were analyzed by immunoblotting with antibodies that detect PKD1 phosphorylated at Ser203 (pSer203). Immunoblotting for β-tubulin (B and C) or GFP (D–F) was also included. G, epithelial cells from the ileum of PKD1 transgenic (Tg) mice and non-transgenic (NTg) littermates were isolated sequentially by timed incubations in a EDTA-PBS solution. The lysates of these cells were used to analyze for PKD1 phosphorylated at either Ser916 (pSer916) or Ser203 (pSer203). Equivalent loading was verified by immunoblotting for β-tubulin. Image editing in D: irrelevant lanes were removed (indicated by a thin, vertical dotted line) from the acquired digital images, and flanking lanes were juxtaposed using Adobe Photoshop.
We next determined whether PKD1 phosphorylation at Ser203 in response to Gq-coupled receptor activation can be demonstrated in other cellular model systems. As shown in Fig. 1C, bombesin induced a rapid, persistent, and striking increase in PKD1 phosphorylation at Ser203 in Swiss 3T3-PKD.GFP cells, which endogenously express GPCRs for the gastrointestinal peptides of the bombesin/GRP family (13, 60, 62–76). We also detected phosphorylation of ectopically expressed PKD1 at Ser203 in COS-7 cells in response to bombesin receptor activation (Fig. 1D) and in HEK 293 cells stimulated with carbachol via endogenously expressed muscarinic receptors (Fig. 1E).
To verify the specificity of the antibody used for detecting the phosphorylated state of PKD1 at Ser203, we assessed its reactivity against GFP-tagged wild-type PKD1 (GFP-PKD1) or a GFP-PKD1 mutant with Ser203 replaced by non-phosphorylatable alanine (S203A). COS-7 cells were co-transfected with constructs encoding the bombesin/GRP receptor and either GFP-PKD1 or GFP-PKDS203A. The phospho-antibody reacted strongly with GFP-PKD1 isolated from bombesin-stimulated cells but did not recognize the PKD1 mutant in which Ser203 was replaced by alanine, i.e. GFP-PKDS203A (Fig. 1D). A similar result was obtained when GFP-PKD1 and GFP-PKDS203A were transfected into HEK 293 cells stimulated with carbachol (Fig. 1E) or IEC-18 cells challenged with ANG II (Fig. 1F). These results confirmed the specificity of the antibody used to detect the phosphorylated state of PKD at Ser203. Collectively, the results presented in Fig. 1 demonstrate that agonist-induced activation of multiple GPCRs induced robust phosphorylation of PKD1 at Ser203 in a variety of model cell systems.
To determine whether PKD1 phosphorylation at Ser203 can be demonstrated in vivo, we used epithelial cells eluted from the ileum of transgenic mice overexpressing PKD1 (3). As shown in Fig. 1G, PKD1 phosphorylation at Ser203 was also detected in lysates of epithelial cells isolated from transgenic mice overexpressing PKD1. Collectively, the results in Fig. 1 identify a novel signal-responsive phosphorylation in the N-terminal regulatory region of PKD1.
Dissociation of PKD1 phosphorylation at Ser203 from PKD1 catalytic activity
As a first step in identifying the protein kinases responsible for PKD1 phosphorylation at Ser203, we considered at least two alternative mechanisms involving autophosphorylation or transphosphorylation. In the first mechanism, PKD1 activation via membrane translocation and activation loop phosphorylation would lead to its autophosphorylation at Ser203. In the second mechanism, PKD1 phosphorylation would result from transphosphorylation by a different upstream protein kinase.
If PKD1 phosphorylation at Ser203 is mediated by autophosphorylation, we expected that this phosphorylation should be prevented by PKD family inhibitors, including kb NB 142-70 (77) and CRT0066101 (78) and abolished in kinase-inactive PKD1 mutants. To test these predictions, IEC-18 cells were treated with increasing concentrations of either CRT0066101 (Fig. 2A) or kb NB 142-70 (Fig. 2B) before stimulation with ANG II. As expected, exposure of intact cells to increasing concentrations of the PKD family inhibitors suppressed PKD1 activation in a dose-dependent manner, as scored by autophosphorylation at Ser916. In contrast, CRT0066101 (Fig. 2A) or kb NB 142-70 (Fig. 2B) did not prevent Ser203 phosphorylation in response to ANGII stimulation, even at 10 μm, the highest concentration tested of each inhibitor (quantification in Fig. 2C).
Figure 2.

PKD1 phosphorylation at Ser203 can be dissociated from PKD or PKC family catalytic activity. A and B, confluent cultures of IEC-18 cells were incubated in the absence (0) or in the presence of increasing concentrations of CRT0066101 (A) or kb NB 142-70 (B) for 1 h and then stimulated without (−) or with 10 nm ANG II for 10 min. Then, the cultures were lysed with 2×SDS-PAGE sample buffer and analyzed by immunoblotting with antibodies that detect PKD1 phosphorylated at either Ser203 (pSer203) or Ser916 (pSer916). Lysates were also analyzed for total PKD1 (PKD). C, quantification of three independent experiments similar to those shown in A and B are included: open circles represent Ser(P)203, whereas closed circles signify Ser(P)916. D and E, confluent cultures of Swiss 3T3.PKD1 cells were incubated in the absence (0) or in the presence of increasing concentrations of CRT0066101 (D) or kb NB 142-70 (E) for 1 h and then stimulated without (−) or with 10 nm bombesin for 10 min. Then the cultures were lysed with 2×SDS-PAGE sample buffer and analyzed by immunoblotting with antibodies that detect PKD1 phosphorylated at either Ser203 (pSer203) or Ser916 (pSer916). Lysates were also analyzed for total PKD1 (PKD). F, quantification of three independent experiments similar to those shown in D and E are included: open circles represent Ser(P)203, whereas closed circles signify Ser(P)916. G, Swiss 3T3.PKD1618N cells were stimulated with bombesin (Bom) for the indicated times. Then the cultures were lysed with 2×SDS-PAGE sample buffer and analyzed by immunoblotting with antibodies that detect PKD1 phosphorylated at Ser203 (pSer203) and GAPDH. H and J, confluent cultures of IEC-18 cells (H) or Swiss 3T3 cells (J) were incubated in the absence (0) or in the presence of increasing concentrations of Go6983 or 3.5 μm GFI (GF) for 1 h as indicated and then stimulated without (−) or with 10 nm ANG II (H) or 10 nm bombesin (J) for 10 min. Then the cultures were lysed with 2×SDS-PAGE sample buffer and analyzed by immunoblotting with antibodies that detect PKD1 phosphorylated at either Ser203 (pSer203) or Ser744 (pSer744) as a marker of PKC activity. Lysates were also analyzed for total PKD1 (PKD).
In consonance with the results obtained with IEC-18 cells, treatment of Swiss 3T3-PKD1.GFP cells with increasing concentrations of CRT0066101 (Fig. 2D) or kb NB 142-70 (Fig. 2E) blocked PKD1 autophosphorylation at Ser916 but did not prevent Ser203 phosphorylation in response to bombesin stimulation (quantification in Fig. 2F). Furthermore, bombesin also induced Ser203 phosphorylation in Swiss 3T3 cells expressing a kinase-inactive PKD1 mutant (Fig. 2G). These results indicate that PKD1 phosphorylation at Ser203 can be dissociated from its catalytic activity implying that the modification of this site is mediated by transphosphorylation through a different upstream protein kinase(s).
Dissociation of PKD1 phosphorylation at Ser203 from PKC family catalytic activity and other basophilic protein kinases
To pursue the transphosphorylation hypothesis, we attempted to identify the protein kinase(s) that phosphorylates PKD1 at Ser203. The multiple basic residues in the sequence surrounding Ser203 (Fig. 1 A) indicated that the upstream kinase must be a basophilic protein kinase. As a first step to identify endogenous PKD1 Ser203 kinase(s), we tested multiple small-molecule inhibitors targeting a variety of protein kinases, especially basophilic kinases.
Given the important role of PKCs in PKD family regulation (11, 12, 79) and the fact that the sequence surrounding Ser203 was recognized as a PKC-putative phosphorylation site by motif-based profile-scanning programs, we initially determined the effect of PKC inhibitors, including GF 109203X, also known as GF I (72, 80, 81), and Go6983 (82) on GPCR-induced PKD Ser203 phosphorylation. Suppression of PKCs with Go6983 or GF I did not prevent PKD1 phosphorylation at Ser203 in response to either ANG II in IEC-18 (Fig. 2H) or bombesin in Swiss 3T3-PKD.GFP (Fig. 2J). The PKC inhibitory activity of Go6983 or GF I in the same cell lysates was verified by abrogation of PKD1 phosphorylation at Ser744 (Fig. 2, H and J), a well established target of PKCs in the activation loop of PKD1 (11, 40). Because Go6983 inhibits all isoforms of the PKC family, these results indicate that PKD1 phosphorylation at Ser203 is not mediated by any of the PKCs.
The absence of proline at the p + 1 position in the sequence surrounding Ser203 (see above) excludes proline-directed kinases, including MAPK families, Cdks (cyclin-dependent kinases) and mTOR (84, 85). Accordingly, the MEK inhibitors U0126 and PD98059 or the active-site mTOR inhibitor KU0063794 did not prevent ANG II-induced phosphorylation of PKD1 at Ser203 in IEC-18 cells (Table 1). Treatment of these cells with multiple chemical inhibitors known to suppress the activity of other basophilic protein kinases, including Akt, AMPK, Aurora A, calmodulin-dependent protein kinase II (CaMKII), Clk family, MSK1, PKA, Pim1, ROCK, RSK, and S6K or tyrosine kinases that function upstream of phosphorylation cascades in GPCR signaling (79), including EGFR, Src, and FAK, did not prevent ANGII-induced PKD1 phosphorylation at Ser203 (Table 1).
Table 1.
