

Abstract
Mutations to the adhesion G protein-coupled receptor ADGRG1 (G1; also known as GPR56) underlie the neurological disorder bilateral frontoparietal polymicrogyria. Disease-associated mutations in G1 studied to date are believed to induce complete loss of receptor function through disruption of either receptor trafficking or signaling activity. Given that N-terminal truncation of G1 and other adhesion G protein-coupled receptors has been shown to significantly increase the receptors' constitutive signaling, we examined two different bilateral frontoparietal polymicrogyria-inducing extracellular loop mutations (R565W and L640R) in the context of both full-length and N-terminally truncated (ΔNT) G1. Interestingly, we found that these mutations reduced surface expression of full-length G1 but not G1-ΔNT in HEK-293 cells. Moreover, the mutations ablated receptor-mediated activation of serum response factor luciferase, a classic measure of Gα12/13-mediated signaling, but had no effect on G1-mediated signaling to nuclear factor of activated T cells (NFAT) luciferase. Given these differential signaling results, we sought to further elucidate the pathway by which G1 can activate NFAT luciferase. We found no evidence that ΔNT activation of NFAT is dependent on Gαq/11-mediated or β-arrestin-mediated signaling but rather involves liberation of Gβγ subunits and activation of calcium channels. These findings reveal that disease-associated mutations to the extracellular loops of G1 differentially alter receptor trafficking, depending on the presence of the N terminus, and differentially alter signaling to distinct downstream pathways.
Keywords: arrestin, G protein-coupled receptor (GPCR), ion channel, receptor, signal transduction
Introduction
Adhesion G protein-coupled receptors (aGPCRs)2 are a family of GPCRs comprising 33 receptors in humans. These receptors are broadly expressed in various tissues and involved in numerous aspects of normal physiology as well as pathological processes (1). Adhesion GPCRs possess extremely long extracellular N termini that are often decorated with multiple protein/protein interaction domains. An unusual feature of these receptors is their ability to autocatalytically cleave into N-terminal fragment (NTF) and C-terminal fragment (CTF) protomers. Autoproteolysis is mediated by the GPCR autoproteolysis-inducing (GAIN) domain, which is found on the receptors' extracellular NT near the first transmembrane domain (2). Postcleavage, the NTF and CTF remain non-covalently associated for at least some period of time. Deletion or removal of the NTF has been demonstrated to strikingly enhance the signaling activity of many aGPCRs, suggesting that the NTF normally suppresses the activity of the CTF when the two protomers are in complex (3–12).
ADGRG1 (G1; also known as GPR56) has been one of the most intensely studied aGPCRs as mutations to G1 were shown more than a decade ago to underlie the human disease bilateral frontoparietal polymicrogyria (BFPP) (13). Subsequent studies have revealed G1 to be involved in many diverse physiological processes including neurodevelopment (13–16), myelination (17), tumorigenesis (18–21), pancreatic function (22), immune function (23, 24), muscle hypertrophy (25, 26), and hematopoietic stem cell maintenance (27). To date, there are more than two dozen distinct BFPP-causing mutations (28). Although most BFPP-causing missense mutations to G1 occur on the NTF, at least five disease-associated missense mutations have been found to occur on the CTF: C418W, S485P, E496K, R565W, and L640R (13, 28, 29). In terms of functional effects, the last of those mutations (L640R) was found to ablate G1-mediated activation of RhoA following stimulation with the G1-interacting protein collagen III (30).
The activation mechanisms of aGPCRs have garnered much attention in recent years (31–33). Studies by several groups have delineated a model of activation termed the cryptic agonist model wherein dissociation of the NTF from the membrane-embedded CTF unveils the agonistic properties of the remaining extracellular stalk (also termed the “stachel”) (9–11, 34).
We previously investigated this model for G1 and found that the stalk is indeed essential for some but not all signaling outputs (35). Moreover, for other aGPCRs, such as ADGRB1 (BAI1), the presence of the extracellular stalk does not appear to matter at all for receptor signaling activity (35). These findings led us to posit that aGPCRs may be capable of at least two distinct modes of signaling activity: stalk-dependent and stalk-independent. For G1, the stalk was found to be required for activation of serum response factor (SRF) luciferase, a traditional measure of activity for Gα12/13-coupled receptors (36). The stalk-independent activation of nuclear factor of activated T cells (NFAT) luciferase, however, was found in those studies to rely on both G protein-dependent and -independent components and has not been clearly defined in terms of the relevant signaling cascade.
In the present study, we investigated the effects of two BFPP-causing mutations, R565W and L640R, on receptor surface expression and signaling. These studies were performed on both full-length G1 and the ΔNT truncated receptor that mimics the cleaved, active receptor. Moreover, the signaling studies assessed both stalk-dependent and stalk-independent signaling activity. The results of these studies have provided new insights into the regulation of G1 signaling and the mechanisms by which these mutations cause human disease.
