

Abstract
Chemokines are essential for antimicrobial host defenses and tissue repair. Herpesviruses and poxviruses also encode chemokines, copied from their hosts and repurposed for multiple functions, including immune evasion. The CC chemokine MCK-2 encoded by mouse CMV (MCMV) has an atypical structure consisting of a classic chemokine domain N-terminal to a second unique domain, resulting from the splicing of MCMV ORFs m131 and m129. MCK-2 is essential for full MCMV infectivity in macrophages and for persistent infection in the salivary gland. However, information about its mechanism of action and specific biochemical roles for the two domains has been lacking. Here, using genetic, chemical, and enzymatic analyses of multiple mouse cell lines as well as primary mouse fibroblasts from salivary gland and lung, we demonstrate that MCK-2 binds glycosaminoglycans (GAGs) with affinities in the following order: heparin > heparan sulfate > chondroitin sulfate = dermatan sulfate. Both MCK-2 domains bound these GAGs independently, and computational analysis together with site-directed mutagenesis identified five basic residues distributed across the N terminus and the 30s and 50s loops of the chemokine domain that are important GAG binding determinants. Both domains were required for GAG-dependent oligomerization of full-length MCK-2. Thus, MCK-2 is an atypical viral chemokine consisting of a CC chemokine domain and a unique non-chemokine domain, both of which bind GAGs and are critical for GAG-dependent oligomerization of the full-length protein.
Keywords: chemokine, glycosaminoglycan, heparin-binding protein, oligomerization, viral protein, cytomegalovirus
Introduction
Chemokines are a family of structurally related chemotactic cytokines that coordinate leukocyte trafficking in the vertebrate immune system in support of antimicrobial host defense and tissue repair (1). Herpesviruses and poxviruses also encode chemokines, which they have copied from their hosts and repurposed for multiple proviral functions, including immune evasion (2). In humans, ∼50 genes encoding chemokines have been identified. Based on the presence and spacing of conserved N-terminal cysteines, they can be classified into one of four subgroups: CC, CXC, C, and XC. Chemokines classically signal by binding to G protein-coupled receptors (GPCRs)2 with a seven-transmembrane domain structure (3). A small group of atypical seven-transmembrane domain chemokine receptors that do not signal through G proteins also exists (3, 4).
Chemokines also interact with glycosaminoglycans (GAGs) on the cell surface and on the extracellular matrix. GAGs are highly sulfated polysaccharides that may be covalently bound to a cell surface protein core (5). This GAG-chemokine interaction is thought to regulate the chemokine bioactivity at multiple levels (6, 7). GAG binding may protect chemokines from proteases and allow them to resist shear forces imposed by blood flow on the luminal side of the endothelium, where they promote local leukocyte adhesion and transendothelial migration (8–10). GAGs are also important for the secretion of chemokines from some tumor cells and T cells and for chemokine transport across the endothelium (11–13). Furthermore, GAGs can mediate chemokine-induced intracellular signaling independently of surface receptors (14). However, the most important consequence of chemokine-GAG interaction is thought to be the formation of chemokine concentration gradients that provide directional signals during cell migration (15, 16). GAG binding determinants are quite diverse across the chemokine family and cannot be reliably predicted from knowledge of conserved sequences, in part because a suitable GAG-binding surface may form only after a chemokine oligomerizes (17, 18). Chemokine oligomerization may occur in multiple modes in solution, and high-order GAG-dependent oligomers may promote the accumulation of chemokines and their presentation to cellular receptors (17, 19). Although chemokines are fully active as monomers in vitro (20, 21), the chemotactic properties of GAG binding- and oligomerization-defective chemokines are impaired in vivo (22–27). Accordingly, targeting chemokine-GAG binding may be a rational strategy for developing new anti-inflammatory therapeutics (28).
In contrast to host chemokines, there is little information available about GAG binding to viral chemokines. In this study, we focus on this question for the mouse CMV (MCMV) CC chemokine MCK-2. A highly unusual feature of MCK-2 is the presence of a second domain located C-terminal to the chemokine domain, resulting from the splicing of the MCMV ORFs m131 and m129 (29). The evolutionary provenance of the C-terminal m129 domain is unclear because it has no sequence homology to other known proteins. Biologically, MCK-2 is required for persistent infection in the mouse salivary gland, but it is dispensable for the infection of other organs (30, 31). MCK-2 also promotes leukocyte recruitment to sites of infection (32–35). However, the biochemical mechanism of action for MCK-2 is poorly understood. In particular, a specific receptor has not yet been identified; instead, surprisingly, it has been reported to be part of a cell entry complex together with the MCMV proteins gH and gL (36). This trimolecular complex is dispensable for viral growth in fibroblasts but required for full infectivity in macrophages in vitro and in alveolar macrophages ex vivo (36, 37). Similarly, UL128, a human CMV (HCMV)-encoded CC chemokine that is 20.1% identical at the amino acid level to MCK-2, is part of an entry complex in the HCMV envelope that also includes gH and gL as well as the HCMV UL130 and UL131 proteins (38). This pentameric complex is essential for HCMV infectivity in endothelial cells and monocytes (38–40), and it has been shown to be an excellent target for new anti-HCMV vaccination strategies (41–44). Importantly, HCMV encodes two other chemokines, UL146 (vCXCL1) and UL147 (vCXCL2), whereas MCK-2 is the only chemokine encoded by MCMV (45).
Here we demonstrate that MCK-2 conserves two typical characteristics of chemokines, GAG binding and the ability to oligomerize on GAGs. In addition, we show that the molecular mechanisms for GAG binding and oligomerization for MCK-2 are distinct from those of host chemokines. This work constitutes the first biochemical analysis of recombinant MCK-2 and could help us to understand the role of this MCMV virulence factor and other viral chemokines in the pathogenesis of herpesviruses.
Results
MCK-2 binds to the cell surface of multiple cell lines and primary mouse fibroblasts
To investigate the mechanism of action of MCK-2, we first constructed C-terminal V5-His6-tagged forms of the full-length protein and both of its subdomains (Fig. 1A). Proteins with the correct size were detected by Western blotting using an anti-His mAb in supernatants of insect cells infected with a corresponding recombinant baculovirus (Fig. 1B). The secreted recombinant proteins—full-length rMCK-2, the MCK-2 chemokine domain rm131, and the C-terminal domain rm129—were then purified from cell supernatants by affinity chromatography (Fig. 1C).
Figure 1.
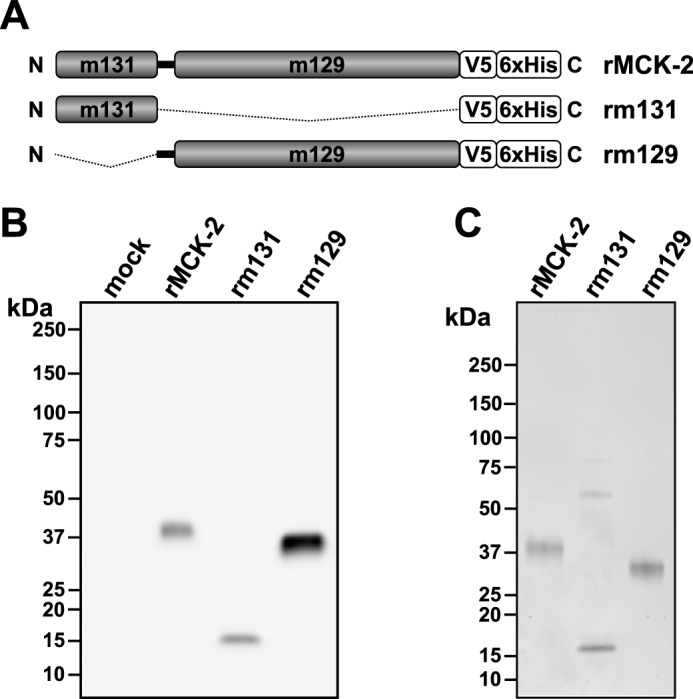
Production of purified recombinant full-length and truncated MCK-2 proteins. A, schematic of recombinant protein domains: rMCK-2, full-length MCK-2; rm131, MCK-2 chemokine domain; rm129, C-terminal domain of MCK-2; N, N terminus; C, C terminus; box, indicated domain of MCK-2; thick black connecting line between the m131 and m129 boxes, sequence contained in the full-length protein encoded by a DNA sequence immediately N-terminal to ORF m129; thin broken line, MCK-2 domain excluded from the construct. All constructs were tagged with a C-terminal V5-His6 epitope. B, production of recombinant MCK-2-derived proteins. Recombinant proteins present in 15 μl of clarified supernatant from insect cells infected or not (mock) with recombinant baculoviruses encoding the construct indicated at the top of each lane were revealed by Western blotting using an anti-His epitope tag mAb. C, purified recombinant MCK-2-derived proteins. His-tagged recombinant proteins were purified by nickel chromatography, separated by polyacrylamide gel electrophoresis, and revealed by Coomassie blue staining of the gel, which was loaded with 0.5 μg of each purified protein. Molecular size markers are indicated in kilodalton.