Multiple protein kinase inhibitors did not prevent PKD1 phosphorylation at Ser203 in IEC-18 cells
Confluent cultures of IEC-18 cells were incubated with each of the inhibitors listed below for 1 h. Then the cells were stimulated with ANG II for 60 min and lysed, and the phosphorylation of PKD1 at Ser203 was determined by immunoblotting using the antibody that detects the phosphorylated state of this residue. The inhibitors listed in this table did not prevent Ser203 phosphorylation at the concentrations tested. cPKC, conventional PKCs; nPKC, novel PKCs; aPKC, atypical PKCs; and mTOR, mechanistic target of rapamycin.
| Inhibitor | Primary target | Concentration |
|---|---|---|
| μm | ||
| kb NB 142–70 | PKD | 1–10 |
| CRT 0066101 | PKD | 1–5 |
| Go6983 | cPKC, nPKC, aPKC | 1–10 |
| GF 109203X | cPKC, nPKC | 3.5 |
| KT 5720 | PKA | 1 |
| KU 0063794 | mTOR | 1–5 |
| GSK 690693 | Akt, PKC, PKA | 1–5 |
| KU 55933 | ATM | 5–25 |
| HA 1077 | ROCK-II, PKA, PKG, PKCα | 10 |
| Y-27632 | ROCK | 10 |
| Compound C | AMPK | 5–25 |
| Aurora kinase inhibitor III | Aurora A | 10 |
| Aurora kinase inhibitor II | Aurora A and B | 10 |
| GSK-3 inhibitor XVI | GSK-3 | 5 |
| LY 294002 | PI3K, mTOR | 10 |
| PP2 | Src | 10 |
| AG1478 | EGFR | 1 |
| U0126 | MEK1/2 | 5 |
| PD98059 | MEK1/2 | 5 |
| PIM-1 inhibitor 2 | Pim-1 kinase | 1–10 |
| TG003 | Clk family | 1–10 |
| Enzastaurin (LY317615) | PKCβ | 1–5 |
| GO6976 | cPKC, PKD | 10 |
| KN92 | CaMKII | 10 |
| KN62 | CaMKII | 10 |
| FAK inhibitor 14 | FAK | 1–5 |
| UCN-01 | Chk1 | 10 |
ATP-competitive inhibitors of PAK prevent PKD1 phosphorylation at Ser203
In striking contrast to the lack of inhibitory effects on PKD1 phosphorylation at Ser203 by multiple inhibitors, the pyrrolopyrazole PAK inhibitor PF-3758309 potently abrogated GPCR-induced PKD1 phosphorylation at Ser203. PF-3758309 is an ATP-competitive inhibitor of both PAK type I and II and also inhibits a number of other protein kinases at higher concentrations (86, 87). Treatment of IEC-18 cells with increasing concentrations of PF-3758309 suppressed GPCR-induced PKD1 Ser203 phosphorylation in a concentration-dependent manner (Fig. 3A). PAK activation leads to autophosphorylation of several sites, including Ser144 in PAK1 (equivalent to Ser141 in PAK2). We verified that treatment with PF-3758309 also blunted PAK1 autophosphorylation, scored by phosphorylation at Ser144. Indeed, treatment with PF-3758309 suppressed GPCR-induced PKD1 Ser203 phosphorylation and PAK1 phosphorylation at Ser144 at similar concentrations in IEC-18 cells (Fig. 3A; quantification in Fig. 3B).
Figure 3.

The increase in PKD1 phosphorylation at Ser203 is prevented by ATP-competitive and allosteric inhibitors of the PAKs. A–D, confluent cultures of IEC-18 cells were incubated in the absence (0) or in the presence of increasing concentrations of PF-3758309 (A) or FRAX597 (C) for 1 h and stimulated without (−) or with 10 nm ANGII for 15 min. Then the cultures were lysed with 2×SDS-PAGE sample buffer and analyzed by immunoblotting with antibodies that detect PKD1 phosphorylated at Ser203 (pSer203) or Ser916 (pSer916) and PAK1/2 at Ser144/141 (PAKpS144/141). Lysates were also analyzed for total PKD1 (PKD). B and D, quantification of four independent experiments similar to those shown in A and C are included. E and F, confluent cultures of IEC-18 cells were incubated in the absence or in the presence of 5 μm PF-3758309 (PF, E) or 5 μm FRAX 486 (F) for 1 h and then stimulated with 10 nm ANGII for the various times as indicated. Then the cultures were lysed with 2×SDS-PAGE sample buffer, and the extracts were analyzed by immunoblotting with antibodies that detect PKD1 phosphorylated at Ser203 (pSer203) and PAK1/2 phosphorylated at Ser144/141 (PAKpS144/141). G, confluent cultures of Swiss 3T3.PKD1 cells were incubated in the absence (0) or in the presence of increasing concentrations of PF-3758309 for 1 h and stimulated without (−) or with 10 nm bombesin for 15 min. Then the cultures were lysed and analyzed by immunoblotting with antibodies that detect PKD1 phosphorylated at Ser203 (pSer203) or Ser916 (pSer916) and PAK1/2 at Ser144/141 (PAKpS141). Lysates were also analyzed for total PKD1 (PKD). H, quantification of three independent experiments similar to those shown in G are included. I and J, confluent cultures of Swiss 3T3.PKD1 cells were incubated in the absence or in the presence of 5 μm PF-3758309 (I) or 5 μm FRAX 486 (J) for 1 h and then stimulated with 10 nm bombesin for various times as indicated. Then, the cultures were lysed with 2×SDS-PAGE sample buffer and analyzed by immunoblotting with antibodies that detect PKD1 phosphorylated at Ser203 (pSer203) and PAK1/2 at Ser144/141 (PAKpS141). Lysates were also analyzed for total PKD1 (PKD). L, confluent cultures of IEC-18 cells were incubated in the absence (0) or in the presence of increasing concentrations of IPA-3 or 30 μm PIRS for 1 h and then stimulated without (−) or with 10 nm ANGII for 15 min. Then the cultures were lysed with 2×SDS-PAGE sample buffer, and the extracts were analyzed by immunoblotting with antibodies that detect PKD1 phosphorylated at Ser203 (pSer203) and PKD1 phosphorylated at Ser916 (pSer916). Lysates were also analyzed for total PKD1 (PKD). M, Quantification of 4 independent experiments similar to those shown in L but including additional concentrations of IPA-3. N, Confluent cultures of 3T3.PKD1 cells were incubated in the absence (0) or in the presence of increasing concentrations of IPA-3 or 20 μm PIRS for 1 h and then stimulated without (−) or with 10 nm bombesin for 15 min. Then, the cultures were lysed with 2×SDS-PAGE sample buffer and analyzed by immunoblotting with antibodies that detect PKD1 phosphorylated at Ser203 (pSer203) or at Ser916 (pSer916). Lysates were also analyzed for total PKD1 (PKD). O, quantification of four independent experiments similar to those shown in N but including additional concentrations of IPA-3.
The results obtained with PF-3758309 prompted us to test other structurally unrelated inhibitors of the PAKs. FRAX597 and FRAX486 are preferential ATP-competitive inhibitors of type I PAKs (87, 88). As shown in Fig. 3C, FRAX597 potently inhibited phosphorylation of PKD1 at Ser203 and PAK1/2 on Ser144/141 induced by ANG II in IEC-18 cells at similar concentrations (quantification in Fig. 3D), suggesting that group I PAKs mediate PKD1 phosphorylation at Ser203.
It is plausible that different upstream kinases phosphorylate PKD at Ser203 at different times of GPCR stimulation. Consequently, we determined whether PAK inhibitors prevented PKD1 Ser203 phosphorylation at various times after stimulation with ANG II in IEC-18 cells. Exposure to PF-3758309 (Fig. 3E) or FRAX486 (Fig. 3F) inhibited phosphorylation of PKD1 Ser203 in response to GPCR stimulation at all times tested. We verified that PF-3758309 and FRAX486 also suppressed GPCR-induced PAK1/2 autophosphorylation at Ser144/141 in IEC-18 at all times tested. The doublet detected in IEC-18 with the PAK1/2 phospho-antibody corresponded to PAK1 Ser(P)144 (Fig. 3, E and F, upper band) and PAK2 Ser(P)141 (Fig. 3, E and F, lower band) as shown by using siRNA-mediated knockdown of each of these PAK formalisms (data not shown).
In agreement with the results obtained with IEC-18 cells, treatment of Swiss 3T3-PKD1.GFP with PF-3758309 before bombesin stimulation prevented phosphorylation of PKD1 at Ser203 and PAK2 (the major isoform expressed in these cells) at Ser141 at similar concentrations (Fig. 3G; quantification in Fig. 3H). We also verified that exposure to PF-3758309 or FRAX 486 inhibited PKD1 Ser203 and PAK2 Ser141 phosphorylation induced by bombesin in Swiss 3T3-PKD.GFP cells at all the times tested (Fig. 3, I and J). Although both FRAX597 and PF-3758309 inhibit other kinases, many of these off-target effects are non-overlapping (87, 89). Therefore, these results imply that type I PAKs mediate PKD1 phosphorylation at Ser203 in response to GPCR stimulation in different cell systems.
IPA-3, an allosteric inhibitor of group I PAKs, also prevented PKD1 phosphorylation at Ser203
To substantiate the results obtained with the ATP-competitive inhibitors, we also tested the compound IPA-3 (90, 91), a specific allosteric inhibitor that binds to the N-terminal regulatory domain of PAK1/2/3 rather than to their kinase catalytic domain. Treatment with IPA-3 inhibited the increase in PKD1 phosphorylation at Ser203 in a dose-dependent manner in IEC-18 cells challenged with ANG II (Fig. 3L; quantification in Fig. 3M). In contrast, a structural isomer of IPA-3 displaying no inhibitory effect toward the PAKs (PIRS) tested at 30 μm, did not interfere with GPCR-induced PKD1 Ser203 phosphorylation in these cells (Fig. 3L; quantification in Fig. 3M).
Treatment with IPA-3 also abolished PKD1 phosphorylation at Ser203 in Swiss 3T3-PKD1.GFP cells stimulated with bombesin (Fig. 3N; quantification in Fig. 3O). Furthermore, IPA-3 also prevented PKD1 phosphorylation at Ser203 in Swiss 3T3 cells expressing a kinase-dead PKD1 mutant (data not shown). Together, the results presented in Fig. 3 obtained with three structurally unrelated inhibitors that act via different mechanisms (i.e. PF-3758309, FRAX597, and IPA-3) indicate that group I PAK mediates PKD1 phosphorylation on Ser203 in intact epithelial and fibroblast cells.
Identification of PAK1 and PAK2 as the upstream protein kinases that phosphorylate PKD1 at Ser203
To corroborate the results obtained with chemical inhibitors, we silenced the expression of PAK1 and PAK2 in IEC-18 cells using RNAi (PAK3, the other member of group I is expressed primarily in neuronal cells). In line with the notion that the type I PAKs mediate PKD1 phosphorylation at Ser203, knockdown of PAK1 and PAK2 prevented the increase in PKD1 phosphorylation at Ser203 in response to ANG II in IEC-18 cells (Fig. 4A; quantification in Fig. 4B). Similarly, knockdown of PAK1 and PAK2 blocked the increase in PKD1 phosphorylation at Ser203 in response to bombesin in Swiss 3T3-PKD.GFP (Fig. 4C; quantification in Fig. 4D).
Figure 4.

Identification of PAK1 and PAK2 as the upstream protein kinases that phosphorylate PKD1 at Ser203. A, cultures of IEC-18 cells were transfected with non-targeting siRNA (Non. Targ) or with a mixture of siRNAs targeting PAK1 and PAK2 (siPAK1+2) for 5 days. Then the cultures were stimulated with 10 nm ANGII for 10 min. C, cultures of 3T3.PKD1 cells were transfected with non-targeting siRNA (Non. Targ) or with a mixture of siRNAs targeting PAK1 and PAK2 (siPAK1+2) for 5 days. Then the cultures were stimulated with 10 nm bombesin (Bom) for 10 min. After stimulation, the cultures in A and C were lysed with 2×SDS-PAGE sample buffer, and the extracts were analyzed by immunoblotting with antibodies that detect phosphorylated PKD1 at Ser203 (pSer203) or Ser916 (pSer916) and PAK1/2 at Ser144/141 (PAK1pS144/141 (A), PAKpS141 (B). Lysates were also analyzed for total PKD1 (PKD). B, quantification of five independent experiments similar to those shown in A are included. D, quantification of three independent experiments similar to those shown in B are included. E, COS-7 cells were co-transfected with either: GFP-PKD and bombesin receptor; GFP-PKD, bombesin receptor, and pCMV6M-PAK1; GFP-PKD, bombesin receptor, and pCMV6M-PAK1T423E; or GFP-PKD, bombesin receptor, and GST-tagged pEBG-PAK1 83–149 (AID). After 4 days the cultures were incubated in the absence (−) or presence of 10 nm bombesin (Bom) for 10 min. Lysates were analyzed for phosphorylated PKD1 at Ser203 (pSer203), PAK1, GST, GFP, and GAPDH. F, quantification of six independent experiments similar to those shown in E; all conditions also included GFP-PKD1 and bombesin receptor. G and H, recombinant PKD1 (200 ng, Sigma-Aldrich) was incubated in the absence or in the presence of 50 ng of recombinant active PAK1 (G) or 50 ng of recombinant active PAK2 (H) in 25 μl of kinase buffer for 5 min at 300 °C. Reactions were terminated by the addition of 2× SDS-PAGE sample buffer and resolved by SDS-PAGE. PKD1 phosphorylation was determined by Western blot analysis using the antibody that detects its phosphorylated state on Ser203 (pSer203). The presence of PAK1, PAK2, and PKD1 was also detected, as indicated. I, quantification of 5 independent experiments similar to those shown in G and 3 independent experiments similar to those shown in H.
