

Abstract
The bifunctional flavoenzyme proline utilization A (PutA) catalyzes the two-step oxidation of proline to glutamate using separate proline dehydrogenase (PRODH) and l-glutamate-γ-semialdehyde dehydrogenase active sites. Because PutAs catalyze sequential reactions, they are good systems for studying how metabolic enzymes communicate via substrate channeling. Although mechanistically similar, PutAs vary widely in domain architecture, oligomeric state, and quaternary structure, and these variations represent different structural solutions to the problem of sequestering a reactive metabolite. Here, we studied PutA from Corynebacterium freiburgense (CfPutA), which belongs to the uncharacterized 3B class of PutAs. A 2.7 Å resolution crystal structure showed the canonical arrangement of PRODH, l-glutamate-γ-semialdehyde dehydrogenase, and C-terminal domains, including an extended interdomain tunnel associated with substrate channeling. The structure unexpectedly revealed a novel open conformation of the PRODH active site, which is interpreted to represent the non-activated conformation, an elusive form of PutA that exhibits suboptimal channeling. Nevertheless, CfPutA exhibited normal substrate-channeling activity, indicating that it isomerizes into the active state under assay conditions. Sedimentation-velocity experiments provided insight into the isomerization process, showing that CfPutA dimerizes in the presence of a proline analog and NAD+. These results are consistent with the morpheein model of enzyme hysteresis, in which substrate binding induces conformational changes that promote assembly of a high-activity oligomer. Finally, we used domain deletion analysis to investigate the function of the C-terminal domain. Although this domain contains neither catalytic residues nor substrate sites, its removal impaired both catalytic activities, suggesting that it may be essential for active-site integrity.
Keywords: analytical ultracentrifugation, dehydrogenase, enzyme kinetics, flavoprotein, oligomerization, X-ray crystallography, bifunctional enzyme, substrate channeling
Introduction
The proline catabolic pathway consists of two enzymes that catalyze the 4-electron oxidation of l-proline to l-glutamate (Fig. 1). The flavoenzyme proline dehydrogenase (PRODH)4 catalyzes the FAD-dependent oxidation of l-proline to the cyclic imine Δ1-pyrroline-5-carboxylate (P5C). Hydrolysis of P5C generates the open-chain aldehyde, l-glutamate-γ-semialdehyde (GSAL), which is the substrate for the second enzyme of the pathway, GSAL dehydrogenase (GSALDH). GSALDH (also known as P5CDH) is an aldehyde dehydrogenase superfamily (ALDHSF) enzyme (also known as ALDH4A1) that catalyzes the NAD+-dependent oxidation of GSAL to l-glutamate.
Figure 1.

The reactions of proline catabolism.
Proline catabolism has emerged as an interesting system for studying the communication between enzymes in metabolic pathways. In some bacteria, PRODH and GSALDH are combined into the bifunctional enzyme proline utilization A (PutA) (1). The covalent linking of the two enzymes suggests a direct mechanism of communication via substrate channeling. Indeed, kinetic studies have shown that PutAs exhibit substrate channeling (2–8). Crystal structures of PutAs showed the basis for substrate channeling; the two active sites are connected by a tunnel through which the intermediate travels (2, 3, 6, 8). The very existence of PutA suggested the hypothesis that substrate channeling might also occur in organisms where PRODH and GSALDH are distinct enzymes encoded by separate genes, as in eukaryotes and some bacteria. This hypothesis was validated recently for Thermus thermophilus PRODH and GSALDH (9). Thus, the available data suggest that communication between PRODH and GSALDH via substrate channeling is a conserved aspect of proline catabolism.
PutAs are also useful for exploring multidomain protein structure. Although all PutAs catalyze the aforementioned reactions using highly conserved active sites, they are surprisingly diverse in terms of domain architecture, sequence similarity, oligomeric state, and quaternary structure. Three PutA domain architectures (A, B, and C) are known (Fig. 2A) (1, 10, 11). Type A PutAs have the minimal set of domains consisting of N-terminal PRODH and C-terminal GSALDH modules (Fig. 2A). Type B and C PutAs have an additional ALDHSF domain following the GSALDH module. The function of this domain is a subject of this report. Finally, type C PutAs have an N-terminal ribbon-helix-helix DNA-binding domain. Type C PutAs are unique in the PutA family in that they regulate transcription of the put regulon, which contains the genes encoding PutA and the proline transporter PutP (12–14). The ribbon-helix-helix domain plays an essential role in the gene regulatory function by binding to multiple operator sites located between the divergently transcribed putA and putP genes.
Figure 2.

Classification and diversity of PutAs. A, the three domain architectures of PutAs. B, phylogenetic tree based on global sequence alignments of PutAs. PutAs with architecture types A, B, and C are indicated by black, blue, and red type, respectively. The four PutAs for which crystal structures have been obtained are listed in large type. The alignments were calculated with Clustal Omega (56) and visualized with DrawTree (57). C, structures of PutA oligomers that have been validated by crystallography and SAXS (PDB codes 4NMA, 3HAZ, and 5KF6).
Sequence alignments reveal another layer of diversity. The PutA phylogenetic tree has three main branches (Fig. 2B). The pairwise sequence identities between PutAs from different branches are typically ∼30%, suggesting substantial differences. Paradoxically, the branches do not correspond exactly to the three domain architectures described above. In fact, all three architectures are represented in branch 1, whereas branch 2 contains only type A PutAs, and branch 3 contains only type B PutAs.
Combining the domain architectures with phylogeny yields five PutA classes designated by the branch number and domain architecture type: 1A, 1B, 1C, 2A, and 3B. This scheme differs slightly from an older one in which 2A and 3B PutAs were grouped in the same branch of the tree (1, 10). Crystal structures are available for the 1A PutA from Bradyrhizobium japonicum (BjPutA) (2), the 2A PutA from Geobacter sulfurreducens (GsPutA) (6), and the 1B PutA from Sinorhizobium meliloti (SmPutA) (8). Also, structures of the PRODH (15–18) and DNA-binding domains (13, 14) of the 1C PutA from Escherichia coli (EcPutA) have been determined.
The diversity of PutAs extends to oligomeric state and quaternary structure. Type A PutAs form domain-swapped dimers (2, 6), and in at least one case (BjPutA), two of the dimers assemble into a ring-shaped tetramer (2) (Fig. 2C). Type B PutAs are monomeric (4) or exist as a concentration-dependent monomer-dimer equilibrium (8), with the dimer having a completely different quaternary structure from type A dimers (Fig. 2C). Type C PutAs form a third distinct type of dimer, which is mediated by the DNA-binding domain (19).
As part of our exploration of PutA structural diversity, we targeted 3B PutAs for analysis. Herein, we report the first structure of a class 3B PutA along with solution oligomeric state studies, kinetic characterization of substrate channeling, and domain deletion analysis probing the role of the C-terminal ALDHSF domain. The structure of the 3B PutA from Corynebacterium freiburgense (CfPutA) exhibits novel features, including an unexpected FAD conformation and extensive conformational disorder, resulting in a highly open PRODH active site. Sedimentation-velocity data show that CfPutA dimerizes in the presence of active-site ligands, identifying CfPutA as a potential morpheein enzyme. Transient time analysis and intermediate trapping assays are consistent with a substrate-channeling mechanism. Deletion of the C-terminal ALDHSF domain abrogates both catalytic activities, suggesting that it is essential for maintaining the integrity of the active sites. The results further highlight the diversity of the PutA family and provide insight into structural basis of hysteretic substrate channeling.
Results
Steady-state kinetic characterization of the PRODH and GSALDH activities of CfPutA
Purified CfPutA is a yellow solution, consistent with a flavoenzyme. The extent of FAD incorporation into the protein was determined. The extinction coefficient of CfPutA·FAD was experimentally estimated to be 13,100 m−1 cm−1. Measurement of the flavin spectrum after size exclusion chromatography indicated 0.3 mol of FAD/mol of CfPutA (30% incorporation). This value is lower than other PutAs, which have FAD incorporation ranging from 60% (20) to > 80% (7, 21, 22).
The steady-state kinetic parameters describing the PRODH activity were measured using the dichlorophenolindophenol assay. When proline is the variable substrate, the kinetic parameters are Km of 145 mm and kcat of 2.6 s−1, which corresponds to a catalytic efficiency (kcat/Km) of 18 m−1 s−1 (Table 1 and Fig. 3A). The PRODH catalytic efficiency of CfPutA is on the low end for PutAs, which have reported values of 30–400 m−1 s−1 (4, 20, 22–24).
Table 1.
Kinetic parameters for wild-type and mutant CfPutA
| Activity | Enzyme | Variable substrate | Km | kcat | kcat/Km |
|---|---|---|---|---|---|
| s−1 | m−1 s−1 | ||||
| PRODH | Wild type | Proline | 145 ± 16 mm | 2.6 ± 0.1 | 17.9 ± 0.1 |
| S916* | Proline | 0.20 ± 0.02a | |||
| Y927* | Proline | 0.20 ± 0.02a | |||
| GSALDH | Wild type | P5C | 54 ± 6 μm | 1.7 ± 0.1 | 32,000 ± 5,000 |
a Calculated from line of best fit over three data points (Fig. 3A).
Figure 3.