Results
BFPP-causing mutations R565W and L640R differentially affect surface expression of full-length versus ΔNT versions of ADGRG1
We generated four mutants version of G1: full-length (FL) and ΔNT receptors harboring either the R565W or L640R mutation (Fig. 1). The ΔNT versions of G1 lack most of the N terminus (Δ amino acids 1–382) up to the site of predicted GAIN domain cleavage and therefore mimic the CTF of G1 that is cleaved at the GAIN domain and undergoes dissociation from the NTF. We assessed the surface trafficking of each mutant in HEK-293T cells in relation to its wild-type counterpart via a cell surface biotinylation approach. As shown in Fig. 2, A and B, we observed that the surface expression and total expression of both full-length mutants were drastically reduced in comparison with the wild-type full-length receptor. Surprisingly, however, the ΔNT mutants displayed no significant deficits in surface expression compared with the wild-type ΔNT receptor (Fig. 2, C and D). It should be noted that the major band observed in Western blots for both full-length and ΔNT G1 exhibits a molecular mass of ∼40–45 kDa. The full-length receptor is efficiently cleaved in HEK-293T cells, and under the denaturing conditions of SDS-PAGE utilized here, the non-covalent associations between the NTF and CTF regions are lost. Thus, Western blotting of the full-length and ΔNT receptors results in detection of the same major band as the C-terminal antibody detects the same CTF species for both versions of the receptor.
Figure 1.
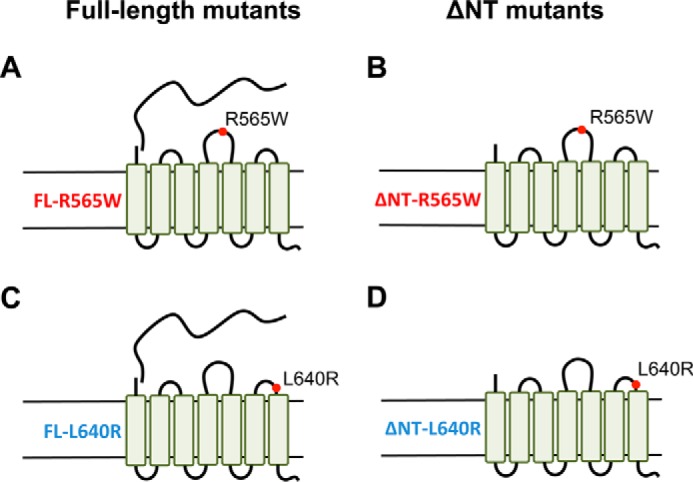
Schematic diagrams of full-length and ΔNT versions of R565W and L640R ADGRG1 mutant receptors. The illustrations depict the predicted transmembrane architecture and relative positions of mutations on the extracellular loops for the FL-R565W G1 mutant (A), the truncated ΔNT-R565W mutant (B), the FL-L640R mutant (C), and the ΔNT-L640R mutant (D).
Figure 2.

R565W and L640R mutations have differential effects on the surface expression of full-length versus ΔNT ADGRG1. A and C, representative Western blots showing surface and total expression of R565W and L640R mutant receptors compared with their wild-type counterparts. The lower blot in each panel represents total receptor expression, and the upper blot in each panel represents the amount of receptor pulled down by streptavidin beads (“Strep”) following biotinylation of surface-expressed proteins. B and D, quantified results of three independent Western blot experiments demonstrating that both full-length mutants exhibit markedly reduced surface and total expression, whereas ΔNT mutants do not, relative to their wild-type counterparts (one-way analysis of variance; **, p < 0.01; ***, p < 0.001 for the indicated comparisons; error bars represent S.E.). IB, immunoblotting; CT, C terminus; NS, not significant.
A key difference between the full-length and ΔNT receptors is that G1-ΔNT has a fully exposed extracellular stalk, whereas the stalk of the full-length receptor is mostly hidden due to either a lack of GAIN cleavage or masking by the associated NTF. The exposed stalk of G1 has agonistic properties (10, 35) and therefore may serve as a pharmacological chaperone for the receptor, counteracting the trafficking deficits conferred by the R565W and L640R mutations in a manner analogous to pharmacological chaperones for other misfolded GPCRs (37). To test the hypothesis of the stalk as a pharmacological chaperone, we generated a stalkless version of the L640R G1 mutant (SL-L640R (Δ amino acids 1–403); Fig. 3B). As shown in Fig. 3, C and D, however, SL-L640R retained normal surface expression and trafficked to the plasma membrane at levels comparable with wild-type ΔNT, L640R-ΔNT and the wild-type stalkless receptor. These data suggest that the extracellular stalk (stachel) of G1 does not act as a pharmacological chaperone as its presence made no difference for trafficking of the L640R mutant.