Further evidence for a role of type I PAKs in PKD1 phosphorylation at Ser203 was obtained by transient co-transfection of COS-7 cells with constructs encoding bombesin receptor, PKD1, and either wild-type or constitutively activated PAK1 in which Thr423 was replaced by Glu (92). Our results demonstrate that expression of PAK1T423E (but not wild-type PAK1) markedly increased the phosphorylation of PKD1 at Ser203 in unstimulated cells to a degree comparable with that produced by agonist stimulation (Fig. 4E; quantification in Fig. 4F). Similar results were obtained when the transient co-transfections were performed using HEK 293 cells instead of COS-7 cells (data not shown). We also used a dominant-negative strategy to test the role of the PAKs in PKD1 phosphorylation at Ser203. We expressed a highly conserved fragment corresponding to the autoinhibitory domain (AID) of PAK 1 (residues 83–149) that inhibits type I PAK activity (93). The expression of GST-AID prevented an agonist-stimulated increase in PKD1 Ser203 phosphorylation (Fig. 4E; quantification in Fig. 4F). On the basis of multiple approaches, we can conclude that PKD1 phosphorylation at Ser203 is mediated through a PAK1/2-dependent pathway.
We next determined whether Ser203 of PKD1 can serve as a direct substrate for activated PAK1 and PAK2. To test this possibility, recombinant PKD1 was incubated in the absence or presence of activated recombinant PAK1 or PAK2. After 5 min, the reactants were analyzed by Western blotting using the antibody that detects the phosphorylated state of PKD1 at Ser203. We found that the phosphorylation of Ser203 was markedly increased in vitro when PKD1 was incubated with either PAK1 (Fig. 4G) or PAK2 (Fig. 4H) in the presence of ATP (quantification in Fig. 4I).
Based on evidence generated via pharmacological, biochemical, and genetic approaches, we conclude that the group I PAKs phosphorylate the N-terminal domain of PKD1 at Ser203 in a stimulus-dependent manner. The identification of PAK-mediated PKD1 phosphorylation reveals a new point of integration in the signal transduction pathways initiated by GPCR agonists and raises important questions concerning its function in cell regulation.
PKD1 phosphorylation at Ser203 regulates its intracellular redistribution in response to GPCR agonists
The CRD domain of PKD1, composed of a tandem repeat of cysteine-rich motifs, mediates its translocation from the cytosol to the plasma membrane in response to GPCR-induced generation of DAG (11). Because Ser203 lies between the cysteine-rich motifs of the CRD (Fig. 1A), we considered the possibility that the phosphorylation of this residue regulates the intracellular localization of PKD1 in response to GPCR stimulation. To explore this mechanism with high resolution, we generated IEC-18 cell lines that express PKD1-EGFP in an inducible manner. After exposure to doxycycline for 24 h to induce the expression of PKD1-EGFP, cells were treated in the absence or presence of PF-3758309, FRAX597, or IPA-3 and subsequently stimulated with ANG II for various times. In line with previous results, stimulation of control IEC-18 cells with ANG II induced a rapid (3 min) and striking translocation of PKD1 from the cytosol to the plasma membrane, causing a localized fluorescence at the cell periphery (Fig. 5A). The translocation of PKD1 was transient as it quickly dissociated from the plasma membrane, returned to the cytoplasm, and accumulated in the nucleus of most cells after 30 min of stimulation (Fig. 5A; quantification in Fig. 5B). In sharp contrast, cells treated with PF-3758309 (Fig. 5, C and D), FRAX597 (Fig. 5, E and F), or IPA-3 (Fig. 5, G and H) displayed PKD1-EFGP at the plasma membrane but not in the nucleus, even after 60 min of ANG II stimulation. As shown in Fig. 5, I and J (quantification in Fig. 5K), the results obtained with chemical inhibitors were corroborated by knockdown of the expression of PAK1 and PAK2 in IEC-18 cells using siRNAs (Fig. 5L). Specifically, cells depleted of PAK1 and PAK2 exhibited PKD1-EFGP at the plasma membrane but not in the nucleus, even after 60 min of ANG II stimulation (Fig. 5, I and J (quantification in Fig. 5K). Collectively, the results implied that PAK-mediated phosphorylation of PKD1 facilitates membrane dissociation and nuclear localization of PKD1 in GPCR-stimulated cells.
Figure 5.

Inducible EGFP-PKD1 remains associated to the plasma membrane in the presence of inhibitors of the group I PAK or in cells transfected with siRNAs targeting PAK1 and PAK2. Confluent cultures of IEC-18 cells that express PKD1-EGFP in an inducible manner were treated with 1 μg/ml doxycycline for 24 h to promote the expression of PKD1-EGFP. Then the cells were treated in the absence (A and B) or presence of 3.5 μm PF-3758309 (C and D), 5 μm FRAX597 (E and F), or 30 μm IPA-3 (G and H) for 1 h and subsequently stimulated with 10 nm ANG II for various times as indicated. I, J, and K, cultures of IEC-18 cells that express inducible PKD1-EGFP were transfected with non-targeting siRNA (Non. Targ) or with a mixture of siRNAs targeting PAK1 and PAK2 (siPAK1+2) for 5 days. Then, the cultures were stimulated with 10 nm ANGII for the indicated times. All other cultures were fixed with 4% paraformaldehyde. Quantitative analysis of the relative change in plasma membrane and cytosol fluorescence intensity (expressed as ratio of membrane/cytosol) is shown in panels B, D, F, H, J, and K. In panel L, cultures of IEC-18 cells that express inducible PKD1-EGFP were transfected with non-targeting siRNA (Non. Targ) or with a mixture of siRNAs targeting PAK1 and PAK2 (siPAK1+2) for 5 days. Then, the cultures were stimulated without (−) or with 10 nm ANGII for 10 min. The extracts were analyzed by immunoblotting with antibodies that detect PAK1, PAK2, and GAPDH.
To rule out possible interference with the intracellular distribution of PKD1 due to the presence of the fluorescent tag, we analyzed the localization of endogenous PKD1. As illustrated by representative cells displayed in Fig. 6, the distribution of endogenous PKD1 was very similar to that detected with EGFP-PKD1. Specifically, endogenous PKD1 was located primarily in the cytoplasm of unstimulated IEC-18 cells. Stimulation of these cells with ANG II induced rapid (3 min) PKD1 translocation to the plasma membrane. Subsequently, PKD1 underwent reverse translocation returning to the cytosol and nucleus 60 min after stimulation. Treatment of the cells with chemical inhibitors of group I PAKs, including PF-3758309 (Fig. 6B), FRAX597 (Fig. 6C), and IPA-3 (Fig. 6D) prevented the dissociation of endogenous PKD1 from the plasma membrane in agonist-stimulated cells. Similarly, siRNA-mediated knockdown of PAK1 and PAK2 also impeded reverse translocation of endogenous PKD1 from the plasma membrane to the cytoplasm and nucleus in cells stimulated with ANG II (Fig. 6, E and F). We verified that transfection of siRNAs targeting PAK1 and PAK 2 resulted in the extensive knockdown of their protein expression (similar to results shown in Fig. 4A and Fig. 5L). The images obtained were quantified, and the results (bars in Fig. 6) substantiated that chemical inhibitors of group I PAKs and siRNA-mediated knockdown of PAK1 and PAK2 prevent the dissociation of endogenous PKD1 from the plasma membrane.
Figure 6.

Endogenous PKD1 remains associated to the plasma membrane in the presence of inhibitors of the group I PAK or in cells transfected with siRNAs targeting PAK1 and PAK2. Confluent cultures of IEC-18 cells were treated in the absence (A) or presence of 3.5 μm PF-3758309 (B), 5 μm FRAX597 (C), or 30 μm IPA-3 (D) for 1 h and subsequently stimulated with 10 nm ANG II for various times, as indicated. E and F, cultures of IEC-18 cells were transfected with non-targeting siRNA (Non. Targ) or with a mixture of siRNAs targeting PAK1 and PAK2 (siPAK1+2) for 5 days. Then, the cultures were stimulated with 10 nm ANGII for the indicated times. The cultures were then washed and fixed with 4% paraformaldehyde and stained with an antibody that detects the PKD1 and Hoechst 33342 stain to visualize the nuclei. The bars represent the analysis of the relative change in plasma membrane fluorescence intensity (expressed as ratio of membrane/cytosol).
Having established that PAK1 and PAK2 regulate the intracellular distribution of PKD1 in agonist-stimulated cells, we next determined the role of Ser203 phosphorylation in this process. Specifically, cultures of IEC-18 cells were transiently transfected with wild-type GFP-PKD1 or a mutant GFP-PKD1 with Ser203 substituted by alanine (PKD1S203A), and then the localization of these PKD1 forms was examined after various times of ANG II stimulation. In unstimulated cells, GFP-PKD1 and GFP-PKD1(S203A) were predominantly in the cytoplasm. As expected, stimulation of IEC-18 cells with ANG II induced a rapid but transient translocation of wild-type GFP-PKD1 to the plasma membrane (Fig. 7A). ANG II also induced rapid translocation of GFP-PKD1(S203A) from the cytosol to the plasma membrane, but this mutant PKD1 remained at the membrane for much longer with significant fluorescence signals localized to the plasma membrane even after 60 min of stimulation (Fig. 7A; quantification in Fig. 7B). These results substantiated the notion that PAK1/2-mediated phosphorylation of PKD1 at Ser203 accelerates its reverse translocation from the membrane to the interior of the cell. Taken together, the results show for the first time that the PAKs of group I regulate the intracellular localization of PKD1. In this manner it was plausible that the PAKs control the access to specific substrates phosphorylated by activated PKD1.
Figure 7.