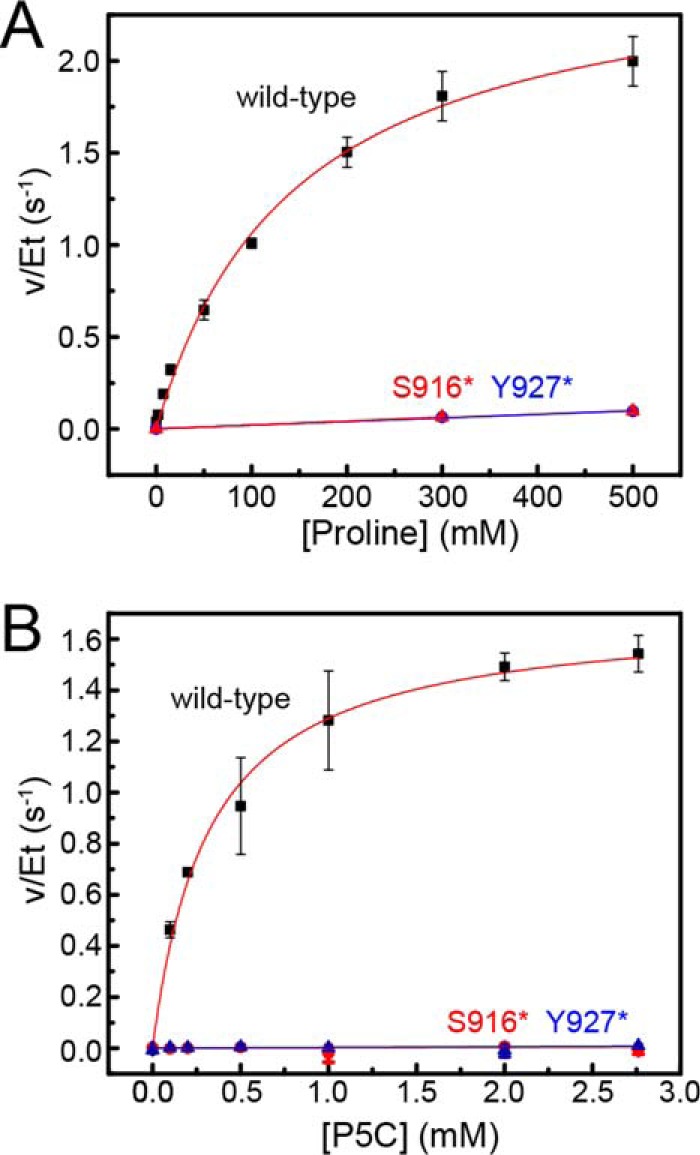
Steady-state kinetics data. A, dependence of PRODH activity on proline concentration. B, dependence of GSALDH activity on P5C concentration. For both panels, results are shown for wild-type CfPutA (black squares), CfPutA S916* (red triangles), and CfPutA Y927* (blue triangles). Fits to the Michaelis-Menten equation for wild-type CfPutA are represented by the red curves. Linear regression was used for the mutant variants to guide the eye and to estimate kcat/Km for PRODH activity. Analysis was done using Origin 2016.
The GSALDH activity of CfPutA using P5C as the variable substrate revealed a Km of 54 μm and a kcat of 1.7 s−1, corresponding to a catalytic efficiency of 32,000 m−1 s−1 (Table 1 and Fig. 3B). For reference, the kcat/Km values of other PutAs span the range of 4800–214,000 m−1 s−1 (4, 6). Thus, the GSALDH activity of CfPutA is not unusual.
Structure of CfPutA
Homolog screening of five class 3B PutAs identified CfPutA as amenable to crystal structure determination. The crystal structure of CfPutA was determined at 2.7 Å resolution in space group P1 (Table 2). It is the first structure of a class 3B PutA and the second of a PutA containing the C-terminal ALDHSF domain. Other PutAs represented in the PDB share < 30% sequence identity with CfPutA. Because of the low sequence identity to other PutAs, molecular replacement required the use of a search model based on a monofunctional GSALDH with only 33% identity to CfPutA over 415 residues (see “Experimental procedures”).
Table 2.
Data collection and refinement statistics
Values for the outer resolution shell of data are given in parentheses. RMSD, root mean square deviation.
| Triclinic | Trigonal | |
|---|---|---|
| Space group | P1 | P3121 |
| Unit cell parameters | ||
| Å | a = 85.0, b = 116.4, c = 140.7 | a = 145.6, c = 214.3 |
| Degrees | α = 71.5, β = 89.7, γ = 83.4 | |
| Wavelength | 1.000 | 1.000 |
| Resolution (Å) | 63.29 to 2.70 (2.75 to 2.70) | 63.07 to 3.30 (3.43 to 3.30) |
| Observations | 260,275 (12,837) | 434,766 (44,854) |
| Unique reflections | 137,220 (6798) | 40,228 (4491) |
| Rmerge(I) | 0.058 (0.488) | 0.177 (1.224) |
| Rmeas(I) | 0.082 (0.689) | 0.186 (1.291) |
| Rpim(I) | 0.058 (0.485) | 0.056 (0.406) |
| Mean I/σ | 8.3 (1.7) | 14.4 (2.1) |
| Mean CC½ | 0.994 (0.620) | 0.997 (0.742) |
| Completeness (%) | 98.5 (98.1) | 100.0 (100.0) |
| Multiplicity | 1.9 (1.9) | 10.8 (10.8) |
| No. of atoms | 29,652 | |
| Protein | 29,280 | |
| FAD | 212 | |
| NAD+ | 140 | |
| Sulfate ion | 20 | |
| Rwork | 0.183 (0.273) | |
| Rfreea | 0.236 (0.349) | |
| RMSD bond lengths (Å) | 0.009 | |
| RMSD bond angles (degrees) | 1.053 | |
| Ramachandran plotb | ||
| Favored (%) | 97.20 | |
| Outliers (%) | 0.00 | |
| Clashscore (PR)b | 5.05 (100) | |
| MolProbity score (PR)b | 1.68 (100) | |
| Average B-factor (Å2) | ||
| Protein | 51.5 | |
| FAD | 56.0 | |
| NAD+ | 60.3 | |
| Sulfate ion | 74.1 | |
| Coordinate error (Å)c | 0.38 | |
| PDB entry | 5UX5 | |
a 5% test set.
b From MolProbity. The percentile ranks (PR) for Clashscore and MolProbity score are given in parenthesis.
c Maximum likelihood-based coordinate error estimate reported by phenix.refine.
Despite the low sequence identity to other PutAs, CfPutA resembles other PutAs in both the folds and spatial arrangement of the catalytic modules (Fig. 4A). For example, analysis of the PDB with PDBeFold (25) identified GsPutA (31% identical) as the closest structural neighbor of CfPutA, with a root mean square deviation of 1.8 Å over 941 residues. The similarity of CfPutA to SmPutA (27% identity) is lower, with a root mean square deviation of 2.6 Å over 1208 residues.
Figure 4.

Structure of the CfPutA monomer and comparison to SmPutA. A, backbone and surface representations of CfPutA. The domains have different colors, with the N-terminal arm in orange, PRODH barrel in cyan, PRODH-GSALDH linker in violet, GSALDH NAD+-binding in red, GSALDH catalytic in blue, and C-terminal ALDHSF in gold. The silver surface represents the substrate-channeling tunnel. The FAD is shown in yellow sticks. The dotted lines indicate disordered residues 33–118 (α-domain) and 421–434 (which connect α8 to the PRODH-GSALDH linker). The sites of domain deletion mutations are indicated as 916* and 927*. B, backbone and surface representations of SmPutA. The domains are colored as in A. The α-domain, which is disordered in CfPutA, is colored green.
Like other PutAs, CfPutA contains a PRODH module in the N-terminal half of the polypeptide chain and a GSALDH module in the C-terminal half (Fig. 4A). The core of the PRODH module is a distorted (βα)8 barrel (residues 130–420) containing the FAD bound at the C termini of the strands of the barrel. The GSALDH module features the two-domain architecture common to all ALDHs. The GSALDH module consists of a Rossmann fold NAD+-binding domain (residues 475–689 and 891–908) and an α/β catalytic domain (residues 690–890), which contains catalytic Cys-723.
As in other PutAs, the PRODH and GSALDH active sites of CfPutA are separated by a linear distance of 45 Å and connected by a curved tunnel that traverses 65 Å and ranges in radius from 1.5 to 4.5 Å (Fig. 4A). The dimensions of the tunnel are consistent with those found in other PutA structures, such as SmPutA (Fig. 4B).
CfPutA also has a domain following the GSALDH module (Fig. 2A). This domain was previously called the C-terminal domain of unknown function, but here we use the term ALDHSF domain in reference to the recently discovered structural similarity to ALDHs (8).
The C-terminal ALDHSF domain (residues 909–1069) consists of an α/β subdomain and protruding β-flap (Fig. 5A). The α/β subdomain resembles the Rossmann dinucleotide-binding domain, yet it does not bind NAD+ in CfPutA or SmPutA. The β-flap resembles the oligomerization domain found in many ALDHs, but it does not mediate oligomerization in CfPutA or SmPutA. Instead, the β-flap appears to facilitate substrate channeling by covering a large section of the tunnel that connects the two active sites, thereby preventing leakage of the intermediate (Fig. 4).
Figure 5.

Comparison of the C-terminal ALDHSF domains of CfPutA and SmPutA. A, the ALDHSF domain of CfPutA. The truncation points for the S916* and Y927* C-terminal deletion mutants are marked by arrowheads. B, the ALDHSF domain of SmPutA. In both panels, the diagram shows the topology of the Rossmann fold part of the ALDHSF domain.
The ALDHSF domains of CfPutA and SmPutA differ in two respects. First, whereas the α/β subdomain of SmPutA is a complete Rossmann domain consisting of five parallel β-strands and three α-helices (Fig. 5B), CfPutA has an abbreviated Rossmann fold, which lacks β3 and its flanking helices (Fig. 5A). Second, the CfPutA ALDHSF domain has an extra β-strand at its N terminus, which forms an interdomain two-stranded antiparallel β-sheet with the NAD+-binding domain (Fig. 5A).
Disorder in the PRODH module
The CfPutA structure is unique for what is unobserved. Electron density for residues 33–118 and 421–434 was very weak in all four chains in the crystallographic asymmetric unit and prevented the inclusion of these residues in the structure (Fig. 4A, dotted lines).
Residues 33–118 correspond to the α-domain, which is a bundle of 4–6 α-helices in other PutAs. As shown for SmPutA, the α-domain contacts the PRODH barrel, GSALDH catalytic domain, and ALDHSF β-flap (Fig. 4B). Thus, the α-domain may be a coupling domain that helps maintain the proper spacing and orientation of the two active sites.