Figure 3.

The exposed stalk of ADGRG1 does not act as a pharmacological chaperone. A and B, to test the idea that the exposed stalk of ΔNT might act as a pharmacological chaperone to counteract surface trafficking deficits conferred by mutations to the G1 extracellular loops, a stalkless version of ΔNT-L640R (B; SL-L640R) was developed. A representative Western blot (C) and the quantified results of three independent experiments (D) demonstrate that deletion of the ΔNT-L640R stalk does not impair receptor surface expression (n = 3; error bars represent S.E.). Strep, streptavidin; IB, immunoblotting; CT, C terminus; NS, not significant.
R565W and L640R mutations disrupt G1-mediated activation of SRF luciferase but not NFAT luciferase
We next assessed the signaling activity of the mutant receptors in HEK-293T cells in two distinct gene reporter assays: SRF luciferase and NFAT luciferase. In the SRF luciferase assay (Fig. 4A), none of the mutant receptors elicited significant levels of activity. In contrast, expression of the ΔNT mutant receptors resulted in substantial activation of NFAT luciferase that was comparable with the activity induced by wild-type G1-ΔNT (Fig. 4B).
Figure 4.

R565W and L640R mutations have differential effects on ADGRG1 signaling. A, FL- and ΔNT-R565W and -L640R mutants failed to elicit significant signaling to SRF luciferase compared with mock-transfected cells, whereas wild-type G1 and ΔNT elicited substantial signaling. B, ΔNT and ΔNT-R565W/L640R displayed signaling to NFAT luciferase comparable with their wild-type counterparts. All signaling data shown here are from at least five independent experiments (**, p < 0.01; ***, p < 0.001; ****, p < 0.0001 versus cells transfected with a mock vector; error bars represent S.E.). A representative Western blot (C) and quantified results from three independent experiments (D) demonstrate that both wild-type ΔNT and ΔNT-L640R robustly co-immunoprecipitate with HA-βArr2. IB, immunoblotting; IP, immunoprecipitation; CT, C terminus; luc, luciferase; NS, not significant.
Another measure of GPCR activity is association with β-arrestins (38). We previously showed that G1-ΔNT associates robustly with β-arrestin2, whereas the full-length receptor does not (4, 35). Therefore we assessed whether ΔNT-L640R could also associate with β-arrestin2 even though it is deficient in signaling to SRF luciferase. As shown in Fig. 4, C and D, co-immunoprecipitation studies revealed that ΔNT and ΔNT-L640R associate with β-arrestin2 to a similar extent, thereby providing further evidence that the mutant receptor is capable of achieving an active conformation.
G1 signaling to NFAT luciferase does not involve β-arrestins or Gαq/11 but does involve Gβγ and calcium channels
Given that the R565W and L640R mutations disrupted signaling to SRF luciferase but preserved signaling to NFAT luciferase and interaction with β-arrestins, we explored whether β-arrestins might be involved in mediating G1 signaling to NFAT luciferase. Overexpression of β-arrestins typically arrests G protein-dependent signaling by GPCRs but enhances β-arrestin-dependent signaling activity (39), and thus we studied G1 signaling in the absence and presence of β-arrestin overexpression. As shown in Fig. 5A, overexpression of β-arrestin2 significantly impaired signaling to SRF luciferase by full-length G1 and G1-ΔNT but had no significant effect on the ability of either version of G1 to activate NFAT luciferase (Fig. 5B). These data suggest that β-arrestins can arrest G1 signaling to SRF luciferase but are not significantly involved in G1 signaling to NFAT luciferase.
Figure 5.