Identification of a novel PAK/PKD/HDAC-signaling pathway. A, IEC-18 cells were transiently transfected with constructs encoding GFP-PKD1 or GFP-PKD1S203A. After 48 h the cultures were incubated in the absence (−) or presence of 10 nm ANG II for various times, as indicated. B, analysis of 70 cells at each time point. Results are expressed as the % of cells with GFP-PKD1 localized on the cell membrane. C, IEC-18 cells were transiently transfected with a plasmid encoding FLAG-tagged HDAC5. The cultures were incubated in the absence (−) or presence of 3.5 μm CRT0066101, 3.5 μm PF-3758309, 3.5 μm FRAX597, or 30 μm IPA-3 for 1 h before stimulation with 10 nm ANG II for 1 h. The cultures were then washed and fixed with 4% paraformaldehyde and stained with an antibody that detects the FLAG tag and Hoechst 33342 stain to visualize the nuclei. D, analysis of 100 cells for each treatment. Results are expressed as the % of cells with FLAG-HDAC5 localized in the nucleus. In each case the closed bars represent cultures stimulated with 10 nm ANG II. E, confluent cultures of IEC-18 cells were incubated in the absence (0) or in the presence of 3.5 μm CRT0066101 (CRT), 3.5 μm PF-3758309 (PF), 3.5 μm FRAX597 (FRAX), or 30 μm IPA-3 (IPA) for 1 h and then stimulated without (−) or with 10 nm ANG II for 15 min. F and G, IEC-18 cells were incubated in the absence (0) or in the presence of increasing concentrations of PF-3758309 (F) or FRAX597 (G) for 1 h and then stimulated without (−) or with 10 nm ANG II for 15 min. H, cultures of IEC-18 cells were transfected with non-targeting siRNA (Non. Targ) or with a mixture of siRNAs targeting PAK1 and PAK2 (siPAK1+2) for 5 days. Then the cultures were stimulated with 10 nm ANG II for 10 min. In E, F, G, and H, all incubations were terminated by the addition of 2× SDS-PAGE sample buffer, and cell lysates were resolved by SDS-PAGE. HDAC5 phosphorylation was determined by Western blot analysis using the antibody that detects its phosphorylated state on Ser498 and GAPDH as a loading control. J, GPCR signaling induces DAG accumulation at the plasma membrane, which mediates the translocation of inactive PKD1 from the cytosol to that cellular compartment. DAG also recruits and activates novel PKCs, which mediate the transphosphorylation of PKD1 on Ser744 (in mouse PKD1). DAG and PKC-mediated transphosphorylation of PKD1 act synergistically to promote PKD1 catalytic activation and autophosphorylation on Ser748. PAK1 and PAK2 also activated in response to GPCR signaling phosphorylate PKD1 at Ser203, thereby facilitating its dissociation from the membrane to the cytosol (PKDcyt) and to the nucleus (PKDnucleus), where PKD1 phosphorylates class IIa HDAC, including HDAC5 and HDAC7. In this manner the PAKs regulate class IIa HDAC phosphorylation and localization through phosphorylation of PKD1 on Ser203. Thus, our results identify a novel PAK/PKD/HDAC5 pathway in signal transduction. For more details, see “Discussion.”
Identification of a novel PAK/PKD/HDAC-signaling pathway
Class IIa HDACs, including HDAC4, HDAC5, HDAC7, and HDAC9, are unique among the HDAC family in that they shuttle between the nucleus and the cytoplasm in response to extracellular signals. Phosphorylation of specific conserved residues in HDAC4, HDAC5, HDAC7, and HDAC9 leads to their nuclear extrusion and sequestration in the cytoplasm. PKD1 has been identified as one of the upstream kinases that mediates the phosphorylation and subcellular localization of class IIa HDACs in a variety of cell types, including intestinal epithelial cells (94). In view of our preceding findings, we hypothesized that PAK-mediated reverse translocation of activated PKD1 and its subsequent entry into the nucleus is necessary for the phosphorylation of nuclear targets, including class IIa HDACs.
To determine whether the PAKs control the access of PKD1 to specific substrates in intact cells, cultures of IEC-18 cells were transfected with epitope (FLAG)-tagged HDAC5. After 48 h, the cells were treated without or with PF-3758309, FRAX597, or IPA-3 to inhibit PAK1/2 and, as a control, with CRT0066101 to inhibit PKDs. Subsequently, the cultures were challenged without or with ANG II for 60 min and fixed. As shown in Fig. 7C, FLAG-tagged HDAC5 was present in the nucleus of unstimulated cells and was strikingly translocated to the cytosol in response to ANG II stimulation. Treatment with CRT0066101 prevented the nuclear extrusion of FLAG-tagged HDAC5, in agreement with previous results (94). A salient feature of our current results is that exposure to the PAK inhibitors PF-3758309, FRAX597, or IPA-3 strikingly inhibited the nuclear extrusion of HDAC5 (Fig. 7C; quantification in Fig. 7D). In line with the notion that nuclear extrusion of HDAC5 is mediated by its phosphorylation, we verified that PF-3758309, FRAX597, and IPA-3 inhibited HDAC5 phosphorylation at Ser498, a site phosphorylated by PKD1. PF-3758309 (Fig. 7F) and FRAX597 (Fig. 7G) inhibited HDAC5 phosphorylation at Ser498 in a concentration-dependent manner. Furthermore, knockdown of PAK1 and PAK2 prevented the increase in HDAC5 phosphorylation at Ser498 in response to ANG II in IEC-18 cells (Fig. 7H). Similar results were obtained when we examined agonist-induced intracellular shuttling of HDAC7 in IEC-18 cells. Because cell treatment with PF-3758309, FRAX597, and IPA-3 or knockdown of PAK1/2 did not interfere with PKD1 activation, as scored by autophosphorylation at Ser916 (results shown in Figs. 3 and 4) or activation loop phosphorylation at Ser744 (data not shown), we conclude that PAK1/2 regulates HDAC5 localization and phosphorylation by facilitating the dissociation of active PKD1 from the plasma membrane and its subsequent entry into the nucleus (Fig. 7J).
Discussion
Although PKC-mediated phosphorylation of PKD has been extensively studied as a critical on/off switch of kinase catalytic activity, little is known about the regulation and function of PKD1 phosphorylation by other upstream kinases. Analysis of phosphoproteome databases indicated that Ser203 of PKD1 has been detected in many studies, but nothing was known about its regulation and function, and the upstream kinase involved had not been identified. Here, we report that stimulation of a variety of cell types with GPCR agonists induced rapid and sustained PKD1 phosphorylation at Ser203, thus identifying a novel signal-dependent phosphorylation site in the N-terminal domain of PKD1.
At least two alternative mechanisms involving autophosphorylation or transphosphorylation could explain PKD1 Ser203 phosphorylation in response to GPCR agonists. Treatment with PKD family inhibitors at concentrations that extinguished PKD autophosphorylation at Ser916 as well as the use of cells transfected with a PKD1 kinase-inactive mutant indicated that the phosphorylation of Ser203 is not mediated by autophosphosphorylation. Consequently, we attempted to identify the protein kinase(s) that transphosphorylates PKD1 at Ser203. The multiple basic residues in the sequence surrounding Ser203 (underlined; GVRRRRLSNVSL), Fig. 1A) indicated that the upstream kinase must be a basophilic protein kinase, whereas the absence of proline at the p+1 position excluded proline-directed kinases, including MAPK families, Cdks (cyclin-dependent kinases), and mTOR (84, 85). Furthermore, multiple small-molecule inhibitors of basophilic protein kinases targeting AKT, AMPK, Aurora A, CaMKII, Clk (Cdc2-like kinase), MSK1, PKA, Pim1, ROCK, RSK, and S6K did not prevent ANG II-induced PKD1 phosphorylation at Ser203. In contrast, several lines of evidence indicated that the phosphorylation of PKD1 at Ser203 is mediated by kinases of the type I PAK subfamily as follows. 1) The pyrrolopyrazole PAK inhibitor PF-3758309 prevented PKD1 phosphorylation at Ser203 induced by either ANG II in epithelial IEC-18 cells or bombesin in Swiss 3T3 fibroblasts. PF-3758309, an ATP-competitive inhibitor of both PAK group I and II (86, 87), impaired PKD1 phosphorylation at Ser203 in a concentration-dependent manner and after various times of stimulation. 2) The results obtained with PF-3758309 were corroborated by results using other structurally unrelated inhibitors. FRAX597 is a novel and potent ATP-competitive inhibitor of group I PAKs (87, 88). FRAX597 inhibited PKD1 Ser203 phosphorylation in epithelial and fibroblastic cells in a concentration-dependent manner and after various times of stimulation. 3) We substantiated the results obtained with the ATP-competitive inhibitors PF-3758309 and FRAX597 using the compound IPA-3 (90, 91), a highly specific allosteric inhibitor that binds covalently to the N-terminal regulatory domain of the PAK group I rather than to the kinase catalytic domain of these enzymes and thereby inhibits PAK activation by GTP-loaded Rac1 and Cdc42. Treatment with IPA-3 blunted PKD1 phosphorylation at Ser203 in intestinal epithelial IEC-18 cells stimulated with ANG II or in Swiss 3T3 fibroblasts challenged with bombesin. Collectively, the results obtained with three structurally unrelated inhibitors that act via different mechanisms (PF-3758309, FRAX597, and IPA-3) indicate that group I PAK mediates PKD1 phosphorylation at Ser203 in intact cells. 4) This conclusion was substantiated by siRNA-mediated knockdown of PAK1 and PAK2 in both IEC-18 and Swiss 3T3 cells and by expression of a dominant negative autoinhibitory fragment of PAK1 in COS-7 cells. 5) Phosphorylation of Ser203 was markedly increased in cells by overexpression of a constitutively activated form of PAK1 (PAK1T423E) and in vitro when recombinant PKD1 was incubated with either PAK1 or PAK2 in the presence of ATP. 6) In kinetic studies we verified that PAK1/2 activation in intact cells (scored by autophosphorylation at Ser144/141) in response to GPCR stimulation occurs as rapidly as expected for an upstream kinase in a PAK/PKD cascade (data not shown). Thus, based on pharmacological, biochemical, and genetic evidence, our results indicate that PAK1/2 directly phosphorylate the N-terminal domain of PKD1 at Ser203 and thus reveal a novel input in PKD1 regulation.
We next explored the possible functional consequence(s) of Ser203 phosphorylation in PKD1. The CRD domain in the N-terminal region of PKD1 is composed of a tandem repeat of cysteine-rich motifs (cys1 and cys2) that mediates rapid PKD1 translocation from the cytosol to the plasma membrane in response to GPCR-induced generation of DAG (Fig. 1A). Interestingly, activated PKD1 quickly dissociates from the plasma membrane, returns to the cytoplasm, and accumulates in the nucleus. Because phosphorylation has been implicated in the regulation of PKD1 intracellular localization (43, 95) and PKD Ser203 lies between cys1 and cys2 of the CRD, we considered the possibility that the PAK-mediated phosphorylation of this residue could play a role in triggering membrane dissociation of PKD1 after GPCR stimulation. In line with this notion, we show that treatment of intact cells with different PAK inhibitors, including PF-3758309, FRAX486, FRAX597, or IPA-3 or knockdown of PAK1 and PAK2 expression strikingly prevented the dissociation of PKD1-EGFP from the plasma membrane. These results were corroborated by detecting the intracellular distribution of endogenous PKD1. The critical role of Ser203 phosphorylation in the regulation of PKD1 reverse translocation was confirmed by using wild-type GFP-PKD1 or a mutant GFP-PKD1 with Ser203 substituted by alanine. We found that the mutation S203A impaired PKD1 dissociation from the plasma membrane after various times of ANG II stimulation. Collectively, the results indicate that PAK-mediated phosphorylation of PKD1 at Ser203 facilitates rapid membrane dissociation of PKD1 and subsequent accumulation in the nucleus in GPCR-stimulated intact intestinal epithelial cells (Fig. 7J).