The second disordered section (residues 421–434) is the beginning of the linker that connects the PRODH barrel to the GSALDH module (Fig. 4A). The disordered section starts just after helix α8 of the PRODH barrel. Helix α8 is notable because it contains several conserved residues that interact with the substrate proline in the Michaelis complex (6, 16, 26) Furthermore, α8 has been shown to move in response to proline binding, giving rise to open (proline-free) and closed (proline-bound) PRODH conformations (6, 26).
Because of the disordered sections, the PRODH site of CfPutA is more open and the FAD is more exposed than in other PutAs (Fig. 4). For example, the reactive surface of the isoalloxazine (si face) is visible in a surface rendering of CfPutA (Fig. 4A) and hidden in SmPutA (Fig. 4B). Analysis by PDBePISA shows the solvent-accessible surface area of the FAD in CfPutA is 179 Å2, which corresponds to 19% of the total area of the free FAD. Thus, the FAD is 81% buried in CfPutA. For comparison, the FAD is 92% buried in the PRODH-open state of GsPutA (PDB code 4NM9) and 95% buried in PRODH-closed states of GsPutA and SmPutA (PDB codes 4NMA and 5KF6; Fig. 4B). Thus, the CfPutA structure reveals a new, more open conformation of the PutA PRODH active site.
New FAD conformation
The FAD in CfPutA has an unexpected conformation, which could be related to the disorder in the surrounding protein. The ribityl 2′-OH group is under the pyrimidine ring of the isoalloxazine, whereas the 3′-OH is directed toward the FAD ribose, and the 4′-OH sits under the dimethylbenzene ring (Fig. 6A). In this conformation, the 2′-OH donates a hydrogen bond to the FAD N1 atom, whereas the 3′-OH forms a hydrogen bond with the FAD ribose. Curiously, this ribityl conformation is diagnostic of the 2-electron-reduced FAD in PutA (Fig. 6B) (6, 18), yet reducing agent was not added during crystallization of CfPutA or cryoprotection of the crystals. We note the ribityl conformation expected for the oxidized PutA FAD is clearly different (Fig. 6C). Interestingly, bona fide PutA FAD reduction results in strong butterfly bending of the isoalloxazine ring system (Fig. 6B, inset). Electron density suggests the isoalloxazine of CfPutA is planar, indicative of the oxidized state (Fig. 6A, inset). In summary, the FAD in CfPutA exhibits the ribityl chain conformation of a reduced PutA FAD, whereas the isoalloxazine conformation is indicative of an oxidized FAD.
Figure 6.
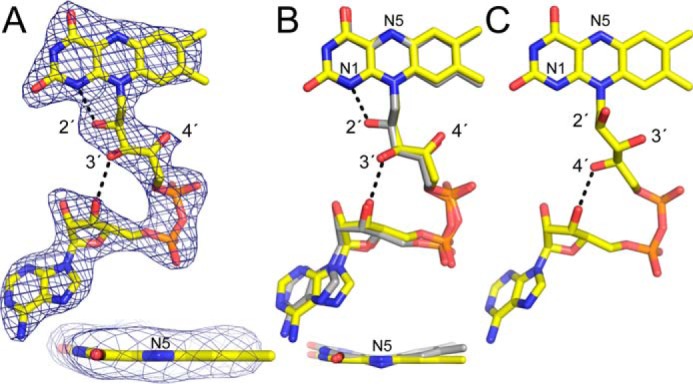
Unexpected FAD conformation. A, 2.7 Å resolution electron density for the FAD in CfPutA. The cage represents a simulated annealing σA-weighted Fo − Fc omit map (3.0σ). The inset shows an edge-on view of the isoalloxazine ring. B, superposition of the FAD in CfPutA (yellow) and the 2-electron-reduced FAD in GsPutA (gray). The inset shows an edge-on view of the isoalloxazine ring. Note that the two FADs have the same ribityl conformation but different butterfly bend angles. C, a typical oxidized PutA FAD (from GsPutA). Note that the ribityl conformation differs from that in B.
Oligomeric state in solution
Sedimentation-velocity experiments were performed on CfPutA to further investigate whether the oligomeric state is conserved across type B PutAs. A sedimentation-velocity study conducted at 0.8 mg/ml (6.7 μm) revealed one major peak with an apparent sedimentation coefficient of 4.4 S, corresponding to ∼129 kDa (Fig. 7A). This molecular mass is within 8% of the predicted mass of a CfPutA monomer of 119 kDa.
Figure 7.
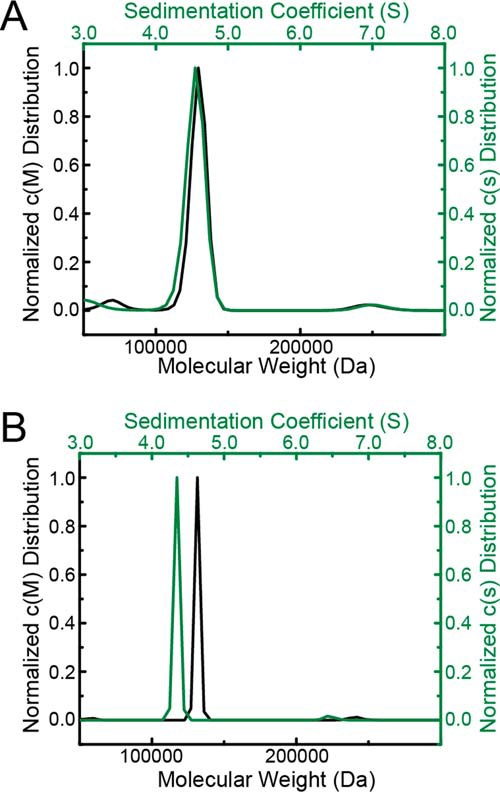
Sedimentation-velocity analysis of CfPutA in the absence of active-site ligands. A, sedimentation coefficient and molecular mass distributions for CfPutA at 0.8 mg/ml (7 μm). B, sedimentation coefficient and molecular mass distributions for CfPutA at 6 mg/ml (50 μm).
A sedimentation-velocity experiment was performed at the higher concentration of 6 mg/ml (50 μm) to check for concentration-dependent oligomerization, as found with SmPutA (8). This analysis also revealed a primary peak at 4.35 S corresponding to ∼130 kDa (Fig. 7B). Taken together, these sedimentation-velocity results suggest that CfPutA is primarily monomeric under the conditions used.
Crystal structure data are consistent with the sedimentation-velocity results. Analysis of protein-protein interfaces in the P1 crystal lattice with PDBePISA revealed no stable assemblies. The largest interface is a two-body assembly formed by chains A and B, which has an interfacial area of 1100 Å2 (Fig. 8A). For reference, the validated dimer of SmPutA (also a type B PutA) has an interface area of 1800 Å2 (Fig. 8C). Similarly, the crystal packing in a low-resolution trigonal CfPutA crystal form (Table 2) implies no stable interfaces; the largest interface in that crystal form buries 1050 Å2 (Fig. 8B). Furthermore, the two CfPutA crystal forms do not share a common assembly, and the SmPutA dimer is not present in either CfPutA lattice. All of these observations are consistent with CfPutA being predominantly monomeric under the solution conditions used in centrifugation and crystallization.
Figure 8.
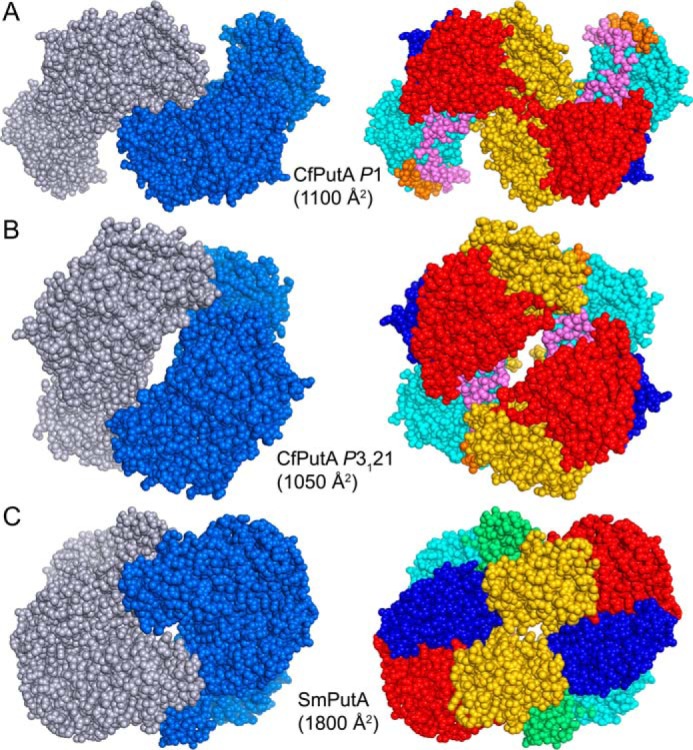
Dimeric assemblies of CfPutA and SmPutA. A, the largest protein-protein interface in the CfPutA P1 crystal form. B, the largest protein-protein interface in the CfPutA trigonal crystal form. C, the bona fide dimer of SmPutA, which has been validated by SAXS and crystallography (PDB code 5KF6). The interfacial surface areas are listed for each assembly. On the left side, the chains have different colors. On the right side, the domains are colored as in Fig. 4: N-terminal arm (orange), α-domain (green), PRODH barrel (cyan), PRODH-GSALDH linker (violet), NAD+-binding (red), GSALDH catalytic (blue), and C-terminal ALDHSF (gold).
Ligand-induced protein dimerization
CfPutA was crystallized with NAD+ in the GSALDH active site and the PRODH proline site unoccupied. We hypothesized that the lack of a PRODH ligand could be responsible for the unprecedented level of disorder observed in the crystal structure near the PRODH active site. To explore this idea, we investigated the solution properties of CfPutA using the proline analog l-tetrahydro-2-furoic acid (THFA), a well-known competitive inhibitor of PRODHs (6, 8, 16, 23, 26). Surprisingly, these studies suggest that THFA may enhance dimerization of CfPutA.