Because MCK-2 is a chemokine, we hypothesized that, like other chemokines, it would signal through one or more GPCRs expressed on the cell surface. To identify a putative MCK-2 receptor, we conducted a flow cytometry-based rMCK-2 binding screen of multiple cell lines, including lymphocytes, fibroblasts, and epithelial cells. rMCK-2 was able to bind to the cell surface of five of the six cell lines tested to some extent, with the highest level of binding to mouse NIH-3T3 fibroblasts and the epithelial lines CHO-K1 and BS-C-1 from hamster and African green monkey, respectively (Fig. 2A). No binding was observed to the mouse B cell line A20. However, rMCK-2 was not associated with calcium flux responses (data not shown), a classic general cell response to chemokine binding. In addition, the binding of rMCK-2 to the NIH-3T3 cell surface did not saturate in the tested concentration range (Fig. 2B). Therefore, we considered whether rMCK-2 binding to these cells might be mediated by GAGs on the cell surface. However, we first verified that rMCK-2 was able to interact not only with the surface of immortalized cell lines but also that of primary cells, including fibroblasts from mouse lung and salivary gland. To extract fibroblasts from mouse salivary gland, we adapted a method used previously for cultured fibroblasts from mouse lung. After three passages, primary lung- and salivary gland-derived fibroblasts were used in binding assays. In this case, we included recombinant CrmE, a poxvirus-encoded soluble TNF decoy receptor that does not bind GAGs, as a negative control.3 CrmE was expressed in insect cells with the same tags and purified following the same protocol used here for rMCK-2. As shown in Fig. 2C, rMCK-2 strongly bound to the cell surface of mouse fibroblasts, whereas CrmE binding was negative. These results demonstrate that, like host chemokines, rMCK-2 can interact with the cell surface and suggest that GAGs could mediate this interaction.
Figure 2.

Extracellular MCK-2 binds to the plasma membrane of epithelial cells and fibroblasts. A, cell lines. B, dose-response binding assay. C, primary mouse fibroblasts from lung and salivary gland. The cell line names (A and B) and primary mouse fibroblast sources (C) are indicated at the top of the corresponding FACS plots. A and C, cells were incubated with 100 nm rMCK-2 (dark gray) or buffer alone (light gray), and cell-associated recombinant proteins were revealed by staining with anti-His mAb under non-permeabilized conditions. C, cells were also incubated with 100 nm purified recombinant His-tagged CrmE protein (white). B, the binding of increasing concentrations (0–200 nm) of rMCK-2, according to the color code show in the inset, was detected as above. Legends are depicted to the right of A and C. Data are from a single experiment representative of three independent experiments.
MCK-2 interaction with the cell surface is mediated by GAGs
To investigate whether rMCK-2 binding to the cell surface is mediated by GAGs, we tested the effect on binding of chemical, genetic, and enzymatic depletion of GAGs in CHO cells. Most protein-GAG interactions are driven by sulfate groups incorporated into GAGs during their synthesis in the Golgi (46, 47). Sodium chlorate impedes sulfation, consequently inhibiting or reducing the interaction of any protein engaging GAG sulfate groups (46). His-tagged B18, the vaccinia virus-encoded interferon inhibitor and a well known GAG-binding protein (48), was included in our experiments as a positive control on the inhibitory effect of chlorate. As shown in Fig. 3A and as reported previously (48), B18 binding was drastically reduced in chlorate-treated CHO-K1 cells, and full binding was recovered when chlorate was competed by the addition of an excess of sodium sulfate to the medium (Fig. 3A). The same pattern was observed for rMCK-2 (Fig. 3A), which indicates that rMCK-2 binding to CHO-K1 cells is mediated by extracellular sulfate groups. There are many different types of cellular GAGs that differ by the extent and pattern of sulfation. The most abundant types in vivo include heparin, heparan sulfate (HS), chrondroitin sulfate (CS), and dermatan sulfate (DS). rMCK-2 failed to bind to the CHO cell line variant CHO-677, which expresses only CS, as well as to the variant cell line CHO-745, which is completely GAG-deficient (Fig. 3B) (49). This demonstrates that GAGs other than CS mediate rMCK-2 binding to the cell surface. Consistent with this, chondroitinase ABC treatment of CHO-K1 cells had no effect on rMCK-2 binding, whereas heparinase II treatment of CHO-K1 cells, which digests HS and heparin, abolished rMCK-2 binding (Fig. 3C). Therefore, at the cellular level, rMCK-2 appears to be a GAG-binding protein specific for HS and heparin.
Figure 3.

GAGs mediate MCK-2 binding to the plasma membrane. A—C, CHO-K1 cells rendered deficient in GAGs either chemically (A), genetically (B), or enzymatically (C) were incubated with recombinant His-tagged proteins (200 nm) and then stained with anti-His mAb under non-permeabilized conditions. The cell line names are indicated at the top of the corresponding FACS plot. A, chemical analysis. Binding of buffer alone (light gray) or recombinant protein (white) was assessed in CHO-K1 cells left untreated or treated with 50 mm sodium chlorate in the presence or absence of 10 mm sodium sulfate, as indicated at the top of the corresponding column of FACS plots. The tested recombinant protein is indicated at the right of the corresponding row of FACS plots. B, genetic analysis. Binding of buffer (light gray), CrmE (white), or rMCK-2 (dark gray) was assessed in parental CHO-K1 cells as well as in the CHO-K1-derived cell lines CHO-745 (GAG-deficient) and CHO-677 (HS-deficient), as indicated at the top of each plot. C, enzymatic analysis. CHO-K1 cells were incubated with the GAG lyase indicated at the top of each plot. Binding key: enzyme-treated cells incubated without rMCK-2, light gray (Buffer); enzyme-treated cells incubated with rMCK-2, white (+ enzyme); cells incubated in enzyme reaction buffer alone and with rMCK-2, dark gray (− enzyme). Data are from a single experiment representative of three independent experiments.
MCK-2 binds directly to GAGs
To assess rMCK-2-GAG binding directly, we analyzed the ability of rMCK-2 to bind heparin immobilized on Sepharose beads or on a surface plasmon resonance (SPR) sensor chip. As shown in Fig. 4A, heparin-coupled beads, but not beads alone, were able to pull down rMCK-2 from solution. In addition, this binding was specific because preincubation of rMCK-2 with soluble heparin completely blocked the interaction with heparin-coupled beads (Fig. 4A, lane 10). The proteins CrmE and B18 were included as negative and positive GAG-binding controls, respectively (Fig. 4A). Accordingly, we confirmed that rMCK-2, but not CrmE, was able to interact with heparin by SPR (Fig. 4B). To analyze whether rMCK-2 was able to bind to soluble GAGs other than heparin, the rMCK-2 binding to the heparin SPR chip was competed with increasing concentrations of soluble heparin, HS, CS, and DS (Fig. 4C). Excess soluble heparin and HS were both able to block binding of rMCK-2 to the heparin surface in a dose-dependent manner, reducing the binding by >50% at 0.1 and 1 μg/ml, respectively. In contrast, although the highest concentration (1000 μg/ml) tested for soluble CS and DS completely blocked the rMCK-2 interaction with the heparin chip, a still massive excess of 100 μg/ml of these two GAGs reduced the binding by only ∼20%, positioning the IC50 of CS and DS in the 100–1000 μg/ml range (Fig. 4C). These results demonstrate that rMCK-2 can interact directly with GAGs and support the rMCK-2 specificity for heparin and HS.
Figure 4.

MCK-2 binds preferentially to heparin and HS. A, heparin-Sepharose pulldown analysis. The indicated recombinant His-tagged proteins were incubated with either uncoupled Sepharose beads (Sepharose) or heparin-coupled Sepharose beads (heparin-Sepharose), as indicated for each lane at the top of the gel. As a control for binding specificity, rMCK2 was also incubated with heparin-Sepharose in the presence of 1 μg of soluble heparin (lane 10). Input protein (lanes 1, 4, and 7) and proteins eluted from the beads (all other lanes) were separated by SDS-PAGE and detected by Western blotting using an anti-His mAb. Molecular size markers are indicated to the left in kilodalton. B and C, SPR analysis. B, time course. The indicated recombinant proteins were injected at 100 nm in HBS-EP buffer onto a heparin-immobilized sensor chip, and the time course of SPR was measured as arbitrary response units. The arrow indicates the end of the injection. C, GAG specificity. The binding of rMCK-2 to a heparin sensor chip in the presence or absence of increasing concentrations of the indicated GAGs was quantitated at 115 s after injection by SPR. Binding is plotted as a percentage of maximum, defined as the SPR signal for rMCK-2 binding to the chip at 115 s after injection in the absence of GAGs. Data represent mean ± S.D. of triplicates. Data are from a single experiment representative of two independent experiments.