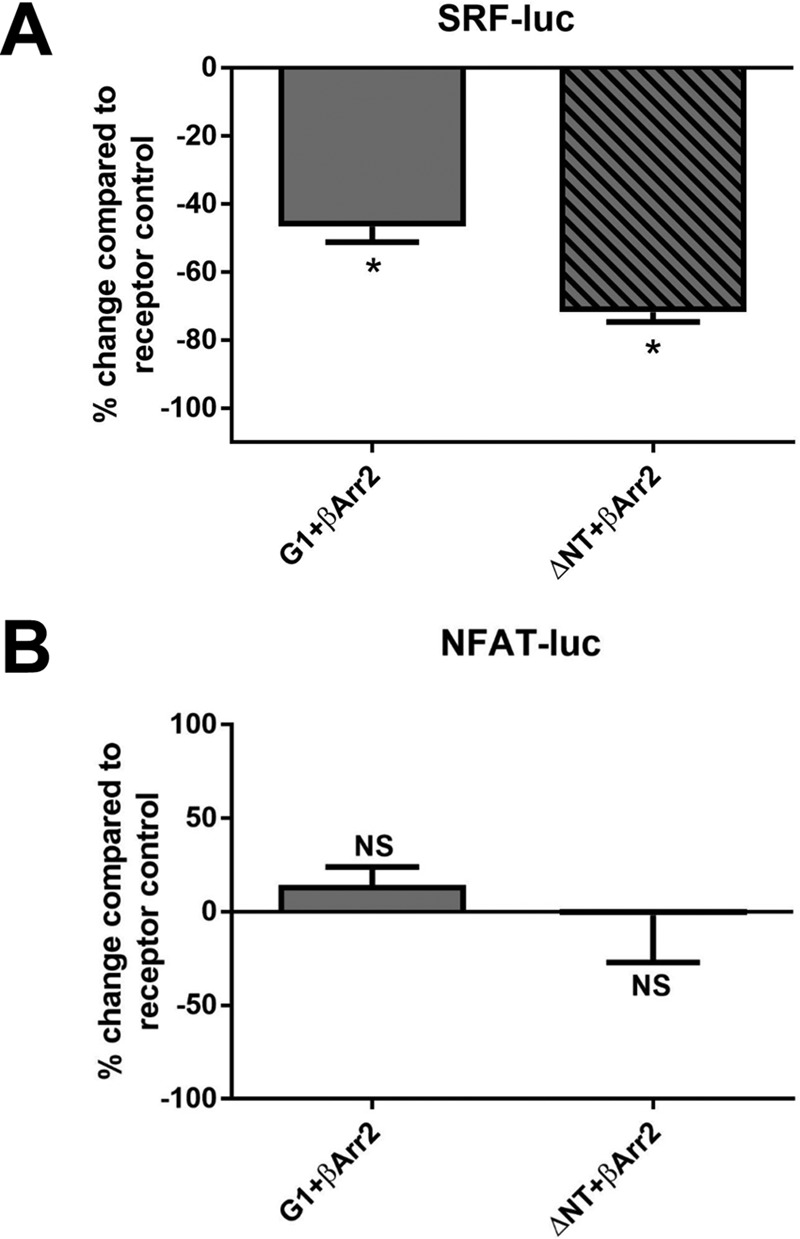
β-Arrestin2 overexpression dampens ADGRG1-mediated activation of SRF but not NFAT luciferase. A, overexpression of FLAG-tagged β-arrestin2 with full-length or ΔNT G1 resulted in significant reductions in receptor-mediated activation of SRF luciferase. B, overexpression of FLAG-β-arrestin2 full-length or ΔNT G1 had no significant effect upon ADGRG1-mediated signaling to NFAT luciferase. Results are from five independent experiments (*, p < 0.05 compared with the corresponding receptor condition without FLAG-β-arrestin2; error bars represent S.E.). luc, luciferase; NS, not significant.
To further explore the potential role of β-arrestins in G1 signaling, we sought to remove key phosphorylation sites from the C terminus of G1 as phosphorylation of GPCR C termini is typically required for β-arrestin association (40). As a starting point for these studies, we focused on Ser-690, which is predicted by phosphorylation motif prediction algorithms to be a GPCR kinase phosphorylation site (41) and has been identified in phosphoproteomic studies to be a highly phosphorylated reside on the G1 C terminus (42). We mutated this serine to an alanine (S690A) in both FL and ΔNT versions of G1, but subsequent co-immunoprecipitation studies revealed that ΔNT-S690A associated with β-arrestin2 to the same extent as wild-type ΔNT (Fig. 6, A and B). These data suggest that this residue is not essential for β-arrestin recruitment. Nonetheless, in the course of performing these experiments, we noted that this mutation markedly enhanced surface expression of the ΔNT mutant (Fig. 6, C and D) and enhanced G1-ΔNT signaling to both SRF and NFAT luciferase (Fig. 6, E and F). Thus, these findings demonstrate that G1-mediated signaling to both SRF and NFAT luciferase is not saturated under our assay conditions, which as discussed below has important implications for interpreting the differential changes in signaling induced by the R565W and L640R mutations in the different pathways downstream of G1.
Figure 6.

Mutation of a putative phosphorylation site (S690A) on the C terminus of ADGRG1 enhances surface expression and signaling by the ΔNT mutant but does not abolish binding to β-arrestin2. A representative Western blot (A) and quantified results from three independent experiments (B) demonstrate that there was no significant difference in co-immunoprecipitation with β-arrestin2 between wild-type G1-ΔNT and ΔNT-S690A. A representative Western blot (C) and quantified results from three independent experiments (D) reveal that the S690A mutation enhanced the surface expression of the ΔNT mutant but not the full-length mutant. The ΔNT-S690A mutant also displayed significantly higher levels of SRF (E) and NFAT luciferase (F) activation compared with the wild-type ΔNT receptor (*, p < 0.05; **, p < 0.01 for the indicated comparisons; error bars represent S.E.). Results shown are from at least four independent experiments. IB, immunoblotting; IP, immunoprecipitation; CT, C terminus; NS, not significant; luc, luciferase; a.u., arbitrary units.