Sedimentation-velocity experiments were used to monitor ligand-induced self-association. Ligand-free CfPutA analyzed at the relatively high protein concentration of 50 μm (6 mg/ml) showed a single peak corresponding to the monomer (Fig. 9, top). In the presence of the THFA (10 mm) and NAD+ (1 mm), a second peak is visible corresponding to a species with an apparent sedimentation coefficient of 5.5–5.6 S. As the protein concentration is raised from 14 to 84 μm, the secondary peak grows while the monomer peak diminishes (Fig. 9, bottom four panels). At 84 μm, the peak with the larger apparent sedimentation coefficient (∼5.5 S) corresponds to a molecular mass of 232.6 kDa, which is within 2% of the theoretical mass of a dimer (238.5 kDa). Overall, these data suggest the oligomeric state of CfPutA in solution is influenced by both the occupancy of the proline substrate site and protein concentration.
Figure 9.
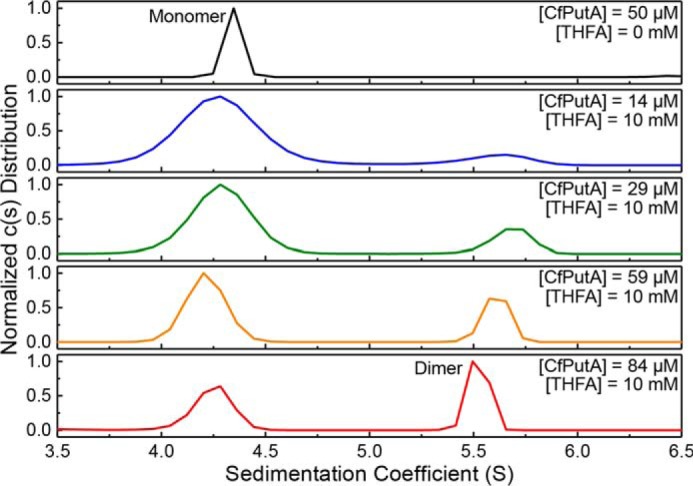
CfPutA undergoes concentration-dependent oligomerization in the presence of active-site ligands. Sedimentation-velocity experiments were performed at the indicated CfPutA concentrations in the absence (top panel) or presence (bottom four panels) of the proline analog THFA (10 mm) and NAD+ (1 mm).
CfPutA exhibits a substrate-channeling mechanism
The observation of a tunnel in the structure motivated studies of substrate channeling. To test whether CfPutA is capable of substrate channeling, a P5C trapping assay was employed. This experiment monitors the formation of a yellow conjugate formed between the intermediate P5C and ortho-aminobenzaldehyde (oAB). Experiments performed in the absence of NAD+ provide the upper limit for the build up of P5C in the bulk solvent. Conversely, experiments performed in the presence of NAD+ should show reduced P5C-oAB conjugate formation if P5C moves to the GSALDH active site without leaving the enzyme. Results of this assay on CfPutA show a 52% decrease in steady-state P5C-oAB conjugate formation in the presence of NAD+, consistent with substrate channeling (Fig. 10A).
Figure 10.
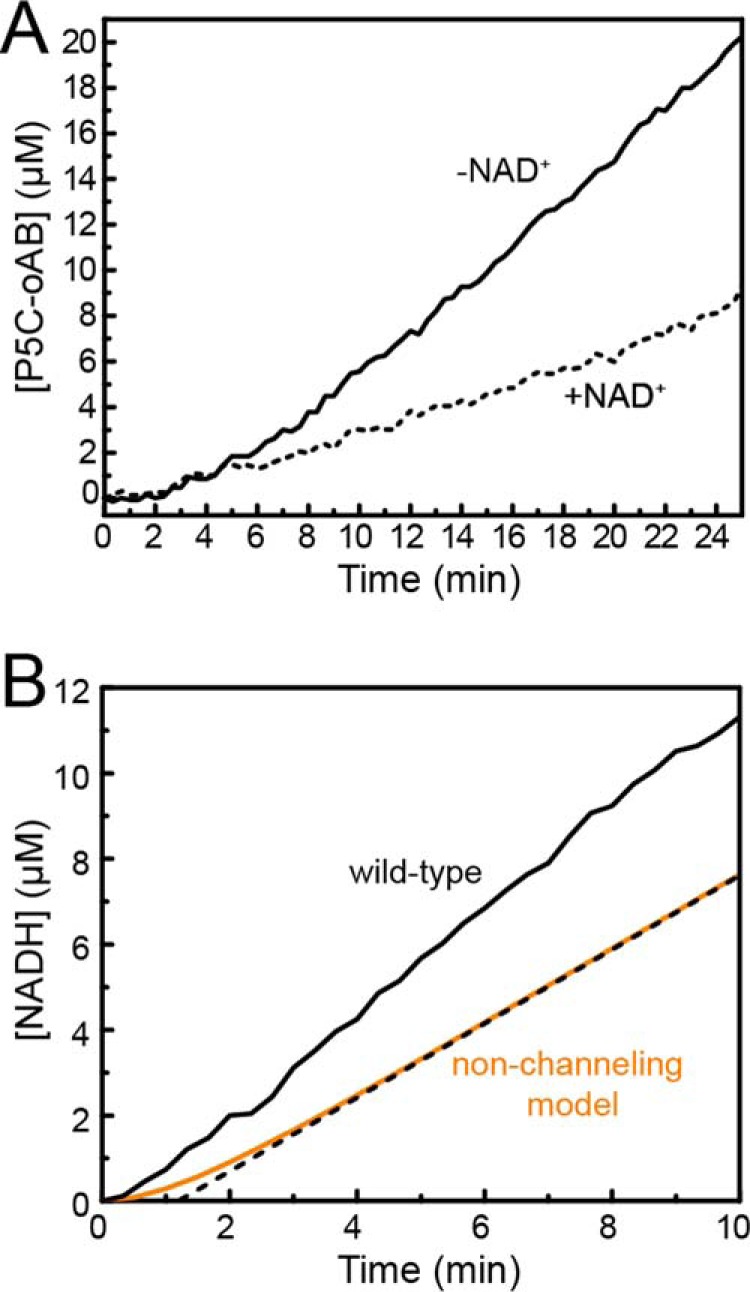
Evidence for substrate channeling in CfPutA. A, P5C-oAB-trapping experiments performed in the absence (solid lines) or presence (dashed lines) of NAD+. B, transient-time analysis of CfPutA. Experimental data from a PRODH-GSALDH-coupled assay are represented in solid black. A non-channeling model calculated from Equation 1 is represented in orange. The dotted black line represents extrapolation of the predicted steady-state rate to estimate a transient time of 1.2 min.
To further investigate whether CfPutA channels substrate, production of NADH was monitored after the addition of proline to determine whether a lag phase occurs, indicative of a non-channeling system (transient time analysis). For wild-type CfPutA, the production of NADH versus time is approximately linear with no substantial lag phase (Fig. 10B). The transient time, τ, for two non-interacting enzymes can be estimated from the free-diffusion model in Equation 1. The calculated theoretical value of τ for CfPutA is 1.2 min (Fig. 10B). A lag time of this magnitude is not evident in the experimental data, which is consistent with a substrate-channeling mechanism (Fig. 10B). Taken together, the P5C-trapping assays and transient time analysis suggest that CfPutA exhibits substrate channeling.
The C-terminal ALDHSF domain is necessary for PRODH and GSALDH activity
Although the structure of the C-terminal ALDHSF domain is now known for two PutAs, its functions have not been fully determined. To gain insight into the functions of this domain, two domain deletion mutants were generated by introducing premature stop codons at Ser-916 (S916*) or Tyr-927 (Y927*) (Fig. 5A).
Because domain deletion is dramatic, a fluorescence-monitored thermal shift assay was used to qualitatively assess whether deletion of the C-terminal domain affects the thermostability of the protein. Wild-type CfPutA displayed an average T0.5 of 47.6 ± 0.1 °C (range of 47.5–47.7 °C; Fig. 11). CfPutA S916* and Y927* mutants displayed a similar unfolding profile with T0.5 values of 48.4 ± 0.2 °C (range of 48.1–48.6 °C) and 48.9 ± 0.2 °C (range of 48.7–49.2 °C), respectively (Fig. 11). This result suggests that the C-terminal truncation mutants display apparent thermostability similar to that of the wild-type protein, consistent with similarly folded proteins. Overall, the thermal shift assay results suggest that the C-terminal domain truncation mutants probably retain significant native structure.
Figure 11.
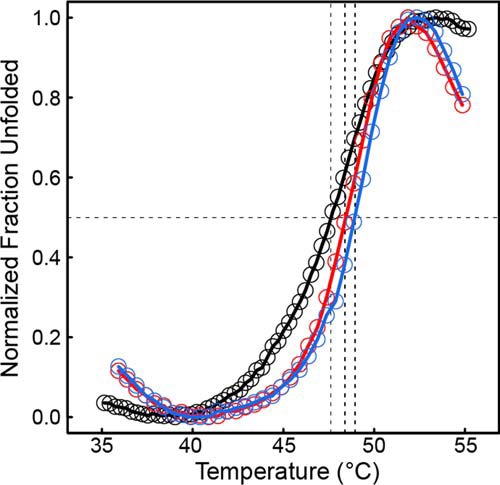
Deletion of the ALDHSF domain does not result in apparent destabilization of CfPutA tertiary structure. Thermal shift assay melt curves are shown for wild-type CfPutA (black), CfPutA S916* (red), and CfPutA Y927* (blue). The horizontal black dashed line represents 0.5 of the total fraction unfolded. The three vertical dashed lines indicate the T0.5 of each protein. The plotted curves are the average of samples measured in triplicate normalized to fraction unfolded.
Steady-state kinetic assays were performed with both truncation mutant variants. PRODH activity was severely decreased in both. Saturation with proline was not possible, indicating a very high Km (Fig. 3A). Estimation of kcat/Km using the linear approximation suggested a 100-fold decrease in catalytic efficiency (Table 1). The impact of the deletion on GSALDH activity was more severe; neither mutant displayed perceptible GSALDH activity under the same reaction conditions as wild type (Fig. 3B).