Both domains of MCK-2 bind to the cell surface via GAGs
To investigate the structural basis of MCK-2 binding to GAGs, we first tested the GAG-binding capacity of each of the two MCK-2 domains separately. The recombinant MCK-2 chemokine domain rm131 was able to bind to the surface of untreated CHO-K1 cells, and binding was abolished when the cells were pretreated with chlorate. Chlorate inhibition of binding was fully reversed by culturing the cells with sodium sulfate (Fig. 5), suggesting that the binding mechanism involves GAGs. This was confirmed by the inability of this domain to bind to the surface of genetically GAG-deficient CHO-745 cells (Fig. 5). The recombinant MCK-2 non-chemokine domain rm129 also bound to CHO-K1 cells in a GAG-dependent manner because no binding was detected to CHO-745 cells or to chlorate-treated CHO-K1 cells (Fig. 5).
Figure 5.

Both the chemokine and non-chemokine domains of MCK-2 bind GAGs on the plasma membrane. The parental CHO-K1 cell line and its GAG-deficient variant CHO-745 were cultured under the conditions indicated above the corresponding column of FACS plots and then incubated with buffer alone (light gray) or 200 nm recombinant protein (dark gray). The tested protein is indicated at the right of the corresponding row of plots. Cell-associated recombinant proteins were identified by staining with anti-His mAb. Data are from a single experiment representative of three independent experiments.
The GAG binding determinants on the MCK-2 chemokine domain include multiple basic amino acids located in the N terminus and the 30s and 50s loops
Protein-GAG interactions are primarily mediated by basic residues in the protein that engage sulfate groups in GAGs. Previous work has shown that a BBXB motif, where B is a basic amino acid (Arg, Lys, or His) conserved in the 40s loop is the main GAG-binding site for human CC chemokines (Fig. 6A) (22, 23, 25, 50, 51). However, this motif is not conserved in MCK-2, and initial analysis of the primary amino acid sequence of the protein did not reveal any obvious conserved GAG binding site in either the m131 or the m129 domains (Fig. 6A). Therefore, we decided to generate a structural model of MCK-2 to identify potential GAG-binding surfaces not detectable in the primary sequence of the protein. For this purpose, we used I-TASSER. The algorithm failed to generate a prediction for the m129 domain, which highlights the lack of primary amino acid sequence homology of this domain with all known proteins. In contrast, we obtained a structural model for m131 that aligned with the classical CC chemokine fold: an N-terminal unordered domain, a 310 turn, three antiparallel β strands, and a C-terminal α helix (Fig. 6B). Interestingly, surface electrostatic potential analysis of the m131 structural model revealed two surfaces containing opposite charge distributions (Fig. 6C). We localized five basic residues (Arg12, His16, Arg35, Lys50, and Arg51) clustered on a positively charged face of the molecule (Fig. 6D). To study their contribution to the binding of m131 with GAGs, we generated seven different recombinant baculovirus-expressing rm131 alanine mutants for combinations of these residues. The mutants were expressed in the supernatants of insect cells infected with the corresponding baculoviruses. The ability of these rm131 mutants to interact with GAGs was analyzed by an ELISA-based heparin-binding assay. All proteins were secreted into the supernatant at comparable levels (Fig. 6E). The concentration of the protein of interest in each supernatant was quantified by ELISA, and a volume equivalent to 4 ng of protein was incubated in heparin-immobilized wells. Supernatants from insect cells infected with baculoviruses encoding wild-type rm131 and vCCI, a vaccinia virus protein unable to interact with GAGs,3 were included as reference and negative control, respectively. As shown in Fig. 6F, all mutants except R35A displayed a significantly reduced capacity to bind heparin. Moreover, binding was increasingly reduced when increasing combinations of mutants were tested, up to a 92% reduction for the variant containing all five mutations. Even though Arg35 was not revealed as a GAG binding determinant by the mutant R35A, it did appear to contribute to binding when analyzed in the context of the other mutations. In particular, adding the R35A mutation to the double mutants K50A/R51A and H12A/R16A further reduced GAG binding from 40% to 20% and from 75% to 38%, respectively (Fig. 6F). Importantly, the GAG binding activity for the H12A/R16A mutant was only 25% less than wild-type rm131 binding, whereas the K50A/R51A mutant had 60% lower GAG binding activity than the wild type. Moreover, the GAG binding activity of the quadruple mutant combining these four mutations was indistinguishable from that of the K50A/R51A double mutant, indicating a bigger contribution of Lys50 and Arg51 to the interaction. Therefore, although all five basic residues appear to contribute to the rm131 binding with heparin, Arg35 in the 30s loop and Lys50 and Arg51 in the 50s loop appear to constitute the main GAG binding determinants of the chemokine domain of MCK-2.
Figure 6.

Identification of GAG binding determinants for chemokine domain m131 of MCK-2. A—D, computational localization of candidate GAG binding determinants. A, identification of conserved basic residues in m131. The indicated chemokine sequences without the signal peptide were aligned using Clustal Omega. Positively charged amino acids are indicated in red, conserved cysteines in blue, and residues known to mediate GAG binding for host chemokines are highlighted in yellow. The BBXB motif conserved in the 40s loop of human CC chemokines is framed. The corresponding predicted secondary structure and positions without considering the signal peptide of basic residues in the m131 sequence are indicated above the alignment. Shown below the alignment is the primary amino acid sequence of the C-terminal non-chemokine m129 domain of MCK-2. A sequence contained in the full-length MCK-2 encoded by a DNA sequence linked to the N terminus of ORF m129 is underlined. B, tertiary structural model of the m131 chemokine domain of MCK-2. A ribbon representation is shown for the superposition of the m131 structural prediction (gray) generated with I-TASSER with the known backbone tertiary structural fold of human CCL2 (red). C, surface electrostatic potential of the m131 structural model (top panel). The model was developed using the PDB2PQR server. Red and blue represent negatively and positively charged surfaces, respectively. Protein folding is represented in ribbons for reference (bottom panel). D, location of basic residues on the surface of the positively charged face of the m131 structural model. E, production of rm131 mutants targeting putative GAG-binding determinants. Recombinant His-tagged WT and mutant proteins are identified at the top of each lane. Recombinant proteins present in 15 μl of cleared supernatants from Hi5 cells infected with the corresponding recombinant baculovirus were separated by SDS-PAGE and revealed by Western blotting using an anti-His mAb. vCCI, poxvirus-encoded viral CC chemokine inhibitor. Molecular size markers are shown at the left in kilodalton. F, heparin-binding determinants of m131. Heparin binding of insect cell supernatants containing 4 ng of each recombinant His-tagged protein indicated on the x axis was determined by ELISA using an anti-His mAb. Values were corrected for background binding to supernatant from mock-infected insect cells, which ranged from 5–8% of wild-type rm131 binding. Data are presented relative to the heparin binding A450 value for WT rm131, which is set at 100%, and are the mean ± S.D. of triplicates of three independent assays. Multiple t tests were performed after logit transformation of the percent values (*, p < 0.05). The horizontal line above the graph indicates rm131 mutants with statistically significant differences to wild-type rm131.
Both domains of MCK2 are required for efficient GAG-dependent oligomerization
With very few exceptions, most chemokines oligomerize upon binding to GAGs (19). This is thought to increase the local chemokine concentration and improve the presentation of receptor-binding epitopes that could be masked in the GAG complex. Importantly, like chemokines defective in GAG-binding, chemokine oligomerization mutants are weak chemotactic factors in vivo (22). Thus, we next addressed whether GAG binding would induce rMCK-2 to form oligomers. For this purpose, we used the zero-length two-step cross-linking protocol, used previously for the study of chemokine oligomerization (25, 52). First, we validated this method with CCL2, which is known to form oligomers upon GAG binding (25). As shown in Fig. 7A, high-order oligomers of CCL2 were detected by Western blotting when the chemokine was incubated with activated heparin (+heparin, +cross-linkers) but not with inactivated heparin (+heparin, −cross-linkers) or cross-linkers alone. By contrast, only the monomer of vCCI, which, as shown in Fig. 6E, does not bind GAGs, was detected under these conditions. Incubation of rMCK-2 with activated heparin resulted in the formation of oligomers of various complexities (Fig. 7B). The bands corresponding to these complexes did not appear when rMCK-2 was incubated with inactivated heparin or cross-linkers alone (Fig. 7B). This result indicates that, like host chemokines, rMCK-2 oligomerizes upon GAG binding. To identify the rMCK-2 domain that mediates this effect, we analyzed the formation of oligomers for rm129 and rm131 when incubated with activated heparin. As shown in Fig. 7B, rm131 was not able to oligomerize independently, and rm129 only displayed a very weak tendency to oligomerize (asterisk, lane 6), suggesting that both domains are required for effective oligomerization of the full-length protein. It is important to note that dimerization of rm129, as CCL2 and full-length rMCK-2, was enhanced in the presence of cross-linkers, whereas these had no effect on dimerization of rm131. This, together with its weak ability to oligomerize by itself in the presence of activated heparin, suggests that rm129 might bear the oligomerization module of rMCK-2.
Figure 7.