It is recognized that the translocation of kinases to specific subcellular locations plays a critical role in the selection of physiological substrates for their phosphorylation. Previous studies demonstrated that active PKD1 acts as a direct class IIa HDAC kinase in a variety of biological systems, inducing their nuclear extrusion and thereby influencing chromatin organization as well as non-canonical functions of these enzymes (96–98). PKD1 also mediates endogenous class IIa HDAC phosphorylation and cytoplasmic localization in response to GPCR activation in intestinal epithelial cells (94). In addition to PKD1, a number of other protein kinases including Ca2+/CaMKs induce class IIa HDAC cytoplasmic accumulation, but none of the previous studies implicated the PAKs in the regulation of the subcellular localization of the class IIa HDACs (98). Indeed, the phosphorylation sites in these HDACs that control their subcellular distribution do not conform to the consensus sequence phosphorylated by PAKs. However, we posited that PAK-mediated dissociation of activated PKD1 from the plasma membrane is necessary for its ability to act as a class IIa HDAC kinase. In line with this hypothesis, we found that treatment of intact IEC-18 cells with different PAK inhibitors, including PF-3758309, FRAX 597, or IPA-3 or knockdown of PAK1 and PAK2 abrogated the cytoplasmic accumulation and phosphorylation of HDAC5 and HDAC7 induced by ANG II as effectively as inhibiting directly PKD1 activity with the preferential PKD inhibitor CRT0066101. Consequently, our findings support the novel notion that the PAKs play a role in the regulation of class IIa HDAC phosphorylation and localization through phosphorylation of PKD1 on Ser203, thereby triggering PKD1 dissociation from the plasma membrane. Thus, our results identify a novel PAK/PKD/HDAC5 pathway in signal transduction (Fig. 7J).
Experimental procedures
Cell culture
The non-transformed rat intestinal epithelial IEC-18 cells (54, 55), originated from intestinal crypt cells, were purchased from ATCC. These cells express Gq-coupled receptors for ANG II and vasopressin (24, 25, 56–60) and have been extensively used as a model system to examine signal transduction pathways in response to GPCR activation. Cultures of IEC-18 cells were maintained as described previously (3, 4). Briefly, cells were cultured in Dulbecco's modified Eagle's medium (DMEM) supplemented with 5% fetal bovine serum (FBS), 100 units/ml penicillin, and 100 μg/ml streptomycin at 37 °C in a humidified atmosphere containing 10% CO2 and 90% air. Stock cultures were subcultured every 3–4 days. For experimental purposes, IEC-18 cells were seeded in 35-mm dishes at a density of 2 × 105 cells/dish.
Stock cultures of Swiss 3T3-PKD.GFP cells, which overexpress wild-type PKD1 and Swiss 3T3-PKDK618N.GFP cells, and overexpress a kinase-deficient PKD were generated as previously described (1, 13). These derivatives of Swiss 3T3 cells, which express endogenously receptors for the peptides of the bombesin family (65), were maintained and transferred for experimental purposes as described previously (14). Human embryonic kidney HEK 293 cells and COS-7 cells were also cultured as described previously (83, 99).
Generation of IEC-18 cells with inducible PKD1-EGFP
An all-in-one Tet-ON inducible system was developed for the expression of PKD1-EGFP. We used the pCD550A-1 lentiviral vector (SystemBio) as a platform and modified it to express the reverse tetracycline-controlled transactivator (rtTA) under the control of the constitutive PGK promoter. The rtTA cDNA is co-expressed with the cDNA encoding DsRed-Fluorescent Protein (Clontech). The two coding sequences are separated by a 2A-like sequence. The “self-cleaving” 2A peptides are used to mediate protein cleavage from a single open reading frame allowing the expression of the two proteins from a single mRNA. We also replaced the original EF1 promoter by an inducible promoter composed of six tetracycline-responsive elements (TRE) followed by the minimal CMV promoter. The resulting lentiviral vector was designated pCD550A1-TRE-rtTA-Dsred and was used to clone PKD1-EGFP or EGFP under the control of the TRE promoter. Lentiviral infections were performed according to standard procedures. Briefly, 293T cells were co-transfected with pMD2.G and psPAX2 (Addgene Inc.) along with either pCD550A1-TRE-EGFP-rtTA-Dsred or pCD550A1-TRE-PKD1-EGFP-rtTA-Dsred vector using JetPrime transfection reagent (Polyplus) according to the manufacturer's protocol. The transfection medium was replaced after 12 h with fresh DMEM medium with 10% FBS, and 48 h later the viral supernatants were collected and concentrated by using Centricon Plus-70 filter units (Millipore). IEC-18 cells (passage 2) were infected overnight with viral supernatants supplemented with 8 μg/ml of Polybrene (Sigma). After 72 h, stably transduced IEC-18 cells were separated by fluorescence-activated cell sorting (FACS) resulting in a homogeneous population of EGFP-labeled cells. The addition of doxycycline to these cells induced PKD1-EGFP expression in a dose-dependent manner, with maximal effect at 1 μg/ml (data not shown).
Imaging of PKD1-EGFP fluorescence
Single cell imaging of the EGFP tagged PKD1 was achieved with a fluorescence microscope. The microscope used was an epifluorescence Zeiss Axioskop and a Zeiss water objective (Achroplan 40/0.75 water Carl Zeiss, Inc.). Images were captured as uncompressed 24-bit TIFF files with a cooled (−12 °C) single CCD color digital camera (Pursuit, Diagnostic Instruments) driven by SPOT version 4.7 software. Quantitative analysis of the relative change in plasma membrane and cytosol fluorescence intensity of individual cells was performed by importing the TIF images into Zeiss LSM 510 software and performing profile scans with the largest line width. Five equally spaced line profiles were taken for each cell or cell pair. Intensities were background-corrected, and the intensities at the membrane were divided by those in the immediately surrounding cytoplasm. We analyzed 30–45 cells in each experiment, and each experiment was performed in duplicate. The selected cells displayed in the figures were representative of 90% of the population of positive cells.
Immunofluorescence
Immunofluorescence of IEC18 cells was performed by fixing the cultures with 4% paraformaldehyde followed by permeabilization with 0.2% Triton X-100. After extensive PBS washing, fixed cells were incubated for 2 h at 25 °C in blocking buffer (BB) consisting of PBS containing 5% FBS and 2% bovine serum albumin and then stained at 4 °C overnight with a polyclonal antibody anti-PKD1 (1:500) or a mouse monoclonal antibody anti-FLAG (1:500) diluted in BB. Subsequently, the cells were washed with PBS at 25 °C and stained at 25 °C for 60 min with either Alexafluor 546- or Alexafluor 488-conjugated goat-anti-rabbit diluted in BB (1:100) and washed again with PBS. Nuclei were stained using Hoechst 33342 (1:10,000). The samples were imaged with an epifluorescence Zeiss Axioskop and a Zeiss water objective (Achroplan 40/0.75 water Carl Zeiss, Inc.). Images were captured as uncompressed 24-bit TIFF files with a cooled (−12 °C) single CCD color digital camera (Pursuit, Diagnostic Instruments) driven by SPOT version 4.7 software. Alexafluor 546 signals were observed with a HI Q filter set for rhodamine/TRITC, and Alexafluor 488 signals were observed with a HI Q filter set 41001 (Chroma Technology). The selected cells displayed in the appropriate figures were representative of 90% of the population.
Immunoblotting and detection of PKD1, PAK1/2, and HDAC5 phosphorylation
Confluent intestinal epithelial IEC-18 cells or Swiss 3T3 fibroblasts, treated as specified in the corresponding figures, were lysed in 2×SDS-polyacrylamide gel electrophoresis (PAGE) sample buffer (20 mm Tris/HCl, pH 6.8, 6% SDS, 2 mm EDTA, 4% 2-mercaptoethanol,10% glycerol) and boiled for 10 min. After SDS-PAGE, proteins were transferred to Immobilon-P membranes. The transfer was carried out at 100 V, 0.4 A at 4 °C for 4 h using a Bio-Rad transfer apparatus. The transfer buffer consisted of 200 mm glycine, 25 mm Tris, 0.01% SDS, and 20% CH3OH. For detection of proteins, membranes were blocked using 5% nonfat dried milk in PBS, pH 7.2, and then incubated for at least 2 h with the desired antibodies diluted in PBS containing 0.1% Tween. Primary antibodies bound to immunoreactive bands were visualized by enhanced chemiluminescence (ECL) detection with horseradish peroxidase-conjugated anti-mouse, anti-rabbit antibody, and a FUJI LAS-4000 Mini Luminescent Image Analyzer.
Knockdown of PAK1/2 via siRNA transfection
Silencer Select siRNA duplexes were purchased from Life Technologies. The siRNAs were designed to target the mRNA of mouse/rat PAK1 and PAK2. GenBankTM mRNA sequences were AF082077.1 (PAK1) and U19967.1 (PAK2). The sequences of the siRNAs were as follows; PAK1 sense (UGACACGCAAGGCTTATGGac), antisense (GUCCAUAAGCCUUGCGUGUca) and PAK2 sense (TTAACTTCTTGGACAGTTAcc) antisense (GGUAACUGUCCAAGAAGUUaa) (lowercase residues represent overhanging residues). For siRNA transfection the reverse transfection method was used. The siRNA pool was mixed with Lipofectamine RNAiMAX (Invitrogen) according to the manufacturer's protocol and added to 35-mm dishes. IEC-18 cells were then plated on top of the siRNA/Lipofectamine RNAiMAX complex at a density of 2 × 105 cells/35-mm dish. Control transfections were carried out with Stealth siRNA negative control (Invitrogen). Five days after transfection, cells were used for experiments and subsequent Western blot analysis.
In vitro kinase assays
For the assays of PAK1 or PAK2 transphosphorylation of PKD1 on Ser203, 200 ng of recombinant PKD1 (Sigma #K4268 or Millipore #14-508) were incubated in the absence or in the presence of 50 ng of recombinant active PAK1 (Sigma, #K2893) or 50 ng of recombinant active PAK2 (Millipore #14-481) in 25 μl of kinase buffer (100 μm ATP, 25 mm MOPS, pH 7.2, 12.5 mm, glycerol 2-phosphate, 25 mm MgCl2, 5 mm EGTA, and 2 mm EDTA, and 0.25 mm DTT) for 5 min at 30 °C. Reactions were terminated by the addition of 2× SDS-PAGE sample buffer and resolved by SDS-PAGE. PKD1 phosphorylation was determined by Western blot analysis using the antibody that detects its phosphorylated state on Ser203. The active PAK1 was generated by incubating a mixture PAK1/CDC42 in 10 μl of kinase buffer with 5 μl of 12.5 mm MnCl2, 0.5 mm GTP solution and incubated for 20 min at 30 °C before the addition of 10 μl of PKD1 (200 ng).
Transient cell transfection and mutagenesis
The single point mutant S203A in the pcDNA3-GFP-PKD construct was made by Retrogen Inc, San Diego, CA. Cultures of IEC-18 cells were transfected with the plasmids containing a cDNA encoding either a GFP PKD WT or a GFP PKD S203A or an epitope (FLAG)-tagged-HDAC5 wild type from Addgene (catalog #32216) by using Lipofectamine 2000 (Invitrogen) as suggested by the manufacturer. Analysis of the cells transiently transfected were performed 48 h after transfection. COS-7 cells were transfected with GFP-PKD WT and bombesin receptor without or with pCMV6M-Pak (Addgene plasmid #12209) or pCMV6M-PAK1 T423E (Addgene plasmid #12208) or GST tagged pEBG-Pak1 83–149 (Addgene plasmid #12214) by using Lipofectamine 3000 (Invitrogen) as suggested by the manufacturer.