Discussion
Our report brings the number of PutA structures to four (Fig. 2B). All three branches are now represented in the PDB, and only the class 1C structure remains unsolved in its full-length form (Fig. 2A). However, the catalytic part of 1C PutA should be very similar to that of SmPutA, based on the high sequence identity (60%) and the fact that the 1C EcPutA PRODH module structure has the conserved (βα)8 barrel fold described here (15).
This catalogue of structures reveals features conserved by the entire PutA family. Although sequence identity between PutAs can be as low as 30%, we now see that the folds of the catalytic modules and dimensions of the intervening tunnel are highly conserved. Also, the two type B PutA structures reveal a shared fold for the C-terminal ALDHSF domain, although the branch 3 ALDHSF domain has a somewhat abbreviated Rossmann subdomain (Fig. 5).
Interestingly, the Conserved Domain Database of NCBI (27), as implemented in BLASTP (28), fails to recognize the C-terminal domain of CfPutA as an ALDHSF domain. Presumably, this is due to the absence of Rossmann β-strand 3 and its flanking helices (Fig. 5A). Nevertheless, the crystal structure supports annotation of the C-terminal domain of CfPutA as an ALDHSF domain. In conclusion, the C-terminal domains of both class 1B and 3B PutAs exhibit the ALDHSF fold (Fig. 5).
The C-terminal ALDHSF domain is the least understood PutA domain, because it contains neither catalytic residues nor substrate-binding sites. We showed here that the ALDHSF domain of CfPutA is critical for both PRODH and GSALDH functions. Essentiality of the ALDHSF domain for GSALDH activity is consistent with the fact that the domain makes intimate contact with the GSALDH module (Fig. 4). Its importance for PRODH activity is more surprising. Because deletion of the ALDHSF domain in CfPutA nearly eliminates PRODH activity, the ALDHSF domain must play a role in promoting the activity of the PRODH active site, presumably by stabilizing the PRODH module via tertiary structural interactions. We note the ALDHSF β-flap does make significant contacts with the PRODH barrel (Fig. 4A).
Comparison of type A and B PutA structures reveals an interesting example of structural mimicry whereby the quaternary structure of one protein (type A PutA) resembles the tertiary structure of another protein (type B PutA). As described previously for SmPutA, the β-flap of the C-terminal ALDHSF domain of type B PutA is structurally similar to the oligomerization flap of type A PutAs (8). In the type B PutA monomer, the ALDHSF β-flap contacts both the PRODH barrel and the GSALDH catalytic domain and also forms a lid that seals the substrate-channeling tunnel (Fig. 4). We showed here using domain deletion analysis that these interactions are apparently needed for both catalytic functions. In type A PutAs, a conserved domain-swapped dimer interface provides analogous structural interactions, thereby enabling catalytic activity in both active sites and substrate channeling in the absence of the ALDHSF domain.
Although CfPutA has the conserved PutA fold, it has unique local structural features. The FAD has an unexpected ribityl conformation, and there is unprecedented in crystallo disorder in the α- and PRODH-barrel domains, resulting in unusually high exposure of the FAD. In contrast, the GSALDH module and active site with NAD+ bound exhibited no disorder and could be completely modeled. Because the PRODH active site is disassembled, we hypothesize that the crystal structure may represent an inactive conformation.
Given the peculiarities of the CfPutA structure (disorder, unusual FAD conformation), it is notable that the enzyme exhibits normal catalytic properties, implying that the active conformation is achieved in solution in the presence of substrates. These results may provide insight into the hysteretic substrate-channeling mechanism of PutA. A previous study of EcPutA showed that the substrate-channeling step is slowest during the first turnover and speeds up 40-fold with subsequent turnovers, implying that channeling is activated (5). Although the structural basis for activation is unknown, it has been proposed that isomerization associated with ligand binding (most likely proline) provokes conformational changes that assemble the PRODH active site and seal leaky parts of the tunnel system (5). The structure reported here could resemble PutA prior to activation.
Importantly, biophysical analysis of CfPutA has expanded the oligomeric diversity of PutA proteins. Previous studies revealed a range of oligomeric states: a monomeric class 1B PutA (4); three different types of dimers in PutA classes 2A (6), 1B (8), and 1C (19); and a tetrameric 1A PutA (2). A recent study of SmPutA, another type B PutA, revealed the first concentration-dependent oligomerization of a PutA (8). Here, we showed that CfPutA is primarily monomeric but is capable of concentration-dependent dimerization in the presence of active site-associated ligands. This result suggests that occupancy of the proline substrate site in the PRODH module may encourage dimerization. This example would add to the list of proteins that experience ligand-induced oligomerization.
The concept of ligand-induced changes in oligomerization is not novel. For example, ligand-induced dimerization of human phenylalanine hydroxylase, the enzyme that catalyzes the first step in the l-phenylalanine to l-tyrosine biosynthetic pathway, was recently reported (29). Further, multiple oligomeric states of DegP, an E. coli periplasmic protease, ranging from a resting hexamer to active higher-order oligomers, are observed in response to various substrate-mimicking peptide fragments (30). Likewise, substrate binding to the catalytic site of porphobilinogen synthase causes conformational changes in the monomeric enzyme allowing for preferential formation of a more active octamer over the less active hexameric form (31). The binding of the substrates to the AMP biosynthetic enzyme adenylosuccinate synthetase decreases the dissociation constant for dimerization by several orders of magnitude (32). Also, ligand-induced dimerization is thought to be a key mechanism in the regulation of MERS coronavirus nsp5 protease activity during virus infection (33). Overall, ligand-induced oligomerization evidently plays an important role in enzyme function, regulation, and stability across kingdoms.
In several of these examples, ligand binding induces formation of a high-activity oligomer, a phenomenon referred to by Jaffe (34, 35) as morpheein equilibrium. Interestingly, the morpheein concept has been used to explain enzyme hysteresis, a form of allosteric regulation that occurs in at least one PutA (5). Whether the morpheein behavior of CfPutA is related to the hysteresis of substrate channeling remains to be investigated.
It is not apparent whether ligand-induced dimerization is specific to CfPutA, branch 3 PutAs, or architecture type B PutAs in general. The other type B PutAs that have been studied are either monomeric (4) or form a monomer-dimer equilibrium (8). Whether ligand binding similarly promotes oligomerization of these PutAs remains to be investigated, as is the underlying functional significance of the phenomenon, vis-à-vis hysteresis.
The quaternary structure of the ligand-bound CfPutA dimer, which is currently unknown, may provide additional information regarding the role of the C-terminal ALDHSF domain. It is clear from analysis of the crystal structures of both CfPutA and SmPutA that the C-terminal ALDHSF domain provides a crucial seal for the tunnel necessary for substrate channeling (Fig. 4). Additionally, the SmPutA dimer shows a novel self-association not seen in any other PutA or ALDHSF enzyme (Fig. 2C). It remains to be investigated whether the CfPutA dimer resembles the SmPutA dimer. If so, this dimer would probably be a hallmark of type B PutAs.
Experimental procedures
Construct generation and site-directed mutagenesis
The gene encoding CfPutA (1074 residues), codon-optimized for expression in E. coli, was synthesized (Bio Basic Inc.) in a pUC57 vector backbone. The synthetic construct includes a 5′ NdeI restriction site, the coding sequence of an 8× His tag, a tobacco etch virus protease site in frame with the CfPutA coding sequence, and a 3′ XhoI restriction site. For protein expression, the construct was subcloned into the pET22b vector backbone.
The CfPutA C-terminal truncation constructs (S916* and Y927*; see Fig. 5A) were generated by introduction of a premature stop codon by site-directed mutagenesis using the QuikChange II XL site-directed mutagenesis kit (Agilent).
Protein expression and purification
Protein expression vectors were transformed into BL21(DE3) cells (Thermo Fisher Scientific) for protein expression. A 20-ml overnight starter culture grown from a single colony was used to inoculate 1 liter of LB supplemented with 100 μg/ml ampicillin and 15 mm riboflavin. Cultures were shaken at 37 °C and 250 rpm until A600 ∼ 0.8. The temperature was then shifted to 18 °C, and protein expression was induced with 0.5 mm isopropyl β-d-1-thiogalactopyranoside. Cultures were shaken overnight. The next day, the cells were pelleted; resuspended in a buffer containing 50 mm Tris, pH 7.8, 300 mm NaCl, and 5% (v/v) glycerol; quick-cooled in liquid nitrogen; and placed at −80 °C for storage.
For purification, the cell suspension was thawed and lysed by sonication in the presence of 0.5 mm phenylmethylsulfonyl fluoride and 0.1 mm FAD. Unbroken cells and cell debris were pelleted by centrifugation at 16,000 rpm for 1 h at 4 °C in an SS-34 rotor. The supernatant was then loaded onto a HisTrap HP nickel-affinity column pre-equilibrated with 50 mm Tris, pH 7.8, 300 mm NaCl, and 5% (v/v) glycerol. Protein was eluted with 300 mm imidazole. Fractions containing protein were pooled and supplemented with 0.1 mm FAD and 0.5 mm Tris(hydroxypropyl)phosphine (THP). The His tag was removed by adding 1 mg of tobacco etch virus protease per 30 mg of target protein, incubating for 2 h at 28 °C, and then dialyzing overnight at 4 °C against buffer containing 50 mm Hepes, pH 7.8, 300 mm NaCl, and 5% (v/v) glycerol. The sample was then passed over a HisTrap HP nickel-affinity column. The eluate from the column was collected, supplemented with 0.1 mm FAD, and dialyzed overnight at 4 °C against a buffer containing 50 mm Tris, pH 7.8, 0.5 mm EDTA, and 5% (v/v) glycerol.