Only full-length MCK-2 oligomerizes in the presence of heparin. A and B, the zero-length two-step method was used to analyze the oligomerization of the indicated recombinant proteins in the presence of inactivated or cross-linking activated heparin or cross-linkers alone, as indicated at the top of each gel. Proteins were separated by SDS-PAGE and revealed by Western blotting using anti-His mAb (A and B) or, in the case of CCL2 in A, by anti-CCL2 antiserum. Black arrowheads point to the monomeric forms of the corresponding protein. White arrowheads point to protein complexes of a higher order than dimers formed by CCL2 and rMCK-2 in the presence of activated heparin. The asterisk in lane 6 of Fig. 7B marks a faint ∼90-kDa band possibly corresponding to an rm129 trimer. Molecular size markers are indicated in kilodalton at the left of each panel.
Discussion
In this study we have demonstrated that the MCMV-encoded chemokine MCK-2 binds GAGs, with a preference for heparin and HS; that GAGs are required for MCK-2 binding to mouse epithelial cells and fibroblasts, including primary fibroblasts from lung and salivary gland; and that the structural basis for MCK-2-GAG binding includes both of the two MCK-2 structural domains, mediated in part by five basic residues distributed across the N terminus and the 30s and 50s loops of the chemokine domain. We have also demonstrated that MCK-2 oligomerizes in a GAG-dependent manner. Our study provides the first analysis of GAG-dependent oligomerization for a viral chemokine as well as the first example of a chemokine that requires a second GAG binding domain in addition to the chemokine domain for oligomerization. Our study is consistent with previous reports indicating that GAG binding may promote chemokine oligomerization (19). Proudfoot et al. (22) were the first to show that mutant recombinant chemokines defective in any of these biochemical properties may be defective or even completely inactive as leukocyte chemotactic factors in vivo despite the fact that these mutants may be fully active in vitro. Several mechanisms have been proposed to explain this paradox, including protection of GAG-bound chemokines from proteolytic activation, retention of chemokines on the luminal surface of endothelial cells, and promotion of chemokine haptotactic gradient formation, which provides directionality for cell migration by GAG-mediated tethering of chemokines to cell surfaces. Meanwhile, chemokine oligomerization may enhance GAG binding affinity as well as the accessibility of receptor-binding epitopes in the GAG-bound chemokine (17, 18).
MCK-2 consists of two spliced domains: m131, which encodes the N-terminal CC-chemokine domain of the protein, and m129, which lacks significant amino acid sequence homology with all known proteins. It is well established that MCK-2 is essential for MCMV persistent infection of host salivary glands in vivo and full infectivity of macrophages in vitro (30, 36, 37). In addition, several studies have proposed that MCK-2 is a proinflammatory factor in the context of MCMV infection (32–34). However, the molecular mechanisms of MCK-2 action have not been well characterized. In particular, the cellular receptor and biochemical characteristics of MCK-2 remain unknown. This work contributes to this gap in knowledge by delineating the molecular and structural determinants of MCK-2 GAG-binding and oligomerization mechanisms.
Since the identification of MCK-2 in 1999 (29), only two previous experiments have been reported using MCK-2 protein. Saederup et al. (53) reported that synthetic MCK-2 induced calcium flux and adherence in mouse peritoneal exudate cells. Later, the same group demonstrated that the injection of recombinant MCK-2 expressed in bacteria produced transient and moderate mouse footpad swelling (32). In contrast, in our hands, rMCK-2 did not trigger intracellular calcium signals in cell lines or in primary mouse peritoneal exudate cells (data not shown). This discrepancy could be explained if the putative MCK-2 cellular receptor is not expressed by the cells used in our study under our conditions. However, the most likely explanation is a three-amino acid difference at the extreme N terminus of the MCK-2 proteins used by us and by Saederup et al. (53). In particular, we introduced a non-native EDV motif at the N terminus of our MCK-2 recombinant proteins as an exigency of the cloning strategy (“Experimental Procedures”). The extreme N terminus of chemokines is well known to be an important determinant of signaling, but it has never been shown previously to affect GAG binding or oligomerization by chemokines, the focus of this study. Furthermore, although rMCK-2, rm131, and mutant rm131 proteins all contain EDV at the N terminus, experimentally only rMCK-2 formed high-order oligomers upon binding to heparin, and the rm131 alanine point mutants displayed major differences in heparin binding activity, which supports that this three-amino acid motif did not interfere in our experiments.
Despite the absence of an obvious chemokine-GAG-binding site in the primary sequence of MCK-2, we were able to predict a novel GAG-binding surface in a structural model of the m131 chemokine domain and to validate it experimentally. Arg35 as well as Lys50 and Arg51 located in the 30s and 50s loops, respectively, were the most important amino acids for the interaction of the chemokine domain with GAGs. A limitation of our interpretation is that there is no validation yet that these mutants were folded correctly, but the fact that they were all efficiently secreted and expressed at comparable levels suggests that this is not likely to be a problem. The MCK-2 GAG-binding mechanisms differ from those of mammalian chemokines not only in the contribution of the C-terminal m129 domain but also in the involvement of different chemokine loops. The rm131 GAG-binding surface is unique compared with that of mammalian CC chemokines, where, instead of the 30s and 50s loops, basic residues located in the N terminus and 40s loop form the primary GAG-binding surfaces (54). These same molecular characteristics were found to be involved in GAG interaction with vCCL2, the only viral chemokine whose GAG binding properties have been reported (55). The rm131 Arg35 residue is conserved in the 30s loop of most CC chemokines; however, it has never been clearly implicated in GAG binding for other chemokines. The 30s loop was shown to establish only complementary GAG interactions in CCL5 (56), and it was proposed to be important for the CX3CL1-GAG interaction; however, this was not demonstrated experimentally (57). On the other hand, the 50s loop has been shown to be critical for the GAG binding of members of other chemokine subgroups and of CC chemokine oligomers (58, 59). Furthermore, we found that the N terminus of the rm131 chemokine domain plays a more limited role in GAG-binding than it does for other CC chemokines. For instance, mutation of the basic residues located in the N terminus of CCL2 reduced GAG binding affinity by more than 4 logs (25). This is important because the N-terminal domain of many, if not all, chemokines is critical for the interaction with GPCRs (60). Therefore, in the GAG-bound form of MCK-2, unlike for other chemokines, the N terminus might be accessible to interact with a cognate GPCR.
Although chemokine monomers are fully functional in vitro, there are examples of forced monomers that are weaker chemoattractants than their wild-type forms, suggesting that oligomerization is essential for the activity of chemokines in vivo (22). We are just beginning to understand how oligomerization can potentiate or regulate chemokine bioactivity. Previous observations suggested that the ability of chemokines to oligomerize and interact with GAGs may actually be interdependent events; GAG binding is known to enhance chemokine oligomerization, and, reciprocally, chemokine oligomers often reveal extended high-affinity GAG-binding surfaces undetectable in the monomers (17). In fact, it has been recently demonstrated that chemokine oligomers have a higher affinity for GAGs than their monomers and that, in some cases, oligomerization can regulate the GAG binding specificity (18). Therefore, the ultimate purpose of chemokine oligomerization might be the establishment of high-affinity GAG interactions. Accordingly, non-oligomeric chemokines such as CCL7 may compensate with an unusually high number of GAG-binding sites (61). Here we demonstrate that MCK-2, like most chemokines, forms self-complexes of high order on GAGs through a mechanism by which the m131 chemokine domain and the C-terminal m129 are required. Three main molecular features have been found to mediate oligomerization in CC chemokines: a Pro residue in position −3 from the CC motif, an aromatic residue in position +3 from the CC motif, and two acidic residues in positions 26 and 66 (21, 62, 63). None of these characteristics are conserved in the chemokine domain of MCK-2, which may explain why the rm131 chemokine domain is unable by itself to oligomerize on GAGs. Similarly, the m129 domain alone showed a very weak tendency to form oligomers when incubated with heparin. Interestingly, as with the well known dimeric chemokine CCL2, we detected a cross-linked dimer of rMCK-2 and rm129 but not of rm131. This suggests that the m129 domain might contain the key molecular determinants for protein oligomerization. The precise molecular basis of rm129 dimerization and why this domain is not able to efficiently oligomerize with GAGs by itself will require further experimentation. Wagner et al. (36) proposed that m129 might be involved in the engagement of MCK-2 to the viral gH-gL complex required for full infectivity of MCMV in macrophages. Here we show that the m129 domain is necessary but not sufficient to promote oligomerization of MCK-2 on GAGs, and therefore, it should be considered as an additional explanation to the attenuated phenotype of MCMV derived from Sm3fr bacmids, which contain a stop codon at the beginning of the m129 domain (30).
In summary, we have demonstrated that the MCMV-encoded chemokine MCK-2 has conserved the GAG interaction and the GAG-dependent oligomerization found in most host chemokines. This is the first biochemical analysis of this important virulence factor of MCMV, a virus used worldwide as a model for HCMV, which constitutes the leading infectious cause of congenital disorders and graft rejection (64, 65). Importantly, the HCMV-encoded chemokine UL128, which displays many similarities to MCK-2, has been shown to be an excellent target for new vaccination strategies (41, 43). However, as in the case of MCK-2, our knowledge of the functional and biochemical properties of UL128 is very limited. The two new biochemical properties for MCK-2 reported here might be fundamental for the activity of the protein in vivo. The generation of recombinant MCMVs expressing mutant MCK-2 lacking one or both of these activities would help to determine their contribution to the role MCK-2 plays in viral pathogenesis. For this purpose, further efforts to decipher the molecular basis of the oligomerization and the GAG binding mediated particularly by the m129 domain will be required. Given the lack of homology of m129 with any other known protein, structural analyses would significantly accelerate progress in this direction.