To shed further light on G1 signaling to NFAT luciferase and how this pathway may be mechanistically distinct from G1 signaling to SRF luciferase, we performed a set of inhibitor studies. First, we assessed whether G1 might be capable of activating the Gαq/11 pathway in addition to coupling to Gα12/13. However, as shown in Fig. 7A, we observed that U71322, an inhibitor of phospholipase Cβ and therefore a blocker of the Gαq/11 signaling cascade, had no effect on G1 activation of NFAT luciferase. Another mechanism by which GPCRs can increase cellular calcium levels to activate NFAT luciferase is via activation of plasma membrane calcium channels. Thus, we assessed G1 signaling to NFAT in the presence of SKF96365, a relatively nonspecific calcium channel inhibitor (43, 44). Treatment with SKF96365 resulted in a dramatic decrease in G1-ΔNT signaling to NFAT luciferase for both G1-ΔNT and G1-ΔNT-L640R (Fig. 7B). Taken together with our previous observations that Gβγ inhibitors antagonize G1-ΔNT-mediated signaling to NFAT luciferase (35), these findings suggest that G1-ΔNT can activate NFAT luciferase via a pathway involving the liberation of Gβγ subunits and activation of calcium channels (Fig. 7C) and moreover demonstrate that the disease-associated mutations to the G1 extracellular loops do not impair this signaling.
Figure 7.

ADGRG1-mediated signaling to NFAT luciferase involves activation of calcium channels but not receptor coupling to Gαq/11. A, treatment with the phospholipase Cβ inhibitor U73122 (50 μm; 8 h) had no effect on ΔNT-mediated activation of NFAT luciferase. B, treatment with the calcium channel inhibitor SKF96365 (SKF) (10 μm; 8 h) ablated activation of NFAT luciferase by both G1-ΔNT and ΔNT-L640R. Results shown are from at least four independent experiments (**, p < 0.01; ***, p < 0.001; ****, p < 0.0001 for the indicated comparisons; error bars represent S.E.). C, schematic model depicting the putative signaling pathways by which G1 stimulates SRF or NFAT luciferase activity. The N-terminal fragment is shown interacting with the extracellular stalk and potentially the extracellular loops of the transmembrane C-terminal fragment to modulate receptor signaling activity. NS, not significant; luc, luciferase; veh, vehicle; GEF, guanine nucleotide exchange factor.
Discussion
In the present study, we assessed the effects of the disease-causing mutations R565W and L640R on G1 surface expression and signaling. One important observation was that the extracellular loop mutations reduced the surface expression of full-length G1 but not the ΔNT receptor, which suggests that the tethered NTF may interact with the extracellular loops of G1. In this scenario, the R565W and L640R mutations may corrupt the normal interaction between the NTF and extracellular loops to cause protein misfolding. It is well accepted that aGPCR NTF and CTF protomers interact via hydrophobic stalk interactions within the cleaved GAIN domain (2, 45), but there has also been speculation that there may be additional NTF/CTF interactions that do not involve the stalk (31). In the case of G1, evidence in support of this idea includes the observation that the presence of the NTF strongly suppresses signaling to NFAT luciferase by the G1 CTF even though this signaling is completely stalk-independent (35). The present study provides additional evidence for NTF/CTF interactions that go beyond stalk/GAIN binding as it is unclear how the effects of extracellular loop mutations on G1 trafficking could be dependent on the presence of the NTF unless the extracellular loops possess the capacity to interact with the NTF in some way.
In addition to the effects of the R565W and L640R mutations on receptor trafficking, we also observed that these mutations ablated G1-ΔNT-mediated signaling to SRF luciferase but not NFAT luciferase. This observation suggests that the pathways by which G1 signals to SRF versus NFAT luciferase are mechanistically distinct. Indeed, we previously reported that G1-ΔNT signaling to SRF luciferase is entirely dependent on the presence of the extracellular stalk, whereas signaling to NFAT luciferase is stalk-independent (35). This previous study also demonstrated that G1 signaling to SRF luciferase was almost entirely blocked by inhibition of Gα12/13, whereas signaling to NFAT luciferase was only partially dependent on Gα12/13 and dependent on liberation of Gβγ subunits (35). The present study provides additional insights into the pathways downstream of G1 as our data revealed that G1 signaling to NFAT luciferase does not involve Gαq/11 or β-arrestins but does involve stimulation of calcium channels in addition to Gβγ subunit liberation. Understanding the mechanism(s) by which G1 can stimulate calcium channel activity will require further elucidation, but it is interesting to note that studies on the Drosophila aGPCR lat-1 have shown that this aGPCR robustly activates transient receptor potential family calcium channels to regulate mechanosensation, perhaps via direct receptor/channel interactions (46).