The following day, the sample was further purified using a HiTrap Q anion-exchange column. Protein was eluted with a linear gradient of NaCl from 0 to 1 m. Fractions containing CfPutA were pooled, supplemented with 0.1 mm FAD, and loaded onto a Superdex 200 16-600 size-exclusion column in the presence of 50 mm Tris, pH 7.8, 100 mm NaCl, 0.5 mm EDTA, 1 mm THP, and 5% (v/v) glycerol. Fractions were pooled and concentrated. Protein concentrations were estimated by the bicinchoninic acid method (Pierce). For analytical ultracentrifugation analysis of ligand-free CfPutA, the protein was concentrated and used immediately. For analytical ultracentrifugation analysis of the CfPutA·THFA·NAD+ complex, eluted protein was supplemented with 0.2 mm FAD, 1 mm NAD+, and 10 mm THFA at pH 8.0 and dialyzed for 4 h with two buffer exchanges against a buffer containing the same components. For all other applications, including crystallization and steady-state kinetics, the protein was quick-cooled in liquid nitrogen in PCR-strip tubes and stored at −80 °C.
Crystallization of CfPutA complexed with NAD+ in space group P1
Homolog screening was performed in search of a 3B PutA that was amenable to crystal structure determination. In addition to CfPutA, the PutAs from Helicobacter pylori, Helicobacter hepaticus, Corynebacterium jeikeium, and Cellulomonas cellasea were screened. Only CfPutA was crystallizable.
Triclinic crystals of CfPutA were grown by sitting-drop vapor diffusion at 20 °C. Sparse matrix screening using commercially available crystallization screens was conducted using purified CfPutA concentrated to 12 mg/ml and supplemented with 10 mm NAD+. The screens produced several crystallization hits in the form of microcrystals. Results from optimization trials led to thin needles/plates grown in 0.2 m magnesium chloride hexahydrate, 0.1 m BisTris, pH 6.5, and 25% (w/v) polyethylene glycol (PEG) 3350. This condition was further optimized by combining it with other crystal screen reagents, essentially using the crystal screen kits as additive screens. The final crystallization reservoir was composed of 3 parts of solution A (0.2 m magnesium chloride hexahydrate, 0.1 m BisTris, pH 6.5, 25% (w/v) PEG 3350) and 1 part of solution B (0.1 m HEPES, pH 7.5, 10% (w/v) PEG 6000, 5% (v/v) (±)-2-methyl-2,4-pentanediol). The crystals were cryoprotected using the reservoir supplemented with 20% (v/v) PEG 200, picked up with nylon loops, and quick-cooled in liquid nitrogen. Approximately 120 crystals were surveyed at beamline 4.2.2 of the Advanced Light Source. Only one crystal diffracted significantly beyond 3.0 Å, from which a 2.70 Å resolution data set was obtained for structure determination (Table 2). The space group is P1 with the unit cell dimensions shown in Table 2. The asymmetric unit contains four protomers, corresponding to a Matthews coefficient of 2.7 Å3/Da and 55% solvent (36). We note that surface mutagenesis or the inclusion of THFA failed to improve crystallization of CfPutA.
Crystallization of CfPutA in space group P3121
The crystal screening trials also produced crystals shaped like hexagonal bipyramids. These crystals were grown by sitting-drop vapor diffusion at 20 °C using CfPutA concentrated to 24 mg/ml. Crystals were obtained from a reservoir solution containing 0.5 m ammonium sulfate, 0.1 m sodium citrate tribasic dihydrate, pH 5.6, and 1.0 m lithium sulfate monohydrate. The crystals were cryoprotected in the crystallization reservoir supplemented with 20% (v/v) (±)-2-methyl-2,4-pentanediol, picked up with nylon loops, and quick-cooled by plunging into liquid nitrogen. Approximately 80 crystals were surveyed at beamline 4.2.2 of the Advanced Light Source. Although the external morphology and typical crystal size of this form were more promising than those of the P1 form, the diffraction was limited to 3.3–3.5 Å resolution. A 3.3 Å resolution data set was collected for the purpose of inspecting oligomeric assemblies. The space group is P3121 with unit cell dimensions a = 146 Å and c = 214 Å. The asymmetric unit contains one CfPutA monomer, which corresponds to a Matthews coefficient of 5.5 Å3/Da and 78% solvent (36).
X-ray diffraction data collection, phasing, and refinement
X-ray diffraction data were collected on Advanced Light Source beamline 4.2.2 at Lawrence Berkeley National Laboratory using a Taurus-1 detector in shutterless mode. All data sets were integrated and scaled with XDS (37). Intensities were merged and converted to amplitudes with Aimless (38). Data-processing statistics for the two crystal forms are listed in Table 2.
Crystallographic phasing by experimental methods was anticipated to be challenging because of the difficulty in reproducing diffraction quality crystals. Phasing by molecular replacement was also challenging due to the low sequence identity of CfPutA with other PutA structures in the PDB (<30%). Nevertheless, phasing by molecular replacement was attempted.
The amino acid sequence of CfPutA and the P1 structure factor amplitudes were input to the BALBES automated molecular replacement pipeline server (39). BALBES (version 1.0.0.Nov_16_2011) generated search models from the structures of BjPutA (PDB code 3HAZ), three bacterial GSALDHs (PDB codes 1UZB, 3QAN, and 3RJL), human succinic semialdehyde dehydrogenase (PDB code 2W8N), the EcPutA PRODH domain (PDB code 4O8A), and a few structures unrelated to PutA. The best solution was obtained with 3QAN and had a Q-factor of 0.55, and 85% probability of being correct. We note that the sequence identity between 3QAN and CfPutA is only 33% over 415 residues, and the search model represented only 46% of CfPutA. Refinement of the molecular replacement solution using REFMAC (40) (as part of the BALBES pipeline) produced a model with Rwork of 0.46 and Rfree of 0.49.
The phases were improved by density modification and automated model building using PHENIX (41). The map coefficient file from BALBES/REFMAC was input to phenix.autobuild for automated model building with density modification. The transformations needed for non-crystallographic symmetry averaging were calculated from the BALBES/REFMAC model (phenix.autobuild input_ncs_file option). The BALBES/REFMAC model was not supplied as the starting model for automated model building, which required ab initio model building in PHENIX. The resulting model from phenix.autobuild included 3184 residues of an expected 4304 (74% complete) and had Rwork of 0.26 and Rfree of 0.31. This model and the corresponding 2Fo − Fc map coefficients were input to phenix.autobuild for a second round of automated building and density modification (rebuild_in_place = True). The resulting model had Rwork of 0.24 and Rfree of 0.31.
The second-generation autobuild model was the starting point for several rounds of model building in COOT (42) and refinement in PHENIX (43). Non-crystallographic symmetry restraints were used during refinement. The B-factor model consisted of TLS refinement (one group/chain) plus an isotropic B-factor for each non-hydrogen atom. FAD and NAD+ were modeled in all four chains with occupancies of 1.0. Electron density for the nicotinamide of NAD+ was weak and diffuse, so this group was not included in the deposited structure. Disorder of the nicotinamide half of the cofactor has been observed in other GSALDH structures (44–46). Electron density maps also indicated the presence of a sulfate ion in the aldehyde site of the GSALDH module. The ion was included in all four chains at an occupancy of 1.0. The binding of sulfate to the aldehyde site has been observed in other GSALDH structures (2, 45). Due to the moderate resolution, water molecules were not modeled. Structure quality was validated using MolProbity (47) and the PDB (48) validation server. Refinement statistics for the final model are listed in Table 2.
Phases for the P3121 data set were obtained by molecular replacement with Phaser (49) using a search model consisting of a monomer from the final P1 structure. A solution with log-likelihood gain of 9000 was obtained. Refinement in PHENIX resulted in Rwork of 0.19 and Rfree of 0.22.
Analytical ultracentrifugation
Sedimentation-velocity experiments were performed at 20 °C in a Beckman XL-I analytical ultracentrifuge using an An50Ti rotor. Aliquots of protein and reference buffer were loaded into a sedimentation-velocity cell equipped with a dual-sector charcoal-Epon centerpiece. The reference buffer used for studies of the ligand-free protein was an eluted fraction lacking protein, as determined by A280, obtained from size-exclusion chromatography of the protein sample. The reference buffer for centrifugation of CfPutA in the presence of active-site ligands was the buffer from dialysis. Following a 2-h temperature equilibration, the sample was centrifuged at 35,000 rpm. The radial distribution of the sample was monitored with Rayleigh interference optics. Data were acquired at 2-min intervals for 300 radial scans. The data set was analyzed globally to obtain the sedimentation coefficient (c(s)) and molecular mass (c(M)) distributions using Sedfit (50).
Steady-state kinetics
Steady-state PRODH activities and apparent kinetic constants for CfPutA and CfPutA C-terminal truncations were determined using dichlorophenolindophenol as the terminal electron acceptor with phenazine methosulfate as a mediator electron acceptor, as described previously (4). Kinetic constants Km and kcat for proline were determined using fixed concentrations of protein (0.42–0.50 μm), excess FAD (20 μm), dichlorophenolindophenol (75 μm), and varying proline concentration (0–500 mm). PRODH kinetics assays were carried out in 20 mm Tris buffer (pH 8.0, 10% (v/v) glycerol). Data were collected using the cuvette port of a NanoDrop 2000c spectrophotometer (Thermo Fisher Scientific). Km and kcat were calculated by fitting observed initial velocities to the Michaelis-Menten equation (Origin 2016). The low activity of the C-terminal truncation mutants prohibited the determination of Km and kcat. For these proteins, the apparent catalytic efficiency (kcat/Km) was estimated by linear regression of rate data measured below saturation (Origin 2016).
Steady-state GSALDH activity was measured in 50 mm potassium phosphate buffer (pH 7.5, 600 mm NaCl) as described previously (5) using enzyme (0.03–0.04 μm) and dl-P5C (0–2.76 mm) in the presence of 200 μm NAD+. GSALDH activity was monitored by following NADH formation at 340 nm (ϵ340 = 6200 m−1 cm−1). Data were collected using a KC4TM plate reader (BioTek). Km and kcat were calculated by fitting observed initial velocities to the Michaelis-Menten equation (SigmaPlot version 12.0).