Experimental procedures
Cells
The NIH-3T3, HEK293, and BS-C-1 cell lines were obtained from the ATCC (Manassas, VA) and cultured in DMEM supplemented with 10% FBS. The cell lines M2–10B4, MOLT4, and A20 were obtained from the ATCC and maintained in RPMI medium supplemented with 10% FBS. The cell line CHO-K1 and its GAG-deficient variants CHO-745 and CHO-677 (gifts from Dr. Antonio Alcami) were cultured in DMEM/F12 supplemented with 10% FBS (Life Technologies). Primary fibroblasts were isolated from mouse lungs and salivary glands following methods published previously with some modifications (66). Briefly, minced organs from two BALB/c mice were digested in DMEM/F12 medium containing 0.14 Wünsch units/ml of Liberase TL (Roche) for 1 h at 37 °C. Tissue fragments were resuspended in DMEM/F12 supplemented with 15% FBS, transferred to a 10-cm-diameter tissue culture plate, and incubated at 37 °C and 5% CO2 under normoxic conditions. After 14 days, adherent cells were collected, and 5 × 105 cells were transferred to a new plate in Eagle's minimum essential medium supplemented with 15% FBS, 1× non-essential amino acids, and 1 mm sodium pyruvate. Fibroblast cultures were split at least three more times before being used for experiments. Recombinant baculoviruses were generated in Hi5 adherent insect cells (a gift from Dr. Antonio Alcami) maintained in TC-100 medium (Sigma) supplemented with 10% FBS and 1× non-essential amino acids. Hi5 suspension insect cells, used for protein expression, were cultured in Express Five medium (Life Technologies). Both adherent and suspension Hi5 cells were grown at 27 °C under atmospheric conditions.
Construction of recombinant baculoviruses
The full-length MCK-2 ORF was amplified by PCR using cDNA from M2–10B4 cells infected with pSM3fr-MCK-2fl-derived MCMV (a gift from Dr. Barbara Adler) as a template and the primers mck2–3F (5′-ccggacgtcaccgtcgcggacctccgc-3′) and mck2–3R (5′-ccgtctagactttcatcggacagtcgttgtagc-3′). These primers exclude the endogenous signal peptide. The amplicon was ligated into the plasmid pAL7 (a gift from Dr. Antonio Alcami), a pFastBac1-derived plasmid bearing the honeybee melittin signal peptide and a V5-His6 tag (48) modified to optimize the signal peptide activity based on analysis using the SignalP 4.1 website. The resulting plasmid was termed pAL7mut. As a consequence of this mutagenesis, the NdeI restriction site of pAL7 was replaced by an AatII site in pAL7mut. The MCK-2 amplicon was cloned between the AatII and XbaI sites of pAL7mut in-frame with the N-terminal melittin signal peptide and a C-terminal V5-His6 tag. The resulting plasmid was named pSP12. The same strategy was followed to generate recombinant baculoviruses for the two individual MCK-2 domains: the chemokine domain encoded by ORF m131 (Thr19-Arg81) and the unique domain encoded by ORF m129 (Ser82-Glu280). The m131 coding sequence was amplified from pSP12 by PCR using the primers mck2–3F and mck2–4R (5′-ccgtctagacttctgaccagacacaagagtc-3′). Similarly, m129 was amplified by PCR with the primers mck2–6F (5′-ccggacgtctctcggtcagattccagac-3′) and mck2–3R. Both PCR products were cloned into pAL7mut as explained above to obtain the plasmids pSP13 and pSP14, respectively. Of note, as an exigency of the cloning strategy, a three-amino acid motif, EDV, was left between the melittin signal peptide and the beginning of the cloned MCK-2 sequences. We confirmed that these three extra amino acids were present at the N terminus of our final protein products by N-terminal sequencing using Edman degradation. Recombinant baculoviruses from pSP12, pSP13, and pSP14 generated using the Bac-to-Bac system (Life Technologies) were designated AcrMCK2, Acrm131, and Acrm129, respectively. Viral stocks were amplified by infection of adherent Hi5 cells at low multiplicities (0.1–0.01 pfu/cell), and high-titer stocks were used for protein production by infection of Hi5 suspension cells at high multiplicities (1–5 pfu/cell).
Protein expression and purification
Recombinant His-tagged proteins (rMCK2, rm131, rm129, and vCCI) were purified from the supernatants of baculovirus-infected Hi5 cells by affinity chromatography. Supernatants were concentrated and dialyzed in phosphate buffer before being incubated for 1 h with nickel-nitrilotriacetic agarose (Qiagen, Valencia, CA). Beads were extensively washed with 20 mm imidazole, and bound protein was eluted with increasing concentrations of imidazole (up to 250 mm). Protein-containing fractions were pooled, dialyzed in PBS, and stored at <−20 °C. Protein concentration was determined by gel densitometry.
Site-directed mutagenesis
Site-directed mutagenesis was performed using the QuikChange Lightning mutagenesis kit (Agilent Technologies, Santa Clara, CA) following the instructions of the manufacturer. The pAL7mut plasmid was generated using the primers pFBmut forward (5′-atacatttcttacatctatgccgaagacgtcggatcccggtccgaagcgcgcgg-3′) and pFBmut reverse (5′-ccgcgcgcttcggaccgggatccgacgtcttcggcatagatgtaagaaatgtat-3′) and the pAL7 plasmid as a template. To identify the GAG binding site in rm131, basic residues were mutated to alanine using pSP13 as template and the following primer pairs for each mutant: R35A, gag1 forward (5-cactaacacttcgtgcggagcagaggtggttttcacta-3′) and gag1 reverse (5′-tagtgaaaaccacctctgctccgcacgaagtgttagtg); K50A/R51A, gag2 forward (5′-ggatgagggtgtgcgctgcggcggcctggtggtcg-3′) and gag2 reverse (5′-cgaccaccaggccgccgcagcgcacaccctcatcc-3′); and R12A/H16A, gag3 forward (5′-atgttgcgcggcgccgcagctggcccctctcccg-3′) and gag3 reverse (5′-cgggagaggggccagctgcggcgccgcgcaacat-3′). For combinations of these mutations, the appropriate primer pairs were used simultaneously or in sequential PCR reactions. Mutagenesis was confirmed by sequencing, and the corresponding recombinant baculoviruses were generated as explained above.
Cell binding experiments
Binding of recombinant proteins to the cell surface was assessed by flow cytometry. 5 × 105 cells were incubated with buffer or different concentrations of rMCK2, rm131, or rm129 in PBS staining buffer (1% FBS and 1% BSA in PBS) on ice for 30 min. In some experiments, the recombinant V5-His-tagged proteins B18 and CrmE (gifts from Dr. Antonio Alcami) were included as positive and negative controls, respectively. Cells were washed twice with PBS staining buffer to remove unbound protein. Recombinant protein retained on the cell surface was detected with a mouse anti-His antibody (Qiagen) and an anti-mouse Alexa Fluor 488 secondary antibody (Life Technologies). 20,000 events were acquired in a FACSCalibur II cytometer (BD Biosciences), and the data were analyzed using FlowJo software (FlowJo, LLC, Ashland, OR). For some assays, cell surface GAGs were depleted by treatment of CHO-K1 cells with GAG lyases. Cells were incubated with 1 unit/ml heparinase II or chondroitinase ABC (Sigma) in reaction buffer (heparinase buffer: 20 mm Tris-HCl (pH 7.5), 4 mm CaCl2, 50 mm NaCl, and 0.01% BSA; chondroitinase buffer: 50 mm Tris-HCl (pH 8.0), 60 mm sodium acetate, and 0.02% BSA) for 30 min at 37 °C. After digestion, cells were washed once with PBS staining buffer and used for binding experiments as above. Protein binding to cells incubated in reaction buffer alone was tested as a reference.
Sodium chlorate treatment
Sulfation of cell surface GAGs was inhibited by sodium chlorate (46). CHO-K1 cells were cultured with 50 mm sodium chlorate in DMEM/F12 supplemented with 10% FBS in the presence or absence of 10 mm sodium sulfate, which reverses the effect of chlorate (48). After overnight incubation at 37 °C, cells were collected and used in binding experiments as explained above.