Previous studies have demonstrated that NT-truncated, constitutively active aGPCRs can robustly associate with β-arrestins (4, 6, 35), but the functional effects of aGPCR interactions with β-arrestins are largely unknown. In the present study, we found evidence that β-arrestins can arrest G protein-mediated signaling by aGPCRs as β-arrestin2 overexpression dramatically inhibited G1 activation of SRF luciferase. Interestingly, however, G1 signaling to NFAT luciferase was unaffected by β-arrestin overexpression, providing yet another mechanistic distinction between these two signaling pathways downstream of G1. We also studied the functional effects of mutating a previously described (42) G1 phosphorylation site (Ser-690). Mutation of this serine residue did not alter β-arrestin association but did increase G1-ΔNT surface expression and signaling to both SRF and NFAT luciferase. These data are important because they demonstrate that G1 signaling to both SRF and NFAT luciferase is not saturated under our assay conditions. A potentially trivial explanation for the differential effects of the R565W and L640R mutations on G1-ΔNT signaling to SRF versus NFAT luciferase would be that one of these pathways was saturated, meaning that even a miniscule amount of activity in the mutant receptors might provoke a maximal amount of signaling. However, the S690A signaling data demonstrate that neither signaling pathway is saturated under the conditions of our experiments, thereby further supporting the idea that the pathways downstream of G1 to SRF versus NFAT luciferase are mechanistically distinct.
Several previous reports have assessed the trafficking and signaling properties of full-length BFPP-associated G1 mutants including the R565W and L640R mutants studied here (30, 47, 48). Lin and co-workers (48) found via confocal immunofluorescence that the NTF protomers for mutants R38W (distal NT), R565W (second extracellular loop), and L640R (third extracellular loop) were sharply reduced at the cell surface. In a separate study, Piao and co-workers (47) demonstrated via a cell surface biotinylation approach that surface expression of both CTF and NTF protomers was sharply reduced for mutants R38Q, R38W, Y88C (distal NT), C91S (distal NT), C346S (GAIN domain), C349S (GAIN domain), and R565W. Mixed results were obtained for the L640R mutant in this study as the L640R CTF displayed a level of surface expression comparable with that of the wild-type receptor, whereas the NTF protomer was reduced at the cell surface (47). In further studies, Piao and co-workers (30) found that the L640R mutant exhibits reduced signaling relative to WT following treatment with the G1-binding protein collagen III. Our findings in the present study are consistent with the trafficking deficits that have been reported previously for the full-length R565W and L640R mutants. However, the present study also significantly extends work in this area with the surprising observation that the deleterious effects of these mutations on G1 trafficking are completely abrogated in the ΔNT form of the receptor. Additionally, we found that, although the activated L640R mutant receptor is deficient in Gα12/13-mediated signaling as Piao and co-workers (30) observed, this receptor still robustly binds to β-arrestins and can activate NFAT luciferase via stimulation of calcium channel activity. Thus, our data suggest that the L640R mutant receptor is not completely inactive but rather selectively deficient in certain aspects of its signaling.
In summary, the present study has provided novel insights into how disease-associated mutations to the G1 extracellular loops can differentially impact G1 trafficking and signaling. Going forward, it will be of interest to study further whether aGPCR extracellular loops do indeed interact with the tethered NTF regions as suggested by the findings reported here and to understand what the structural basis of these interactions may be. Additionally, another point of interest will be to further dissect stalk-dependent versus stalk-independent modes of aGPCR signaling and to understand how aGPCR extracellular regions (the NTF and extracellular loops) can differentially modulate distinct aspects of receptor signaling. Finally, a major goal of fundamental studies into aGPCR signaling like those reported here is to set the stage for the future pharmacological targeting of aGPCRs with small molecule agonists, antagonists, and modulators. Given the importance of this family of receptors for human health and disease (1), the members of this family may prove to be important drug targets for novel classes of therapeutics in the treatment of many different human diseases.
Experimental procedures
Constructs
Human G1-ΔNT (amino acids 383–693) and G1-SL (amino acids 404–693) were subcloned into pcDNA3.1 between 5′ HindIII (G1ΔNT, GCAAAGAAGCTTATGACCTACTTTGCAGTGCTGATG; G1-SL, GCAAAGAAGCTTATGAGCCTCCTCTCCTACGTGGG) and 3′ XbaI (GCAAAGTCTAGACTAGATGCGGCTGGACGAGGT). FLAG- and HA-β-arrestin2 constructs have been described previously (35). R565W mutant receptors were generated using the following primers: 5′-CCATGTGCTGGATCTGGGACTCCCTGGTC-3′ and 5′-GACCAGGGAGTCCCAGATCCAGCACATGG-3′. L640R mutant receptors were generated using the following primers: 5′-TGATGCTGAAAAGGTAGCGGACGACAAGCTGGAAG-3′ and 5′-CTTCCAGCTTGTCGTCCGCTACCTTTTCAGCATCA-3′. S690A mutant receptors were generated using the following primers: 5′-AGATGCGGCTGGCCGAGGTGCTGCC-3′ and 5′-GGCAGCACCTCGGCCAGCCGCATCT-3′. All mutant receptors were generated using the QuikChange Lightning site-directed mutagenesis kit (Agilent, catalog number 210519) according to the manufacturer's protocol.