PKD Ser203 phosphorylation in intestinal epithelial cells in vivo
To verify that PKD1 Ser203 phosphorylation occurs in vivo, we used transgenic mice that express elevated PKD1 protein in the ileal epithelium and control non-transgenic littermates. The generation of PKD1 transgenic mice was described elsewhere (3). To perform anatomical dissection and tissue collection, mice were euthanized in a CO2 chamber. Epithelial cells were isolated sequentially along the crypt-villus axis by timed incubations in EDTA-PBS solutions (3). To measure active PKD1 expression and PKD1 Ser203 phosphorylation, lysates of intestinal cells isolated from gender- and age-matched mice were subjected to immunoblotting as described above. This study was carried out in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. The protocol was approved by the Animal Research Committee of the University of California, Los Angeles (protocol number 2014-070-01).
Materials
DMEM, FBS, goat anti-mouse IgG secondary antibody conjugated to Alexa Fluor 546, and Silencer Select siRNAs were obtained from Invitrogen. ANG II, bombesin, and carbachol were obtained from Sigma. The kinase inhibitors kb NB 142-70, CRT0066101, Gö6983, GF 109203X, and IPA-3 were from R&D Systems (Minneapolis, MN), and FRAX597 and FRAX486 were from Selleckchem (Boston, MA). PF-3758309 was purchased from EMD Millipore (Billerica, MA). Primary antibodies used were: phospho-PKD Ser203 (ab59413, final dilution 1:1000) and phospho-PAK1/2 on Ser144/141 (ab40795, final dilution 1:1000) from Abcam: PKD C-20 (sc-639; final dilutions 1:400), β-tubulin antibody (sc-36579, final dilution 1:400), and GAPDH (sc-365062; final dilution 1:400) from Santa Cruz Biotechnology, Inc. Dallas, TX); phospho-PKD Ser916 (#2051; final dilution 1:1000), phospho-PKD Ser744/748 (#2054, final dilution 1:1000), PKD1 (#2052; final dilution 1:1000 for Western blotting and 1:500 for immunofluorescence), phospho-HDAC5 Ser498 (#3424, final dilution 1:1000), and DYKDDDDK (FLAG) tag (#8146, final dilution 1:500 for immunofluorescence) from Cell Signaling Technology (Danvers, MA). The inhibitors used in Table 1 were purchased either from R&D Systems (Minneapolis, MN) or EMD Millipore (Billerica, MA), or Sigma). FLAG HDAC5 and FLAG HDAC7 were gifts from Reuben Shaw (Addgene plasmid #32213), and pCMV6M-PAK1 (Addgene plasmid #12209), pCMV6M-PAK1 T423E (Addgene plasmid #12208), and pEBG-PAK1 83–149 (Addgene plasmid # 12214) were all gifts from Jonathan Chernoff. Bombesin receptor (pcDNA3-GRPR) was purchased from cDNA Resource Center (Rolla, MO). All other reagents were of the highest grade available.
Author contributions
E. R. and J. S. S. were responsible for the conception and design of the research. J.-K. C., Y. N., L. H., J. S. S., R. J., O. R., and S. H. Y. performed the experiments. J.-K. C., J. S. S., S. H. Y., and E. R. analyzed the data. E. R. and J. S. S. interpreted the results of the experiments. J. S. S. and J.-K. C. prepared the figures. E. R. drafted the manuscript. J. S. S., J.-K. C., R. J., O. R., and E. R. edited and revised the manuscript. E. R. approved the final version of the manuscript.
Acknowledgment
We thank the Imaging and Stem Cell Biology Core of the CURE: Digestive Diseases Research Center (P30DK41301) for assistance.
This work was supported by National Institutes of Health Grants R01DK100405, P30DK41301, and P01CA163200. This work was also supported by the Department of Veterans Affair Merit Award 1I01BX001473 (to E. R.). Additional funding came from the Ronald S. Hirshberg Endowed Chair of Pancreatic Cancer Research (to E. R.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
- PKD
- protein kinase D
- GPCR
- G protein-coupled receptor
- PAK
- p21-activated kinase
- HDAC
- histone deacetylase
- ANG II
- angiotensin II
- CRD
- cysteine-rich domain
- AID
- autoinhibitory domain
- CaMK
- calmodulin-dependent protein kinase
- rtTA
- reverse tetracycline-controlled transactivator
- TRE
- tetracycline-responsive element
- EGFP
- enhanced GFP
- BB
- blocking buffer
- TRITC
- tetramethylrhodamine isothiocyanate
- DAG
- diacylglycerol
- AMPK
- adenosine monophosphate-activated protein kinase
- ROCK
- rho-associated protein kinase
- RSK
- p90rsk
- S6K
- p70S6 kinase
- EGFR
- epidermal growth factor receptor.
References
- 1. Sinnett-Smith J., Zhukova E., Hsieh N., Jiang X., and Rozengurt E. (2004) Protein kinase D potentiates DNA synthesis induced by Gq-coupled receptors by increasing the duration of ERK signaling in swiss 3T3 cells. J. Biol. Chem. 279, 16883–16893 [DOI] [PubMed] [Google Scholar]
- 2. Wang Y., Waldron R. T., Dhaka A., Patel A., Riley M. M., Rozengurt E., and Colicelli J. (2002) The RAS effector RIN1 directly competes with RAF and is regulated by 14-3-3 proteins. Mol. Cell. Biol. 22, 916–926 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Sinnett-Smith J., Rozengurt N., Kui R., Huang C., and Rozengurt E. (2011) Protein kinase D1 mediates stimulation of DNA synthesis and proliferation in intestinal epithelial IEC-18 cells and in mouse intestinal crypts. J. Biol. Chem. 286, 511–520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Ni Y., Sinnett-Smith J., Young S. H., and Rozengurt E. (2013) PKD1 mediates negative feedback of PI3K/Akt activation in response to G protein-coupled receptors. PLoS ONE 8, e73149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Wang J., Han L., Sinnett-Smith J., Han L.-L., Stevens J. V., Rozengurt N., Young S. H., and Rozengurt E. (2016) Positive cross talk between protein kinase D and β-catenin in intestinal epithelial cells: impact on β-catenin nuclear localization and phosphorylation at Ser552. Am. J. Physiol. Cell Physiol. 310, C542–C557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Wang J., Sinnett-Smith J., Stevens J. V., Young S. H., and Rozengurt E. (2016) Biphasic regulation of Yes-associated protein (YAP) cellular localization, phosphorylation, and activity by G protein-coupled receptor agonists in intestinal epithelial cells: a novel role for protein kinase D (PKD). J. Biol. Chem. 291, 17988–18005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Matthews S. A., Liu P., Spitaler M., Olson E. N., McKinsey T. A., Cantrell D. A., and Scharenberg A. M. (2006) Essential role for protein kinase D family kinases in the regulation of class II histone deacetylases in B lymphocytes. Mol. Cell. Biol. 26, 1569–1577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Storz P., and Toker A. (2003) Protein kinase D mediates a stress-induced NF-κB activation and survival pathway. EMBO J. 22, 109–120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Mihailovic T., Marx M., Auer A., Van Lint J., Schmid M., Weber C., and Seufferlein T. (2004) Protein kinase D2 mediates activation of nuclear factor κB by Bcr-Abl in Bcr-Abl+ human myeloid leukemia cells. Cancer Res. 64, 8939–8944 [DOI] [PubMed] [Google Scholar]
- 10. Chiu T. T., Leung W. Y., Moyer M. P., Strieter R. M., and Rozengurt E. (2007) Protein kinase D2 mediates lysophosphatidic acid-induced interleukin 8 production in nontransformed human colonic epithelial cells through NF-κB. Am. J. Physiol. Cell Physiol 292, C767–C777 [DOI] [PubMed] [Google Scholar]
- 11. Rozengurt E., Rey O., and Waldron R. T. (2005) Protein kinase D Signaling. J. Biol. Chem. 280, 13205–13208 [DOI] [PubMed] [Google Scholar]
- 12. Rozengurt E. (2011) Protein kinase D signaling: multiple biological functions in health and disease. Physiology 26, 23–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Zhukova E., Sinnett-Smith J., and Rozengurt E. (2001) Protein kinase D potentiates DNA synthesis and cell proliferation induced by bombesin, vasopressin, or phorbol esters in Swiss 3T3 cells. J. Biol. Chem. 276, 40298–40305 [DOI] [PubMed] [Google Scholar]
- 14. Sinnett-Smith J., Jacamo R., Kui R., Wang Y. M., Young S. H., Rey O., Waldron R. T., and Rozengurt E. (2009) Protein kinase D mediates mitogenic signaling by Gq-coupled receptors through protein kinase c-independent regulation of activation loop Ser-744 and Ser-748 phosphorylation. J. Biol. Chem. 284, 13434–13445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Young S. H., Rozengurt N., Sinnett-Smith J., and Rozengurt E. (2012) Rapid protein kinase D1 signaling promotes migration of intestinal epithelial cells. Am. J. Physiol. Gastrointest Liver Physiol 303, G356–G366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Fielitz J., Kim M. S., Shelton J. M., Qi X., Hill J. A., Richardson J. A., Bassel-Duby R., and Olson E. N. (2008) Requirement of protein kinase D1 for pathological cardiac remodeling. Proc. Natl. Acad. Sci. U.S.A. 105, 3059–3063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hollenbach M., Stoll S. J., Jörgens K., Seufferlein T., and Kroll J. (2013) Different regulation of physiological and tumor angiogenesis in zebrafish by protein kinase D1 (PKD1). PLoS ONE 8, e68033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ishikawa E., Kosako H., Yasuda T., Ohmuraya M., Araki K., Kurosaki T., Saito T., and Yamasaki S. (2016) Protein kinase D regulates positive selection of CD4+ thymocytes through phosphorylation of SHP-1. Nat. Commun. 7, 12756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Weinreb I., Piscuoglio S., Martelotto L. G., Waggott D., Ng C. K., Perez-Ordonez B., Harding N. J., Alfaro J., Chu K. C., Viale A., Fusco N., da Cruz Paula A., Marchio C., Sakr R. A., Lim R., et al. (2014) Hotspot activating PRKD1 somatic mutations in polymorphous low-grade adenocarcinomas of the salivary glands. Nat. Genet. 46, 1166–1169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Zugaza J. L., Sinnett-Smith J., Van Lint J., and Rozengurt E. (1996) Protein kinase D (PKD) activation in intact cells through a protein kinase C-dependent signal transduction pathway. EMBO J. 15, 6220–6230 [PMC free article] [PubMed] [Google Scholar]