Heparin-Sepharose pulldown assay
Recombinant proteins were incubated with heparin-Sepharose or Sepharose beads (bioWORLD, Dublin, OH) in a rotator for 1 h at room temperature. Before the experiment, beads were equilibrated by washing once with water and twice with binding buffer (1× PBS, 0.05% Tween 20, and 0.2% FBS). rmMCK2 protein at 20 nm was incubated with 10 μl of washed beads in 400 μl of binding buffer. The recombinant V5-His-tagged proteins CrmE and B18 were included as negative and positive controls, respectively. The beads were washed three times with binding buffer before eluting the bound protein by boiling in 25 μl of Laemmli loading buffer. Protein in the inputs and eluates was analyzed by Western blotting with an anti-His antibody (Qiagen). To demonstrate the specificity of the interaction, rmMCK2 binding to heparin-Sepharose was competed with 1 μg of soluble heparin (Sigma) where indicated.
SPR assays
SPR experiments were performed in a Biacore 3000 biosensor (GE Healthcare) using a heparin-coupled SA chip (GE Healthcare). One flow cell of the SA sensor chip was immobilized with 50 response units of biotinylated heparin (EMD Millipore, Bedford, MA) following the instructions of the manufacturer. For binding experiments, 100 nm of rmMCK2 or CrmE (negative control) was injected in HBS-EP buffer (10 mm HEPES, 150 mm NaCl, 3.4 mm EDTA, and 0.005% P20 (pH 7.4)) at a flow rate of 10 μl/min. For binding competition with soluble GAGs, 100 nm of rmMCK2 was incubated with increasing concentrations of heparin, heparan sulfate, chondroitin sulfate, or dermatan sulfate (Sigma) in HBS-EP for 15 min on ice before being injected onto the heparin-coupled SA chip at 10 μl/min. The association was recorded during 120 s, and the response units bound at 115 s were compared with the binding response in the absence of soluble GAGs.
ELISA
The GAG-binding capacity of rm131 mutants expressed in the supernatants of infected insect cells was assessed by an ELISA-based heparin binding experiment. First, the concentration of the protein of interest in each supernatant was determined by ELISA. For this purpose, Nunc MaxiSorp 96-well plates were coated with 1 μg/ml of a mouse anti-V5 antibody (Life Technologies) in bicarbonate/carbonate buffer (pH 9.6) overnight at 4 °C. Plates were blocked with 3% milk in PBS containing 0.05% Tween 20 for 1 h at room temperature before being incubated with serial dilutions of baculovirus supernatants in blocking buffer. After a 1-h incubation at room temperature, plates were washed three times with PBS containing 0.05% Tween 20, and bound protein was detected with a rabbit anti-His antibody (Cell Signaling Technology, Beverly, MA) and an HRP-coupled anti-rabbit secondary antibody (Santa Cruz Biotechnology, Santa Cruz, CA) in blocking buffer. Plates were developed with TMB One Component (SurModics, Eden Prairie, MN). The reaction was stopped with sulfuric acid, and absorbance at 450 nm (A450) was measured in a FlexStation3 reader (Molecular Devices, Sunnyvale, CA). Protein concentration was calculated by interpolation in a standard curve generated with purified rm131. For the heparin-binding ELISA, 10 μg/ml of biotinylated heparin (EMD Millipore) was immobilized on streptavidin-coated ELISA plates (Life Technologies) following the instructions of the manufacturer. Plates were blocked with TBS containing 5% BSA and 0.05% Tween 20. The supernatant volume equivalent to 4 ng of rm131 or mutant m131 proteins was incubated in triplicate in blocking buffer for 1 h at room temperature. Supernatants from mock-infected cells were used to eliminate the background. The supernatant from cells infected with a V5-His-tagged vCCI-expressing baculovirus (a gift from Dr. Antonio Alcami) was used as a negative control. After extensive washing with TBS containing 0.05% Tween 20, bound protein was detected as explained above.
Computational modeling of the rm131 protein structure
The three-dimensional structure of MCK-2 was predicted using I-TASSER (67). The corresponding amino acid sequence excluding the signal peptide was submitted to the I-TASSER web server without any restriction or suggested templates. The server selected the structure of human CCL2 as the best template to model the structure of the m131-encoded chemokine moiety of MCK-2, whereas it was unable to generate a prediction for the unique m129-encoded domain of the protein. The molecular structure of CCL2 (PDB code 3IFD) and the m131 model were superimposed in the UCSF Chimera package (68). The surface electrostatic potential of the m131 model was generated using the online PDB2PQR server and the APBS plug-in of Chimera (69, 70).
Heparin cross-linking
The ability of full-length rMCK-2 and the rm131 and rm129 domain proteins to oligomerize upon heparin binding was assessed using the zero-length two-step method as described previously (71). Briefly, unfractionated heparin (Sigma) was activated with 6 mm 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide (Life Technologies) and 15 mm N-hydroxy-sulfosuccinimide (Life Technologies) in reaction buffer (10 mm MES (pH 6.0) and 50 mm NaCl) for 1 h at room temperature. Subsequently, 20 mm β-mercaptoethanol was added to inactivate the cross-linker excess. 20 min after inactivation, 100 ng of each protein was incubated in the presence or absence of activated heparin in a final 1:4 molar ratio. After a 2-h reaction at room temperature, Laemmli loading buffer was added, and samples were analyzed by an anti-His Western blotting. Recombinant vCCI and human CCL2 (Peprotech, Rocky Hill, NJ) were included as negative and positive controls, respectively. vCCI samples were analyzed as before, whereas a rabbit polyclonal anti-CCL2 antiserum (Peprotech) was used to analyze CCL2-containing reactions. Blot images were captured using an Omega Lum C imager (Aplegen, San Francisco, CA).
Author contributions
P. M. M. and S. M. P. conceived and designed the experiments and wrote the paper. S. M. P. performed the experiments. Both authors read and approved the final manuscript.
This work was supported by the Intramural Research Program of the NIAID, National Institutes of Health. The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
A. Alcami, personal communication.
- GPCR
- G protein-coupled receptor
- GAG
- glycosaminoglycan
- MCMV
- mouse CMV
- HCMV
- human CMV
- HS
- heparan sulfate
- CS
- chondroitin sulfate
- DS
- dermatan sulfate
- SPR
- surface plasmon resonance.
References
- 1. Rot A., and von Andrian U. H. (2004) Chemokines in innate and adaptive host defense: basic chemokinese grammar for immune cells. Annu. Rev. Immunol. 22, 891–928 [DOI] [PubMed] [Google Scholar]
- 2. Alcami A. (2003) Viral mimicry of cytokines, chemokines and their receptors. Nat. Rev. Immunol. 3, 36–50 [DOI] [PubMed] [Google Scholar]
- 3. Bachelerie F., Ben-Baruch A., Burkhardt A. M., Combadiere C., Farber J. M., Graham G. J., Horuk R., Sparre-Ulrich A. H., Locati M., Luster A. D., Mantovani A., Matsushima K., Murphy P. M., Nibbs R., Nomiyama H., et al. (2014) International Union of Basic and Clinical Pharmacology [corrected]: LXXXIX: Update on the extended family of chemokine receptors and introducing a new nomenclature for atypical chemokine receptors. Pharmacol Rev. 66, 1–79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Graham G. J., Locati M., Mantovani A., Rot A., and Thelen M. (2012) The biochemistry and biology of the atypical chemokine receptors. Immunol. Lett. 145, 30–38 [DOI] [PubMed] [Google Scholar]
- 5. Iozzo R. V., and Schaefer L. (2015) Proteoglycan form and function: A comprehensive nomenclature of proteoglycans. Matrix Biol. 42, 11–55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Kuschert G. S., Coulin F., Power C. A., Proudfoot A. E., Hubbard R. E., Hoogewerf A. J., and Wells T. N. (1999) Glycosaminoglycans interact selectively with chemokines and modulate receptor binding and cellular responses. Biochemistry 38, 12959–12968 [DOI] [PubMed] [Google Scholar]
- 7. Handel T. M., Johnson Z., Crown S. E., Lau E. K., and Proudfoot A. E. (2005) Regulation of protein function by glycosaminoglycans as exemplified by chemokines. Annu. Rev. Biochem. 74, 385–410 [DOI] [PubMed] [Google Scholar]
- 8. Ellyard J. I., Simson L., Bezos A., Johnston K., Freeman C., and Parish C. R. (2007) Eotaxin selectively binds heparin. An interaction that protects eotaxin from proteolysis and potentiates chemotactic activity in vivo. J. Biol. Chem. 282, 15238–15247 [DOI] [PubMed] [Google Scholar]