Reagents
SKF 96365 was purchased from Cayman Chemicals (catalog number 10009312), and U73122 was purchased from Tocris Biosciences (catalog number 1268). All other general reagents were from Sigma.
Cell culture
HEK-293T/17 cells were acquired from ATCC (Manassas, VA) and maintained in DMEM (Life Technologies) supplemented with 10% fetal bovine serum and 1% penicillin/streptomycin in a humid, 5% CO2, 37 °C incubator. Cells were transfected using Mirus (Madison, WI) TransIT-LT1 according to the manufacturer's protocol.
Western blotting
Protein samples were reduced and denatured in Laemmli buffer, loaded into 4–20% Tris-glycine gels (Bio-Rad) for SDS-PAGE, and then transferred to nitrocellulose membranes (Bio-Rad). Blots were blocked with 5% milk (in 50 mm NaCl, 10 mm HEPES, pH 7.3, with 0.1% Tween 20) and incubated with primary antibodies for 1 h at room temperature or overnight at 4 °C. The anti-GPR56 C-terminal antibody was developed by Orbigen, Inc. via injection of rabbits with a peptide (CSNSDSARLPISSGSTSSSRI) derived from the GPR56 C terminus and has been characterized previously (4). Rat anti-HA (Roche Applied Science) and mouse HRP-conjugated anti-FLAG (Sigma) antibodies were used to detect epitope-tagged proteins. HRP-conjugated secondary antibodies were purchased from GE Healthcare, and antibody labeling of specific bands was visualized using Thermo Scientific SuperSignal West solutions.
Cell surface biotinylation
HEK-293T cells were transfected with 2 μg of DNA (empty vector or receptor). At 24-h post-transfection, cells were placed on ice and washed with ice-cold PBS + Ca2+ three times. Cells were then incubated with 10 mm Sulfo-NHS-Biotin (Thermo Scientific) in PBS + Ca2+ on ice for 30 min and then washed three more times with PBS + Ca2+ + 100 mm glycine. Cells were resuspended in 250 μl of lysis buffer (1% Triton X-100, 25 mm HEPES, 150 mm NaCl, 10 mm MgCl2, 1 mm EDTA, protease inhibitor mixture (Roche Diagnostics), 2% glycerol) and lysed by slowly rotating on a spinning wheel for 30 min at 4 °C. Cell debris was cleared by centrifugation, and soluble cell lysates were incubated with 50 μl of streptavidin-agarose beads (Thermo Scientific) for 1 h at 4 °C. Beads were washed three times with lysis buffer and resuspended in 60 μl of Laemmli buffer. Biotinylated proteins were detected via Western blotting as described above. Western blot bands were quantified using Image Studio software (LI-COR Biosciences, Lincoln, NE).
β-Arrestin binding assay
HEK-293T cells were transfected with a total of 6 μg of DNA (empty vector, receptor, and/or HA-β-arrestin2 (βArr2) or FLAG-βArr2). The next day, cells were washed with cold PBS + Ca2+ and lysed in harvest buffer (150 mm NaCl, 25 mm HEPES, pH 7.3, 1 mm EDTA, 10 mm MgCl2, 1% Triton X-100, Roche EDTA-free Complete protease inhibitor mixture tablet). Lysates were rotated at 4 °C for 45 min to solubilize integral membrane proteins, and membranes were cleared by centrifugation (15 min at 17,000 × g at 4 °C). Solubilizates were added to anti-HA- (Sigma) or anti-FLAG-agarose beads (Sigma) and rotated at 4 °C for 1 h. Beads were washed three times in harvest buffer, and proteins were eluted in Laemmli buffer at 37 °C for 10–15 min and loaded in 4–20% Tris-glycine gels for SDS-PAGE and Western blotting. Western blot bands were quantified using Image Studio software.