- 21. Valverde A. M., Sinnett-Smith J., Van Lint J., and Rozengurt E. (1994) Molecular cloning and characterization of protein kinase D: a target for diacylglycerol and phorbol esters with a distinctive catalytic domain. Proc. Natl. Acad. Sci. U.S.A. 91, 8572–8576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Zugaza J. L., Waldron R. T., Sinnett-Smith J., and Rozengurt E. (1997) Bombesin, vasopressin, endothelin, bradykinin, and platelet-derived growth factor rapidly activate protein kinase D through a protein kinase C-dependent signal transduction pathway. J. Biol. Chem. 272, 23952–23960 [DOI] [PubMed] [Google Scholar]
- 23. Zhukova E., Sinnett-Smith J., Wong H., Chiu T., and Rozengurt E. (2001) CCKB/gastrin receptor mediates synergistic stimulation of DNA synthesis and cyclin D1, D3, and E expression in Swiss 3T3 cells. J. Cell. Physiol. 189, 291–305 [DOI] [PubMed] [Google Scholar]
- 24. Chiu T., and Rozengurt E. (2001) PKD in intestinal epithelial cells: rapid activation by phorbol esters, LPA, and angiotensin through PKC. Am. J. Physiol. Cell Physiol. 280, C929–C942 [DOI] [PubMed] [Google Scholar]
- 25. Chiu T., Wu S. S., Santiskulvong C., Tangkijvanich P., Yee H. F. Jr, and Rozengurt E. (2002) Vasopressin-mediated mitogenic signaling in intestinal epithelial cells. Am. J. Physiol. Cell Physiol. 282, C434–C450 [DOI] [PubMed] [Google Scholar]
- 26. Guha S., Rey O., and Rozengurt E. (2002) Neurotensin induces protein kinase C-dependent protein kinase D activation and DNA synthesis in human pancreatic carcinoma cell line PANC-1. Cancer Res. 62, 1632–1640 [PubMed] [Google Scholar]
- 27. Waldron R. T., Rey O., Iglesias T., Tugal T., Cantrell D., and Rozengurt E. (2001) Activation loop Ser-744 and Ser-748 in protein kinase D are transphosphorylated in vivo. J. Biol. Chem. 276, 32606–32615 [DOI] [PubMed] [Google Scholar]
- 28. Yuan J., Slice L. W., and Rozengurt E. (2001) Activation of protein kinase D by signaling through Rho and the α subunit of the heterotrimeric G protein G13. J. Biol. Chem. 276, 38619–38627 [DOI] [PubMed] [Google Scholar]
- 29. Yuan J., Slice L. W., Gu J., and Rozengurt E. (2003) Cooperation of Gq, Gi, and G12/13 in protein kinase D activation and phosphorylation induced by lysophosphatidic acid. J. Biol. Chem. 278, 4882–4891 [DOI] [PubMed] [Google Scholar]
- 30. Yuan J., Slice L., Walsh J. H., and Rozengurt E. (2000) Activation of protein kinase D by signaling through the α subunit of the heterotrimeric G protein Gq. J. Biol. Chem. 275, 2157–2164 [DOI] [PubMed] [Google Scholar]
- 31. Paolucci L., Sinnett-Smith J., and Rozengurt E. (2000) Lysophosphatidic acid rapidly induces protein kinase D activation through a pertussis toxin-sensitive pathway. Am. J. Physiol. Cell Physiol 278, C33–C39 [DOI] [PubMed] [Google Scholar]
- 32. Yuan J., Rey O., and Rozengurt E. (2006) Activation of protein kinase D3 by signaling through Rac and the α subunits of the heterotrimeric G proteins G12 and G13. Cell. Signal. 18, 1051–1062 [DOI] [PubMed] [Google Scholar]
- 33. Abedi H., Rozengurt E., and Zachary I. (1998) Rapid activation of the novel serine/threonine protein kinase, protein kinase D by phorbol esters, angiotensin II, and PDGF-BB in vascular smooth muscle cells. FEBS Lett. 427, 209–212 [DOI] [PubMed] [Google Scholar]
- 34. Matthews S. A., Iglesias T., Rozengurt E., and Cantrell D. (2000) Spatial and temporal regulation of protein kinase D (PKD). EMBO J. 19, 2935–2945 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Matthews S. A., Rozengurt E., and Cantrell D. (2000) Protein kinase D: a selective target for antigen receptors and a downstream target for protein kinase C in lymphocytes. J. Exp. Med. 191, 2075–2082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Matthews S. A., Rozengurt E., and Cantrell D. (1999) Characterization of serine 916 as an in vivo autophosphorylation site for protein kinase D/protein kinase Cμ. J. Biol. Chem. 274, 26543–26549 [DOI] [PubMed] [Google Scholar]
- 37. Waldron R. T., and Rozengurt E. (2000) Oxidative stress induces protein kinase D activation in intact cells: involvement of Src and dependence on protein kinase C. J. Biol. Chem. 275, 17114–17121 [DOI] [PubMed] [Google Scholar]
- 38. Waldron R. T., Rey O., Zhukova E., and Rozengurt E. (2004) Oxidative stress induces protein kinase C-mediated activation loop phosphorylation and nuclear redistribution of protein kinase D. J. Biol. Chem. 279, 27482–27493 [DOI] [PubMed] [Google Scholar]
- 39. Iglesias T., Waldron R. T., and Rozengurt E. (1998) Identification of in vivo phosphorylation sites required for protein kinase D activation. J. Biol. Chem. 273, 27662–27667 [DOI] [PubMed] [Google Scholar]
- 40. Waldron R. T., and Rozengurt E. (2003) Protein kinase C phosphorylates protein kinase D activation loop Ser-744 and Ser-748 and releases autoinhibition by the pleckstrin homology domain. J. Biol. Chem. 278, 154–163 [DOI] [PubMed] [Google Scholar]
- 41. Jacamo R., Sinnett-Smith J., Rey O., Waldron R. T., and Rozengurt E. (2008) Sequential protein kinase C (PKC)-dependent and PKC-independent protein kinase D catalytic activation via Gq-coupled receptors: differential regulation of activation loop Ser-744 and Ser-748 phosphorylation. J. Biol. Chem. 283, 12877–12887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Rey O., Sinnett-Smith J., Zhukova E., and Rozengurt E. (2001) Regulated nucleocytoplasmic transport of protein kinase D in response to G protein-coupled receptor activation. J. Biol. Chem. 276, 49228–49235 [DOI] [PubMed] [Google Scholar]
- 43. Rey O., Young S. H., Cantrell D., and Rozengurt E. (2001) Rapid protein kinase D translocation in response to G protein-coupled receptor activation: dependence on protein kinase C. J. Biol. Chem. 276, 32616–32626 [DOI] [PubMed] [Google Scholar]
- 44. Rey O., Papazyan R., Waldron R. T., Young S. H., Lippincott-Schwartz J., Jacamo R., and Rozengurt E. (2006) The nuclear import of protein kinase D3 requires its catalytic activity. J. Biol. Chem. 281, 5149–5157 [DOI] [PubMed] [Google Scholar]
- 45. Hornbeck P. V., Zhang B., Murray B., Kornhauser J. M., Latham V., and Skrzypek E. (2015) PhosphoSitePlus, 2014: mutations, PTMs, and recalibrations. Nucleic Acids Res. 43, D512–D520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Bokoch G. M. (2003) Biology of the p21-activated kinases. Annu. Rev. Biochem. 72, 743–781 [DOI] [PubMed] [Google Scholar]
- 47. Ha B. H., Morse E. M., Turk B. E., and Boggon T. J. (2015) Signaling, regulation, and specificity of the type II p21-activated kinases. J. Biol. Chem. 290, 12975–12983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Carter J. H., Douglass L. E., Deddens J. A., Colligan B. M., Bhatt T. R., Pemberton J. O., Konicek S., Hom J., Marshall M., and Graff J. R. (2004) Pak-1 expression increases with progression of colorectal carcinomas to metastasis. Clin. Cancer Res. 10, 3448–3456 [DOI] [PubMed] [Google Scholar]
- 49. Fernandez-Zapico M. E., Gonzalez-Paz N. C., Weiss E., Savoy D. N., Molina J. R., Fonseca R., Smyrk T. C., Chari S. T., Urrutia R., and Billadeau D. D. (2005) Ectopic expression of VAV1 reveals an unexpected role in pancreatic cancer tumorigenesis. Cancer Cell 7, 39–49 [DOI] [PubMed] [Google Scholar]
- 50. Molli P. R., Li D. Q., Murray B. W., Rayala S. K., and Kumar R. (2009) PAK signaling in oncogenesis. Oncogene 28, 2545–2555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Zhu G., Wang Y., Huang B., Liang J., Ding Y., Xu A., and Wu W. (2012) A Rac1/PAK1 cascade controls [beta]-catenin activation in colon cancer cells. Oncogene 31, 1001–1012 [DOI] [PubMed] [Google Scholar]
- 52. Radu M., Semenova G., Kosoff R., and Chernoff J. (2014) PAK signalling during the development and progression of cancer. Nat. Rev. Cancer 14, 13–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Dammann K., Khare V., and Gasche C. (2014) Tracing PAKs from GI inflammation to cancer. Gut 63, 1173–1184 [DOI] [PubMed] [Google Scholar]
- 54. Quaroni A., Wands J., Trelstad R. L., and Isselbacher K. J. (1979) Epithelioid cell cultures from rat small intestine: characterization by morphologic and immunologic criteria. J. Cell Biol. 80, 248–265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Quaroni A., and May R. J. (1980) Establishment and characterization of intestinal epithelial cell cultures. Methods Cell Biol. 21B, 403–427 [PubMed] [Google Scholar]
- 56. Wu S. S., Chiu T., and Rozengurt E. (2002) ANG II and LPA induce Pyk2 tyrosine phosphorylation in intestinal epithelial cells: role of Ca2+, PKC, and Rho kinase. Am. J. Physiol. Cell Physiol 282, C1432–C1444 [DOI] [PubMed] [Google Scholar]
- 57. Rey O., Zhukova E., Sinnett-Smith J., and Rozengurt E. (2003) Vasopressin-induced intracellular redistribution of protein kinase D in intestinal epithelial cells. J. Cell. Physiol. 196, 483–492 [DOI] [PubMed] [Google Scholar]
- 58. Chiu T., Santiskulvong C., and Rozengurt E. (2003) ANG II stimulates PKC-dependent ERK activation, DNA synthesis, and cell division in intestinal epithelial cells. Am. J. Physiol. Gastrointest. Liver Physiol. 285, G1–G11 [DOI] [PubMed] [Google Scholar]
- 59. Chiu T., Santiskulvong C., and Rozengurt E. (2005) EGF receptor transactivation mediates ANG II-stimulated mitogenesis in intestinal epithelial cells through the PI3-kinase/Akt/mTOR/p70S6K1 signaling pathway. Am. J. Physiol. Gastrointest. Liver Physiol. 288, G182–G194 [DOI] [PubMed] [Google Scholar]
- 60. Young S. H., and Rozengurt E. (2006) Qdot nanocrystal conjugates conjugated to bombesin or ANG II label the cognate G protein-coupled receptor in living cells. Am. J. Physiol. Cell Physiol. 290, C728–C732 [DOI] [PubMed] [Google Scholar]
- 61. Slice L. W., Chiu T., and Rozengurt E. (2005) Angiotensin II and epidermal growth factor induce cyclooxygenase-2 expression in intestinal epithelial cells through small GTPases using distinct signaling pathways. J. Biol. Chem. 280, 1582–1593 [DOI] [PubMed] [Google Scholar]
- 62. Rozengurt E., and Sinnett-Smith J. (1983) Bombesin stimulation of DNA synthesis and cell division in cultures of Swiss 3T3 cells. Proc. Natl. Acad. Sci. U.S.A. 80, 2936–2940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Zachary I., and Rozengurt E. (1985) High-affinity receptors for peptides of the bombesin family in Swiss 3T3 cells. Proc. Natl. Acad. Sci. U.S.A. 82, 7616–7620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Zachary I., and Rozengurt E. (1987) Internalization and degradation of peptides of the bombesin family in Swiss 3T3 cells occurs without ligand-induced receptor down-regulation. EMBO J. 6, 2233–2239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Zachary I., and Rozengurt E. (1987) Identification of a receptor for peptides of the bombesin family in Swiss 3T3 cells by affinity cross-linking. J. Biol. Chem. 262, 3947–3950 [PubMed] [Google Scholar]