- 9. Tanaka Y., Adams D. H., and Shaw S. (1993) Proteoglycans on endothelial cells present adhesion-inducing cytokines to leukocytes. Immunol. Today 14, 111–115 [DOI] [PubMed] [Google Scholar]
- 10. Middleton J., Patterson A. M., Gardner L., Schmutz C., and Ashton B. A. (2002) Leukocyte extravasation: chemokine transport and presentation by the endothelium. Blood 100, 3853–3860 [DOI] [PubMed] [Google Scholar]
- 11. Soria G., Lebel-Haziv Y., Ehrlich M., Meshel T., Suez A., Avezov E., Rozenberg P., and Ben-Baruch A. (2012) Mechanisms regulating the secretion of the promalignancy chemokine CCL5 by breast tumor cells: CCL5's 40s loop and intracellular glycosaminoglycans. Neoplasia 14, 1–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Wagner L., Yang O. O., Garcia-Zepeda E. A., Ge Y., Kalams S. A., Walker B. D., Pasternack M. S., and Luster A. D. (1998) β-Chemokines are released from HIV-1-specific cytolytic T-cell granules complexed to proteoglycans. Nature 391, 908–911 [DOI] [PubMed] [Google Scholar]
- 13. Wang L., Fuster M., Sriramarao P., and Esko J. D. (2005) Endothelial heparan sulfate deficiency impairs L-selectin- and chemokine-mediated neutrophil trafficking during inflammatory responses. Nat. Immunol. 6, 902–910 [DOI] [PubMed] [Google Scholar]
- 14. Chang T. L., Gordon C. J., Roscic-Mrkic B., Power C., Proudfoot A. E., Moore J. P., and Trkola A. (2002) Interaction of the CC-chemokine RANTES with glycosaminoglycans activates a p44/p42 mitogen-activated protein kinase-dependent signaling pathway and enhances human immunodeficiency virus type 1 infectivity. J. Virol. 76, 2245–2254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Weber M., Hauschild R., Schwarz J., Moussion C., de Vries I., Legler D. F., Luther S. A., Bollenbach T., and Sixt M. (2013) Interstitial dendritic cell guidance by haptotactic chemokine gradients. Science 339, 328–332 [DOI] [PubMed] [Google Scholar]
- 16. Patel D. D., Koopmann W., Imai T., Whichard L. P., Yoshie O., and Krangel M. S. (2001) Chemokines have diverse abilities to form solid phase gradients. Clin. Immunol. 99, 43–52 [DOI] [PubMed] [Google Scholar]
- 17. Salanga C. L., and Handel T. M. (2011) Chemokine oligomerization and interactions with receptors and glycosaminoglycans: the role of structural dynamics in function. Exp. Cell Res. 317, 590–601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Dyer D. P., Salanga C. L., Volkman B. F., Kawamura T., and Handel T. M. (2016) The dependence of chemokine-glycosaminoglycan interactions on chemokine oligomerization. Glycobiology 26, 312–326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Hoogewerf A. J., Kuschert G. S., Proudfoot A. E., Borlat F., Clark-Lewis I., Power C. A., and Wells T. N. (1997) Glycosaminoglycans mediate cell surface oligomerization of chemokines. Biochemistry 36, 13570–13578 [DOI] [PubMed] [Google Scholar]
- 20. Rajarathnam K., Sykes B. D., Kay C. M., Dewald B., Geiser T., Baggiolini M., and Clark-Lewis I. (1994) Neutrophil activation by monomeric interleukin-8. Science 264, 90–92 [DOI] [PubMed] [Google Scholar]
- 21. Paavola C. D., Hemmerich S., Grunberger D., Polsky I., Bloom A., Freedman R., Mulkins M., Bhakta S., McCarley D., Wiesent L., Wong B., Jarnagin K., and Handel T. M. (1998) Monomeric monocyte chemoattractant protein-1 (MCP-1) binds and activates the MCP-1 receptor CCR2B. J. Biol. Chem. 273, 33157–33165 [DOI] [PubMed] [Google Scholar]
- 22. Proudfoot A. E., Handel T. M., Johnson Z., Lau E. K., LiWang P., Clark-Lewis I., Borlat F., Wells T. N., and Kosco-Vilbois M. H. (2003) Glycosaminoglycan binding and oligomerization are essential for the in vivo activity of certain chemokines. Proc. Natl. Acad. Sci. U.S.A. 100, 1885–1890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ali S., Robertson H., Wain J. H., Isaacs J. D., Malik G., and Kirby J. A. (2005) A non-glycosaminoglycan-binding variant of CC chemokine ligand 7 (monocyte chemoattractant protein-3) antagonizes chemokine-mediated inflammation. J. Immunol. 175, 1257–1266 [DOI] [PubMed] [Google Scholar]
- 24. Peterson F. C., Elgin E. S., Nelson T. J., Zhang F., Hoeger T. J., Linhardt R. J., and Volkman B. F. (2004) Identification and characterization of a glycosaminoglycan recognition element of the C chemokine lymphotactin. J. Biol. Chem. 279, 12598–12604 [DOI] [PubMed] [Google Scholar]
- 25. Lau E. K., Paavola C. D., Johnson Z., Gaudry J.-P., Geretti E., Borlat F., Kungl A. J., Proudfoot A. E., and Handel T. M. (2004) Identification of the glycosaminoglycan binding site of the CC chemokine, MCP-1: implications for structure and function in vivo. J. Biol. Chem. 279, 22294–22305 [DOI] [PubMed] [Google Scholar]
- 26. Campanella G. S., Grimm J., Manice L. A., Colvin R. A., Medoff B. D., Wojtkiewicz G. R., Weissleder R., and Luster A. D. (2006) Oligomerization of CXCL10 is necessary for endothelial cell presentation and in vivo activity. J. Immunol. 177, 6991–6998 [DOI] [PubMed] [Google Scholar]
- 27. Gangavarapu P., Rajagopalan L., Kolli D., Guerrero-Plata A., Garofalo R. P., and Rajarathnam K. (2012) The monomer-dimer equilibrium and glycosaminoglycan interactions of chemokine CXCL8 regulate tissue-specific neutrophil recruitment. J. Leukocyte Biol. 91, 259–265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Johnson Z., Proudfoot A. E., and Handel T. M. (2005) Interaction of chemokines and glycosaminoglycans: a new twist in the regulation of chemokine function with opportunities for therapeutic intervention. Cytokine Growth Factor Rev. 16, 625–636 [DOI] [PubMed] [Google Scholar]
- 29. MacDonald M. R., Burney M. W., Resnick S. B., Virgin H. W. 4th (1999) Spliced mRNA encoding the murine cytomegalovirus chemokine homolog predicts a β chemokine of novel structure. J. Virol. 73, 3682–3691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Jordan S., Krause J., Prager A., Mitrovic M., Jonjic S., Koszinowski U. H., and Adler B. (2011) Virus progeny of murine cytomegalovirus bacterial artificial chromosome pSM3fr show reduced growth in salivary glands due to a fixed mutation of MCK-2. J. Virol. 85, 10346–10353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Fleming P., Davis-Poynter N., Degli-Esposti M., Densley E., Papadimitriou J., Shellam G., and Farrell H. (1999) The murine cytomegalovirus chemokine homolog, m131/129, is a determinant of viral pathogenicity. J. Virol. 73, 6800–6809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Noda S., Aguirre S. A., Bitmansour A., Brown J. M., Sparer T. E., Huang J., and Mocarski E. S. (2006) Cytomegalovirus MCK-2 controls mobilization and recruitment of myeloid progenitor cells to facilitate dissemination. Blood 107, 30–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Daley-Bauer L. P., Wynn G. M., and Mocarski E. S. (2012) Cytomegalovirus impairs antiviral CD8+ T cell immunity by recruiting inflammatory monocytes. Immunity 37, 122–133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Daley-Bauer L. P., Roback L. J., Wynn G. M., and Mocarski E. S. (2014) Cytomegalovirus hijacks CX3CR1hi patrolling monocytes as immune-privileged vehicles for dissemination in mice. Cell Host Microbe 15, 351–362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Saederup N., Aguirre S. A., Sparer T. E., Bouley D. M., and Mocarski E. S. (2001) Murine cytomegalovirus CC chemokine homolog MCK-2 (m131-129) is a determinant of dissemination that increases inflammation at initial sites of infection. J. Virol. 75, 9966–9976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Wagner F. M., Brizic I., Prager A., Trsan T., Arapovic M., Lemmermann N. A., Podlech J., Reddehase M. J., Lemnitzer F., Bosse J. B., Gimpfl M., Marcinowski L., MacDonald M., Adler H., Koszinowski U. H., and Adler B. (2013) The viral chemokine MCK-2 of murine cytomegalovirus promotes infection as part of a gH/gL/MCK-2 complex. PLoS Pathog. 9, e1003493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Stahl F. R., Keyser K. A., Heller K., Bischoff Y., Halle S., Wagner K., Messerle M., and Förster R. (2015) Mck2-dependent infection of alveolar macrophages promotes replication of MCMV in nodular inflammatory foci of the neonatal lung. Mucosal Immunol. 8, 57–67 [DOI] [PubMed] [Google Scholar]