The coupled PRODH-GSALDH assay was carried out as described previously (6). A fixed concentration of enzyme (0.42–0.50 μm) was mixed with 0.1 mm menadione bisulfite, 0.2 mm NAD+, and 40 mm proline. The reaction was performed in 50 mm potassium phosphate buffer (pH 7.5, 25 mm NaCl). Reduction of NAD+ was followed at 340 nm (ϵ340 = 6200 m−1 cm−1) using the cuvette port of a NanoDrop 2000c spectrophotometer (Thermo Fisher Scientific). Spectral interference of menadione bisulfite was subtracted from all curves by performing the assays in the presence and absence of NAD+, as described for GsPutA (6).
The coupled PRODH-GSALDH assay can be used to detect substrate channeling by comparing the observed progress curve with a theoretical prediction for non-interacting PRODH and GSALDH enzymes calculated from the free diffusion model (Equation 1) (51, 52).
| (Eq. 1) |
In this equation, t is time, v1 is the rate of PRODH activity under the reaction conditions (1.03 μm·min−1), and the terms v2 and Km are the steady-state kinetic parameters for the GSALDH activity of wild-type CfPutA (v2 = 43 μm·min−1, Km = 54 μm). In the absence of substrate channeling, the transient time (τ) to achieve steady-state production of NADH is the ratio of the GSALDH kinetic parameters, Km/v2. If substrate channeling occurs, the observed τ will be smaller than the limiting value of Km/v2.
P5C intermediate trapping assays
Substrate channeling was also assessed by chemical trapping of the intermediate P5C by oAB (2, 9). oAB traps P5C as a dihydroquinazolinium compound that can be detected by absorbance at 443 nm (ϵ443 = 2590 m−1 cm−1). A fixed concentration of enzyme (0.03–0.04 μm) was mixed with 0.1 mm menadione bisulfite, 4 mm oAB, and 40 mm proline in 20 mm Hepes buffer (pH 7.5, 10 mm MgCl2). The absorbance was measured using an Epoch2 plate reader (BioTek). The assay was conducted both in the presence of 0.2 mm NAD+ or the absence of NAD+, with the latter experiment defining the upper limit for P5C release in the absence of channeling.
Measurement of flavin incorporation
The extinction coefficient of CfPutA·FAD was determined as described previously (53) by measuring the absorption spectra of CfPutA in the absence and presence of 0.5% (w/v) SDS. Incubation with 0.5% (w/v) SDS was performed for 1 h prior to recording the UV-visible spectrum. All measurements were performed in 0.1 m phosphate buffer, pH 7.0. Spectra were measured in a CARY 50 UV-visible spectrophotometer (Agilent) at 25 °C. The percentage of flavin incorporation was then determined from the concentration ratio of FAD-bound CfPutA to total CfPutA.
Thermal shift assays
The melting temperatures of CfPutA and the C-terminal truncation variants were measured in a QuantStudio version 3 real-time PCR system (Thermo Fisher Scientific). Proteins were loaded into a MicroAmp 96-well optical plate (Thermo Fisher Scientific) at a concentration of 0.5 mg/ml in a buffer containing 50 mm Tris, pH 7.8, 100 mm NaCl, 0.5 mm EDTA, 1 mm THP, and 5% (v/v) glycerol. The protein solutions were diluted 1:1 with a 10× solution of SYPRO Orange (Thermo Fisher Scientific; 5000× stock in DMSO) in 50 mm Tris, pH 7.8, 100 mm NaCl, 0.5 mm EDTA, 1 mm THP, and 5% (v/v) glycerol such that the final reaction concentration of SYPRO Orange was 5×. The plate was sealed with optical film and allowed to incubate in the dark at 4 °C for 1 h prior to the experiment. The incubated plate was subjected to a protocol in which the plate was heated from 4 to 95 °C in 0.5 °C steps with a 20-s hold following each step. Data were exported using QuantStudio version 3 software and then normalized using the minimum and maximum relative fluorescence of the transition, as described previously (54), and plotted in R (55).
Author contributions
D. A. K., T. T. G., S. C., M. T. H., D. F. B., and J. J. T. designed experiments. D. A. K., T. T. G., and S. C. performed experiments. D. A. K., T. T. G., S. C., K. M. S., L. J. B., M. T. H., D. F. B., and J. J. T. analyzed the data. D. A. K, S. C., D. F. B., and J. J. T. wrote the paper.
Acknowledgments
We thank Dr. Jay Nix for help with X-ray diffraction data collection. Part of this work was conducted at the Advanced Light Source, which is supported by the Director, Office of Science, Office of Basic Energy Sciences, of the United States Department of Energy under Contract DE-AC02-05CH11231.
This work was supported by NIGMS, National Institutes of Health, Grants R01GM065546, R01GM061068, and P30GM103335. The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
The atomic coordinates and structure factors (code 5UX5) have been deposited in the Protein Data Bank (http://wwpdb.org/).
- PRODH
- proline dehydrogenase
- P5C
- Δ1-pyrroline-5-carboxylate
- GSAL
- l-glutamate-γ-semialdehyde
- GSALDH
- l-glutamate-γ-semialdehyde dehydrogenase
- ALDHSF
- aldehyde dehydrogenase superfamily
- PutA
- proline utilization A
- BjPutA
- B. japonicum proline utilization A
- GsPutA
- G. sulfurreducens proline utilization A
- SmPutA
- S. meliloti proline utilization A
- EcPutA
- E. coli proline utilization A
- CfPutA
- C. freiburgense proline utilization A
- PDB
- Protein Data Bank
- THFA
- l-tetrahydrofuroic acid
- THP
- Tris(hydroxypropyl)phosphine
- oAB
- ortho-aminobenzaldehyde
- BisTris
- 2-[bis(2-hydroxyethyl)amino]-2-(hydroxymethyl)propane-1,3-diol.
References
- 1. Tanner J. J., and Becker D. F. (2013) PutA and proline metabolism. in Handbook of Flavoproteins, Vol. 1 (Hille R., Miller S. M., and Palfey B. A., eds) pp. 31–56, De Gruyter, Berlin [Google Scholar]
- 2. Srivastava D., Schuermann J. P., White T. A., Krishnan N., Sanyal N., Hura G. L., Tan A., Henzl M. T., Becker D. F., and Tanner J. J. (2010) Crystal structure of the bifunctional proline utilization A flavoenzyme from Bradyrhizobium japonicum. Proc. Natl. Acad. Sci. U.S.A. 107, 2878–2883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Arentson B. W., Luo M., Pemberton T. A., Tanner J. J., and Becker D. F. (2014) Kinetic and structural characterization of tunnel-perturbing mutants in Bradyrhizobium japonicum proline utilization A. Biochemistry 53, 5150–5161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Luo M., Christgen S., Sanyal N., Arentson B. W., Becker D. F., and Tanner J. J. (2014) Evidence that the C-terminal domain of a type B PutA protein contributes to aldehyde dehydrogenase activity and substrate channeling. Biochemistry 53, 5661–5673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Moxley M. A., Sanyal N., Krishnan N., Tanner J. J., and Becker D. F. (2014) Evidence for hysteretic substrate channeling in the proline dehydrogenase and Δ1-pyrroline-5-carboxylate dehydrogenase coupled reaction of proline utilization A (PutA). J. Biol. Chem. 289, 3639–3651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Singh H., Arentson B. W., Becker D. F., and Tanner J. J. (2014) Structures of the PutA peripheral membrane flavoenzyme reveal a dynamic substrate-channeling tunnel and the quinone-binding site. Proc. Natl. Acad. Sci. U.S.A. 111, 3389–3394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Arentson B. W., Hayes E. L., Zhu W., Singh H., Tanner J. J., and Becker D. F. (2016) Engineering a trifunctional proline utilization A chimaera by fusing a DNA-binding domain to a bifunctional PutA. Biosci. Rep. 36, e00413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Luo M., Gamage T. T., Arentson B. W., Schlasner K. N., Becker D. F., and Tanner J. J. (2016) Structures of proline utilization A (PutA) reveal the fold and functions of the aldehyde dehydrogenase superfamily domain of unknown function. J. Biol. Chem. 291, 24065–24075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Sanyal N., Arentson B. W., Luo M., Tanner J. J., and Becker D. F. (2015) First evidence for substrate channeling between proline catabolic enzymes: a validation of domain fusion analysis for predicting protein-protein interactions. J. Biol. Chem. 290, 2225–2234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Singh R. K., and Tanner J. J. (2012) Unique structural features and sequence motifs of proline utilization A (PutA). Front Biosci. 17, 556–568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Tanner J. J. (2017) Structural biology of proline catabolic enzymes. Antioxid. Redox Signal., in press [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Gu D., Zhou Y., Kallhoff V., Baban B., Tanner J. J., and Becker D. F. (2004) Identification and characterization of the DNA-binding domain of the multifunctional PutA flavoenzyme. J. Biol. Chem. 279, 31171–31176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Larson J. D., Jenkins J. L., Schuermann J. P., Zhou Y., Becker D. F., and Tanner J. J. (2006) Crystal structures of the DNA-binding domain of Escherichia coli proline utilization A flavoprotein and analysis of the role of Lys9 in DNA recognition. Protein Sci. 15, 2630–2641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Zhou Y., Larson J. D., Bottoms C. A., Arturo E. C., Henzl M. T., Jenkins J. L., Nix J. C., Becker D. F., and Tanner J. J. (2008) Structural basis of the transcriptional regulation of the proline utilization regulon by multifunctional PutA. J. Mol. Biol. 381, 174–188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lee Y. H., Nadaraia S., Gu D., Becker D. F., and Tanner J. J. (2003) Structure of the proline dehydrogenase domain of the multifunctional PutA flavoprotein. Nat. Struct. Biol. 10, 109–114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Zhang M., White T. A., Schuermann J. P., Baban B. A., Becker D. F., and Tanner J. J. (2004) Structures of the Escherichia coli PutA proline dehydrogenase domain in complex with competitive inhibitors. Biochemistry 43, 12539–12548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ostrander