- 66. Coffer A., Fabregat I., Sinnett-Smith J., and Rozengurt E. (1990) Solubilization of the bombesin receptor from Swiss 3T3 cell membranes: functional association to a guanine nucleotide regulatory protein. FEBS Lett. 263, 80–84 [DOI] [PubMed] [Google Scholar]
- 67. Coffer A., Sinnett-Smith J., and Rozengurt E. (1990) Bombesin receptor from Swiss 3T3 cells: affinity chromatography and reconstitution into phospholipid vesicles. FEBS Lett. 275, 159–164 [DOI] [PubMed] [Google Scholar]
- 68. Millar J. B., and Rozengurt E. (1990) Chronic desensitization to bombesin by progressive down-regulation of bombesin receptors in Swiss 3T3 cells: distinction from acute desensitization. J. Biol. Chem. 265, 12052–12058 [PubMed] [Google Scholar]
- 69. Sinnett-Smith J., Lehmann W., and Rozengurt E. (1990) Bombesin receptor in membranes from Swiss 3T3 cells: binding characteristics, affinity labelling, and modulation by guanine nucleotides. Biochem. J. 265, 485–493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Zachary I., Gil J., Lehmann W., Sinnett-Smith J., and Rozengurt E. (1991) Bombesin, vasopressin, and endothelin rapidly stimulate tyrosine phosphorylation in intact Swiss 3T3 cells. Proc. Natl. Acad. Sci. U.S.A. 88, 4577–4581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Walsh J. H., Bouzyk M., and Rozengurt E. (1993) Homologous desensitization of bombesin-induced increases in intracellular Ca2+ in quiescent Swiss 3T3 cells involves a protein kinase C-independent mechanism. J. Cell. Physiol. 156, 333–340 [DOI] [PubMed] [Google Scholar]
- 72. Sinnett-Smith J., Zachary I., Valverde A. M., and Rozengurt E. (1993) Bombesin stimulation of p125 focal adhesion kinase tyrosine phosphorylation: role of protein kinase C, Ca2+ mobilization, and the actin cytoskeleton. J. Biol. Chem. 268, 14261–14268 [PubMed] [Google Scholar]
- 73. Rodríguez-Fernández J. L., and Rozengurt E. (1996) Bombesin, bradykinin, vasopressin, and phorbol esters rapidly and transiently activate Src family tyrosine kinases in Swiss 3T3 cells: dissociation from tyrosine phosphorylation of p125 focal adhesion kinase. J. Biol. Chem. 271, 27895–27901 [DOI] [PubMed] [Google Scholar]
- 74. Casamassima A., and Rozengurt E. (1997) Tyrosine phosphorylation of p130(cas) by bombesin, lysophosphatidic acid, phorbol esters, and platelet-derived growth factor: signaling pathways and formation of a p130(cas)-Crk complex. J. Biol. Chem. 272, 9363–9370 [DOI] [PubMed] [Google Scholar]
- 75. Salazar E. P., and Rozengurt E. (2001) Src family kinases are required for integrin-mediated but not for G protein-coupled receptor stimulation of focal adhesion kinase autophosphorylation at Tyr-397. J. Biol. Chem. 276, 17788–17795 [DOI] [PubMed] [Google Scholar]
- 76. Fan R. S., Jácamo R. O., Jiang X., Sinnett-Smith J., and Rozengurt E. (2005) G protein-coupled receptor activation rapidly stimulates focal adhesion kinase phosphorylation at Ser-843: mediation by Ca2+, calmodulin, and Ca2+/calmodulin-dependent kinase II. J. Biol. Chem. 280, 24212–24220 [DOI] [PubMed] [Google Scholar]
- 77. Lavalle C. R., Bravo-Altamirano K., Giridhar K. V., Chen J., Sharlow E., Lazo J. S., Wipf P., and Wang Q. J. (2010) Novel protein kinase D inhibitors cause potent arrest in prostate cancer cell growth and motility. BMC Chem. Biol. 10, 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Harikumar K. B., Kunnumakkara A. B., Ochi N., Tong Z., Deorukhkar A., Sung B., Kelland L., Jamieson S., Sutherland R., Raynham T., Charles M., Bagherzadeh A., Bagherazadeh A., Foxton C., Boakes A., Farooq, et al. (2010) A novel small-molecule inhibitor of protein kinase D blocks pancreatic cancer growth in vitro and in vivo. Mol. Cancer Ther. 9, 1136–1146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Rozengurt E. (2007) Mitogenic signaling pathways induced by G protein-coupled receptors. J. Cell. Physiol. 213, 589–602 [DOI] [PubMed] [Google Scholar]
- 80. Toullec D., Pianetti P., Coste H., Bellevergue P., Grand-Perret T., Ajakane M., Baudet V., Boissin P., Boursier E., and Loriolle F. (1991) The bisindolylmaleimide Gf-109203x is a potent and selective inhibitor of protein kinase C. J. Biol. Chem. 266, 15771–15781 [PubMed] [Google Scholar]
- 81. Martiny-Baron G., Kazanietz M. G., Mischak H., Blumberg P. M., Kochs G., Hug H., Marmé D., and Schächtele C. (1993) Selective inhibition of protein kinase C isozymes by the indolocarbazole Gö 6976. J. Biol. Chem. 268, 9194–9197 [PubMed] [Google Scholar]
- 82. Gschwendt M., Dieterich S., Rennecke J., Kittstein W., Mueller H. J., and Johannes F. J. (1996) Inhibition of protein kinase Cμ by various inhibitors: differentiation from protein kinase C isoenzymes. FEBS Lett. 392, 77–80 [DOI] [PubMed] [Google Scholar]
- 83. Waldron R. T., Innamorati G., Torres-Marquez M. E., Sinnett-Smith J., and Rozengurt E. (2012) Differential PKC-dependent and -independent PKD activation by G protein α subunits of the Gq family: selective stimulation of PKD Ser748 autophosphorylation by Gαq. Cell. Signal. 24, 914–921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Zhu G., Fujii K., Belkina N., Liu Y., James M., Herrero J., and Shaw S. (2005) Exceptional disfavor for proline at the P+1 position among AGC and CAMK kinases establishes reciprocal specificity between them and the proline-directed kinases. J. Biol. Chem. 280, 10743–10748 [DOI] [PubMed] [Google Scholar]
- 85. Hsu P. P., Kang S. A., Rameseder J., Zhang Y., Ottina K. A., Lim D., Peterson T. R., Choi Y., Gray N. S., Yaffe M. B., Marto J. A., and Sabatini D. M. (2011) The mTOR-regulated phosphoproteome reveals a mechanism of mTORC1-mediated inhibition of growth factor signaling. Science 332, 1317–1322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Murray B. W., Guo C., Piraino J., Westwick J. K., Zhang C., Lamerdin J., Dagostino E., Knighton D., Loi C. M., Zager M., Kraynov E., Popoff I., Christensen J. G., Martinez R., Kephart S. E., et al. (2010) Small-molecule p21-activated kinase inhibitor PF-3758309 is a potent inhibitor of oncogenic signaling and tumor growth. Proc. Natl. Acad. Sci. U.S.A. 107, 9446–9451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Rudolph J., Crawford J. J., Hoeflich K. P., and Wang W. (2015) Inhibitors of p21-activated kinases (PAKs). J. Med. Chem. 58, 111–129 [DOI] [PubMed] [Google Scholar]
- 88. Licciulli S., Maksimoska J., Zhou C., Troutman S., Kota S., Liu Q., Duron S., Campbell D., Chernoff J., Field J., Marmorstein R., and Kissil J. L. (2013) FRAX597, a Small Molecule inhibitor of the p21-activated kinases, inhibits tumorigenesis of neurofibromatosis Type 2 (NF2)-associated Schwannomas. J. Biol. Chem. 288, 29105–29114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Chow H. Y., Jubb A. M., Koch J. N., Jaffer Z. M., Stepanova D., Campbell D. A., Duron S. G., O'Farrell M., Cai K. Q., Klein-Szanto A. J., Gutkind J. S., Hoeflich K. P., and Chernoff J. (2012) p21-activated kinase 1 is required for efficient tumor formation and progression in a Ras-mediated skin cancer model. Cancer Res. 72, 5966–5975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Deacon S. W., Beeser A., Fukui J. A., Rennefahrt U. E., Myers C., Chernoff J., and Peterson J. R. (2008) An isoform-selective, small-molecule inhibitor targets the autoregulatory mechanism of p21-activated kinase. Chem. Biol. 15, 322–331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Viaud J., and Peterson J. R. (2009) An allosteric kinase inhibitor binds the p21-activated kinase autoregulatory domain covalently. Mol. Cancer Ther. 8, 2559–2565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Sells M. A., Knaus U. G., Bagrodia S., Ambrose D. M., Bokoch G. M., and Chernoff J. (1997) Human p21-activated kinase (Pak1) regulates actin organization in mammalian cells. Curr. Biol. 7, 202–210 [DOI] [PubMed] [Google Scholar]
- 93. Xiao G.-H., Beeser A., Chernoff J., and Testa J. R. (2002) p21-activated kinase links Rac/Cdc42 signaling to Merlin. J. Biol. Chem. 277, 883–886 [DOI] [PubMed] [Google Scholar]
- 94. Sinnett-Smith J., Ni Y., Wang J., Ming M., Young S. H., and Rozengurt E. (2014) Protein kinase D1 mediates class IIa histone deacetylase phosphorylation and nuclear extrusion in intestinal epithelial cells: role in mitogenic signaling. Am. J. Physiol. Cell Physiol. 306, C961–C971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Rey O., Yuan J., Young S. H., and Rozengurt E. (2003) Protein kinase Cν/protein kinase D3 nuclear localization, catalytic activation, and intracellular redistribution in response to G protein-coupled receptor agonists. J. Biol. Chem. 278, 23773–23785 [DOI] [PubMed] [Google Scholar]
- 96. Vega R. B., Harrison B. C., Meadows E., Roberts C. R., Papst P. J., Olson E. N., and McKinsey T. A. (2004) Protein kinases C and D mediate agonist-dependent cardiac hypertrophy through nuclear export of histone deacetylase 5. Mol. Cell. Biol. 24, 8374–8385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Ha C. H., Wang W., Jhun B. S., Wong C., Hausser A., Pfizenmaier K., McKinsey T. A., Olson E. N., and Jin Z.-G. (2008) Protein kinase D-dependent phosphorylation and nuclear export of histone deacetylase 5 mediates vascular endothelial growth factor-induced gene expression and angiogenesis. J. Biol. Chem. 283, 14590–14599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Parra M., and Verdin E. (2010) Regulatory signal transduction pathways for class IIa histone deacetylases. Curr. Opin. Pharmacol. 10, 454–460 [DOI] [PubMed] [Google Scholar]
- 99. Young S. H., Rey O., Sinnett-Smith J., and Rozengurt E. (2014) Intracellular Ca2+ oscillations generated via the Ca2+-sensing receptor are mediated by negative feedback by PKCα at Thr-888. Am. J. Physiol. Cell Physiol. 306, C298–C306 [DOI] [PMC free article] [PubMed] [Google Scholar]