- 38. Ryckman B. J., Rainish B. L., Chase M. C., Borton J. A., Nelson J. A., Jarvis M. A., and Johnson D. C. (2008) Characterization of the human cytomegalovirus gH/gL/UL128–131 complex that mediates entry into epithelial and endothelial cells. J. Virol. 82, 60–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Straschewski S., Patrone M., Walther P., Gallina A., Mertens T., and Frascaroli G. (2011) Protein pUL128 of human cytomegalovirus is necessary for monocyte infection and blocking of migration. J. Virol. 85, 5150–5158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Nogalski M. T., Chan G. C., Stevenson E. V., Collins-McMillen D. K., and Yurochko A. D. (2013) The HCMV gH/gL/UL128–131 complex triggers the specific cellular activation required for efficient viral internalization into target monocytes. PLoS Pathog. 9, e1003463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Chiuppesi F., Wussow F., Johnson E., Bian C., Zhuo M., Rajakumar A., Barry P. A., Britt W. J., Chakraborty R., and Diamond D. J. (2015) Vaccine-derived neutralizing antibodies to the human cytomegalovirus gH/gL pentamer potently block primary cytotrophoblast infection. J. Virol. 89, 11884–11898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Wen Y., Monroe J., Linton C., Archer J., Beard C. W., Barnett S. W., Palladino G., Mason P. W., Carfi A., and Lilja A. E. (2014) Human cytomegalovirus gH/gL/UL128/UL130/UL131A complex elicits potently neutralizing antibodies in mice. Vaccine 32, 3796–3804 [DOI] [PubMed] [Google Scholar]
- 43. Wussow F., Chiuppesi F., Martinez J., Campo J., Johnson E., Flechsig C., Newell M., Tran E., Ortiz J., La Rosa C., Herrmann A., Longmate J., Chakraborty R., Barry P. A., and Diamond D. J. (2014) Human cytomegalovirus vaccine based on the envelope gH/gL pentamer complex. PLoS Pathog. 10, e1004524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Ciferri C., Chandramouli S., Leitner A., Donnarumma D., Cianfrocco M. A., Gerrein R., Friedrich K., Aggarwal Y., Palladino G., Aebersold R., Norais N., Settembre E. C., and Carfi A. (2015) Antigenic characterization of the HCMV gH/gL/gO and pentamer cell entry complexes reveals binding sites for potently neutralizing human antibodies. PLoS Pathog. 11, e1005230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Penfold M. E., Dairaghi D. J., Duke G. M., Saederup N., Mocarski E. S., Kemble G. W., and Schall T. J. (1999) Cytomegalovirus encodes a potent α chemokine. Proc. Natl. Acad. Sci. U.S.A. 96, 9839–9844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Safaiyan F., Kolset S. O., Prydz K., Gottfridsson E., Lindahl U., and Salmivirta M. (1999) Selective effects of sodium chlorate treatment on the sulfation of heparan sulfate. J. Biol. Chem. 274, 36267–36273 [DOI] [PubMed] [Google Scholar]
- 47. Lindahl U., Kusche-Gullberg M., and Kjellén L. (1998) Regulated diversity of heparan sulfate. J. Biol. Chem. 273, 24979–24982 [DOI] [PubMed] [Google Scholar]
- 48. Montanuy I., Alejo A., and Alcami A. (2011) Glycosaminoglycans mediate retention of the poxvirus type I interferon binding protein at the cell surface to locally block interferon antiviral responses. FASEB J. 25, 1960–1971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Zhang L., Lawrence R., Frazier B. A., and Esko J. D. (2006) CHO glycosylation mutants: proteoglycans. Methods Enzymol. 416, 205–221 [DOI] [PubMed] [Google Scholar]
- 50. Koopmann W., and Krangel M. S. (1997) Identification of a glycosaminoglycan-binding site in chemokine macrophage inflammatory protein-1α. J. Biol. Chem. 272, 10103–10109 [DOI] [PubMed] [Google Scholar]
- 51. Proudfoot A. E., Fritchley S., Borlat F., Shaw J. P., Vilbois F., Zwahlen C., Trkola A., Marchant D., Clapham P. R., and Wells T. N. (2001) The BBXB motif of RANTES is the principal site for heparin binding and controls receptor selectivity. J. Biol. Chem. 276, 10620–10626 [DOI] [PubMed] [Google Scholar]
- 52. Vivès R. R., Sadir R., Imberty A., Rencurosi A., and Lortat-Jacob H. (2002) A kinetics and modeling study of RANTES(9–68) binding to heparin reveals a mechanism of cooperative oligomerization. Biochemistry 41, 14779–14789 [DOI] [PubMed] [Google Scholar]
- 53. Saederup N., Lin Y. C., Dairaghi D. J., Schall T. J., and Mocarski E. S. (1999) Cytomegalovirus-encoded β chemokine promotes monocyte-associated viremia in the host. Proc. Natl. Acad. Sci. U.S.A. 96, 10881–10886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Monneau Y., Arenzana-Seisdedos F., and Lortat-Jacob H. (2016) The sweet spot: how GAGs help chemokines guide migrating cells. J. Leukocyte Biol. 99, 935–953 [DOI] [PubMed] [Google Scholar]
- 55. Zhao B., and Liwang P. J. (2010) Characterization of the interactions of vMIP-II, and a dimeric variant of vMIP-II, with glycosaminoglycans. Biochemistry 49, 7012–7022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Shaw J. P., Johnson Z., Borlat F., Zwahlen C., Kungl A., Roulin K., Harrenga A., Wells T. N., and Proudfoot A. E. (2004) The x-ray structure of RANTES: heparin-derived disaccharides allows the rational design of chemokine inhibitors. Structure 12, 2081–2093 [DOI] [PubMed] [Google Scholar]
- 57. Lortat-Jacob H., Grosdidier A., and Imberty A. (2002) Structural diversity of heparan sulfate binding domains in chemokines. Proc. Natl. Acad. Sci. U.S.A. 99, 1229–1234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Severin I. C., Gaudry J.-P., Johnson Z., Kungl A., Jansma A., Gesslbauer B., Mulloy B., Power C., Proudfoot A. E., and Handel T. (2010) Characterization of the chemokine CXCL11-heparin interaction suggests two different affinities for glycosaminoglycans. J. Biol. Chem. 285, 17713–17724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Liang W. G., Triandafillou C. G., Huang T.-Y., Zulueta M. M., Banerjee S., Dinner A. R., Hung S.-C., and Tang W.-J. (2016) Structural basis for oligomerization and glycosaminoglycan binding of CCL5 and CCL3. Proc. Natl. Acad. Sci. U.S.A. 113, 5000–5005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Kufareva I., Salanga C. L., and Handel T. M. (2015) Chemokine and chemokine receptor structure and interactions: implications for therapeutic strategies. Immunol. Cell Biol. 93, 372–383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Salanga C. L., Dyer D. P., Kiselar J. G., Gupta S., Chance M. R., and Handel T. M. (2014) Multiple glycosaminoglycan-binding epitopes of monocyte chemoattractant protein-3/CCL7 enable it to function as a non-oligomerizing chemokine. J. Biol. Chem. 289, 14896–14912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Czaplewski L. G., McKeating J., Craven C. J., Higgins L. D., Appay V., Brown A., Dudgeon T., Howard L. A., Meyers T., Owen J., Palan S. R., Tan P., Wilson G., Woods N. R., Heyworth C. M., et al. (1999) Identification of amino acid residues critical for aggregation of human CC chemokines macrophage inflammatory protein (MIP)-1α, MIP-1β, and RANTES: characterization of active disaggregated chemokine variants. J. Biol. Chem. 274, 16077–16084 [DOI] [PubMed] [Google Scholar]
- 63. Laurence J. S., Blanpain C., Burgner J. W., Parmentier M., and LiWang P. J. (2000) CC chemokine MIP-1β can function as a monomer and depends on Phe13 for receptor binding. Biochemistry 39, 3401–3409 [DOI] [PubMed] [Google Scholar]
- 64. Britt W. (2008) Manifestations of human cytomegalovirus infection: proposed mechanisms of acute and chronic disease. Curr. Top. Microbiol. Immunol. 325, 417–470 [DOI] [PubMed] [Google Scholar]
- 65. Streblow D. N., Orloff S. L., and Nelson J. A. (2007) Acceleration of allograft failure by cytomegalovirus. Curr. Opin. Immunol. 19, 577–582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Seluanov A., Vaidya A., and Gorbunova V. (2010) Establishing primary adult fibroblast cultures from rodents. J. Vis. Exp. 10.3791/2033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Yang J., Yan R., Roy A., Xu D., Poisson J., and Zhang Y. (2015) The I-TASSER suite: protein structure and function prediction. Nat. Methods 12, 7–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Pettersen E. F., Goddard T. D., Huang C. C., Couch G. S., Greenblatt D. M., Meng E. C., and Ferrin T. E. (2004) UCSF Chimera: a visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612 [DOI] [PubMed] [Google Scholar]
- 69. Dolinsky T. J., Czodrowski P., Li H., Nielsen J. E., Jensen J. H., Klebe G., and Baker N. A. (2007) PDB2PQR: expanding and upgrading automated preparation of biomolecular structures for molecular simulations. Nucleic Acids Res. 35, W522-W525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Baker N. A., Sept D., Joseph S., Holst M. J., and McCammon J. A. (2001) Electrostatics of nanosystems: application to microtubules and the ribosome. Proc. Natl. Acad. Sci. U.S.A. 98, 10037–10041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Grabarek Z., and Gergely J. (1990) Zero-length crosslinking procedure with the use of active esters. Anal. Biochem. 185, 131–135 [DOI] [PubMed] [Google Scholar]