E. L., Larson J. D., Schuermann J. P., and Tanner J. J. (2009) A conserved active site tyrosine residue of proline dehydrogenase helps enforce the preference for proline over hydroxyproline as the substrate. Biochemistry 48, 951–959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Srivastava D., Zhu W., Johnson W. H. Jr, Whitman C. P., Becker D. F., and Tanner J. J. (2010) The structure of the proline utilization a proline dehydrogenase domain inactivated by N-propargylglycine provides insight into conformational changes induced by substrate binding and flavin reduction. Biochemistry 49, 560–569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Singh R. K., Larson J. D., Zhu W., Rambo R. P., Hura G. L., Becker D. F., and Tanner J. J. (2011) Small-angle X-ray scattering studies of the oligomeric state and quaternary structure of the trifunctional proline utilization A (PutA) flavoprotein from Escherichia coli. J. Biol. Chem. 286, 43144–43153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Krishnan N., and Becker D. F. (2005) Characterization of a bifunctional PutA homologue from Bradyrhizobium japonicum and identification of an active site residue that modulates proline reduction of the flavin adenine dinucleotide cofactor. Biochemistry 44, 9130–9139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Becker D. F., and Thomas E. A. (2001) Redox properties of the PutA protein from Escherichia coli and the influence of the flavin redox state on PutA-DNA interactions. Biochemistry 40, 4714–4721 [DOI] [PubMed] [Google Scholar]
- 22. Krishnan N., and Becker D. F. (2006) Oxygen reactivity of PutA from Helicobacter species and proline-linked oxidative stress. J. Bacteriol. 188, 1227–1235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Zhu W., Gincherman Y., Docherty P., Spilling C. D., and Becker D. F. (2002) Effects of proline analog binding on the spectroscopic and redox properties of PutA. Arch. Biochem. Biophys. 408, 131–136 [DOI] [PubMed] [Google Scholar]
- 24. Zhu W., Haile A. M., Singh R. K., Larson J. D., Smithen D., Chan J. Y., Tanner J. J., and Becker D. F. (2013) Involvement of the β3-α3 loop of the proline dehydrogenase domain in allosteric regulation of membrane association of proline utilization A. Biochemistry 52, 4482–4491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Krissinel E., and Henrick K. (2004) Secondary-structure matching (SSM), a new tool for fast protein structure alignment in three dimensions. Acta Crystallogr. D Biol. Crystallogr. 60, 2256–2268 [DOI] [PubMed] [Google Scholar]
- 26. Luo M., Arentson B. W., Srivastava D., Becker D. F., and Tanner J. J. (2012) Crystal structures and kinetics of monofunctional proline dehydrogenase provide insight into substrate recognition and conformational changes associated with flavin reduction and product release. Biochemistry 51, 10099–10108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Marchler-Bauer A., Derbyshire M. K., Gonzales N. R., Lu S., Chitsaz F., Geer L. Y., Geer R. C., He J., Gwadz M., Hurwitz D. I., Lanczycki C. J., Lu F., Marchler G. H., Song J. S., Thanki N., et al. (2015) CDD: NCBI's conserved domain database. Nucleic Acids Res. 43, D222–D226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Altschul S. F., Gish W., Miller W., Myers E. W., and Lipman D. J. (1990) Basic local alignment search tool. J. Mol. Biol. 215, 403–410 [DOI] [PubMed] [Google Scholar]
- 29. Patel D., Kopec J., Fitzpatrick F., McCorvie T. J., and Yue W. W. (2016) Structural basis for ligand-dependent dimerization of phenylalanine hydroxylase regulatory domain. Sci. Rep. 6, 23748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Thompson N. J., Merdanovic M., Ehrmann M., van Duijn E., and Heck A. J. (2014) Substrate occupancy at the onset of oligomeric transitions of DegP. Structure 22, 281–290 [DOI] [PubMed] [Google Scholar]
- 31. Tang L., Stith L., and Jaffe E. K. (2005) Substrate-induced interconversion of protein quaternary structure isoforms. J. Biol. Chem. 280, 15786–15793 [DOI] [PubMed] [Google Scholar]
- 32. Honzatko R. B., and Fromm H. J. (1999) Structure-function studies of adenylosuccinate synthetase from Escherichia coli. Arch. Biochem. Biophys. 370, 1–8 [DOI] [PubMed] [Google Scholar]
- 33. Tomar S., Johnston M. L., St John S. E., Osswald H. L., Nyalapatla P. R., Paul L. N., Ghosh A. K., Denison M. R., and Mesecar A. D. (2015) Ligand-induced dimerization of Middle East respiratory syndrome (MERS) coronavirus nsp5 protease (3CLpro): implications for nsp5 regulation and the development of antivirals. J. Biol. Chem. 290, 19403–19422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Jaffe E. K. (2005) Morpheeins: a new structural paradigm for allosteric regulation. Trends Biochem. Sci. 30, 490–497 [DOI] [PubMed] [Google Scholar]
- 35. Selwood T., and Jaffe E. K. (2012) Dynamic dissociating homo-oligomers and the control of protein function. Arch. Biochem. Biophys. 519, 131–143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Matthews B. W. (1968) Solvent content of protein crystals. J. Mol. Biol. 33, 491–497 [DOI] [PubMed] [Google Scholar]
- 37. Kabsch W. (2010) XDS. Acta Crystallogr. D Biol. Crystallogr. 66, 125–132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Evans P. R., and Murshudov G. N. (2013) How good are my data and what is the resolution? Acta Crystallogr. D Biol. Crystallogr. 69, 1204–1214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Long F., Vagin A. A., Young P., and Murshudov G. N. (2008) BALBES: a molecular-replacement pipeline. Acta Crystallogr. D Biol. Crystallogr. 64, 125–132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Murshudov G. N., Vagin A. A., and Dodson E. J. (1997) Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. D Biol. Crystallogr. 53, 240–255 [DOI] [PubMed] [Google Scholar]
- 41. Terwilliger T. C., Grosse-Kunstleve R. W., Afonine P. V., Moriarty N. W., Zwart P. H., Hung L.-W., Read R. J., and Adams P. D. (2008) Iterative model building, structure refinement and density modification with the PHENIX AutoBuild wizard. Acta Crystallogr. D Biol. Crystallogr. 64, 61–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Emsley P., Lohkamp B., Scott W. G., and Cowtan K. (2010) Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Afonine P. V., Grosse-Kunstleve R. W., Echols N., Headd J. J., Moriarty N. W., Mustyakimov M., Terwilliger T. C., Urzhumtsev A., Zwart P. H., and Adams P. D. (2012) Towards automated crystallographic structure refinement with phenix.refine. Acta Crystallogr. D Biol. Crystallogr. 68, 352–367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Inagaki E., Ohshima N., Takahashi H., Kuroishi C., Yokoyama S., and Tahirov T. H. (2006) Crystal structure of Thermus thermophilus Δ1-pyrroline-5-carboxylate dehydrogenase. J. Mol. Biol. 362, 490–501 [DOI] [PubMed] [Google Scholar]
- 45. Srivastava D., Singh R. K., Moxley M. A., Henzl M. T., Becker D. F., and Tanner J. J. (2012) The three-dimensional structural basis of type II hyperprolinemia. J. Mol. Biol. 420, 176–189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Pemberton T. A., Srivastava D., Sanyal N., Henzl M. T., Becker D. F., and Tanner J. J. (2014) Structural Studies of yeast Δ(1)-pyrroline-5-carboxylate dehydrogenase (ALDH4A1): active site flexibility and oligomeric state. Biochemistry 53, 1350–1359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Chen V. B., Arendall W. B. 3rd, Headd J. J., Keedy D. A., Immormino R. M., Kapral G. J., Murray L. W., Richardson J. S., and Richardson D. C. (2010) MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr. 66, 12–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Berman H. M., Westbrook J., Feng Z., Gilliland G., Bhat T. N., Weissig H., Shindyalov I. N., and Bourne P. E. (2000) The Protein Data Bank. Nucleic Acids Res. 28, 235–242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. McCoy A. J., Grosse-Kunstleve R. W., Adams P. D., Winn M. D., Storoni L. C., and Read R. J. (2007) Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Schuck P. (2000) Size-distribution analysis of macromolecules by sedimentation velocity ultracentrifugation and lamm equation modeling. Biophys. J. 78, 1606–1619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Anderson K. S. (1999) Fundamental mechanisms of substrate channeling. Methods Enzymol. 308, 111–145 [DOI] [PubMed] [Google Scholar]
- 52. Arentson B. W., Sanyal N., and Becker D. F. (2012) Substrate channeling in proline metabolism. Front. Biosci. 17, 375–388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. de Jong E., van Berkel W. J., van der Zwan R. P., and de Bont J. A. (1992) Purification and characterization of vanillyl-alcohol oxidase from Penicillium simplicissimum: a novel aromatic alcohol oxidase containing covalently bound FAD. Eur. J. Biochem. 208, 651–657 [DOI] [PubMed] [Google Scholar]
- 54. Andreotti G., Monticelli M., and Cubellis M. V. (2015) Looking for protein stabilizing drugs with thermal shift assay. Drug Test. Anal. 7, 831–834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. R Core Team (2013) R: A Language and Environment for Statistical Computing, R Foundation for Statistical Computing, Vienna, Austria [Google Scholar]
- 56. Sievers F., Wilm A., Dineen D., Gibson T. J., Karplus K., Li W., Lopez R., McWilliam H., Remmert M., Söding J., Thompson J. D., and Higgins D. G. (2011) Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 7, 539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Dereeper A., Guignon V., Blanc G., Audic S., Buffet S., Chevenet F., Dufayard J. F., Guindon S., Lefort V., Lescot M., Claverie J. M., and Gascuel O. (2008) Phylogeny.fr: robust phylogenetic analysis for the non-specialist. Nucleic Acids Res. 36, W465–W469 [DOI] [PMC free article] [PubMed] [Google Scholar]