Luciferase reporter assays
HEK-293T cells were seeded in 96-well plates 20–24 h prior to transfection. Each well was transfected with 50 ng of firefly reporter, 1 ng of Renilla luciferase, and 10 ng of receptor or empty plasmid DNA. All reporter constructs (NFAT, pGL4.30; SRF, pGL4.34; Renilla, pRLSV40) were acquired from Promega (Madison, WI). At 24–48 h later, Dual-Glo luciferase assays (Promega) were performed according to the manufacturer's protocol, and plates were read on a BMG Omega plate reader. For inhibitor studies, U73122 (10 μm; diluted from a stock in DMSO) or SKF96365 (50 μm; diluted from a DMSO stock) were added to wells for 8 h before the plates were read. Vehicle control wells received an equivalent amount of DMSO (0.1% final). Results were calculated for each assay by determining the luminescence ratio of firefly:Renilla luciferase counts normalized to empty vector-transfected wells. Error bars for all empty vector-transfected conditions were represented as the standard errors of the normalized raw value means.
Author contributions
A. K. and R. A. H. designed experiments and wrote the manuscript. A. K. performed the experiments. A. K. and R. A. H. analyzed the data and created the figures. All authors made intellectual contributions to the paper and take responsibility for the data.
Acknowledgments
We thank Dr. Thomas Kukar (Emory University) for helpful comments on this work and for kindly providing the use of imaging instruments to facilitate these studies. We also thank members of the Hall laboratory for helpful comments and discussion about the studies described here with special thanks to Anqi Gao for technical assistance in addition to discussion.
This work was supported by National Institutes of Health Grant R01-NS72394 (to R. A. H.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
- aGPCR
- adhesion G protein-coupled receptor
- GPCR
- G protein-coupled receptor
- ADGRG1
- adhesion G protein-coupled receptor G1
- BFPP
- bilateral frontoparietal polymicrogyria
- CTF
- C-terminal fragment
- FL
- full-length
- GAIN
- GPCR autoproteolysis-inducing domain
- G1
- ADGRG1
- NFAT
- nuclear factor of activated T cells
- NTF
- N-terminal fragment
- SL
- stalkless
- SRF
- serum response factor
- ΔNT
- N-terminally truncated
- NT
- N terminus
- βArr2
- β-arrestin2.
References
- 1. Hamann J., Aust G., Araç D., Engel F. B., Formstone C., Fredriksson R., Hall R. A., Harty B. L., Kirchhoff C., Knapp B., Krishnan A., Liebscher I., Lin H. H., Martinelli D. C., Monk K. R., et al. (2015) International Union of Basic and Clinical Pharmacology. XCIV. Adhesion G protein-coupled receptors. Pharmacol. Rev. 67, 338–367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Araç D., Boucard A. A., Bolliger M. F., Nguyen J., Soltis S. M., Südhof T. C., and Brunger A. T. (2012) A novel evolutionarily conserved domain of cell-adhesion GPCRs mediates autoproteolysis. EMBO J. 31, 1364–1378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Okajima D., Kudo G., and Yokota H. (2010) Brain-specific angiogenesis inhibitor 2 (BAI2) may be activated by proteolytic processing. J. Recept. Signal Transduct. Res. 30, 143–153 [DOI] [PubMed] [Google Scholar]
- 4. Paavola K. J., Stephenson J. R., Ritter S. L., Alter S. P., and Hall R. A. (2011) The N terminus of the adhesion G protein-coupled receptor GPR56 controls receptor signaling activity. J. Biol. Chem. 286, 28914–28921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Ward Y., Lake R., Yin J. J., Heger C. D., Raffeld M., Goldsmith P. K., Merino M., and Kelly K. (2011) LPA receptor heterodimerizes with CD97 to amplify LPA-initiated RHO-dependent signaling and invasion in prostate cancer cells. Cancer Res. 71, 7301–7311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Stephenson J. R., Paavola K. J., Schaefer S. A., Kaur B., Van Meir E. G., and Hall R. A. (2013) Brain-specific angiogenesis inhibitor-1 signaling, regulation, and enrichment in the postsynaptic density. J. Biol. Chem. 288, 22248–22256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Paavola K. J., Sidik H., Zuchero J. B., Eckart M., and Talbot W. S. (2014) Type IV collagen is an activating ligand for the adhesion G protein-coupled receptor GPR126. Sci. Signal. 7, ra76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Hu Q. X., Dong J. H., Du H. B., Zhang D. L., Ren H. Z., Ma M. L., Cai Y., Zhao T. C., Yin X. L., Yu X., Xue T., Xu Z. G., and Sun J. P. (2014) Constitutive Gαi coupling activity of very large G protein-coupled receptor 1 (VLGR1) and its regulation by PDZD7 protein. J. Biol. Chem. 289, 24215–24225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Liebscher I., Schön J., Petersen S. C., Fischer L., Auerbach N., Demberg L. M., Mogha A., Cöster M., Simon K. U., Rothemund S., Monk K. R., and Schöneberg T. (2014) A tethered agonist within the ectodomain activates the adhesion G protein-coupled receptors GPR126 and GPR133. Cell Rep. 9, 2018–2026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Stoveken H. M., Hajduczok A. G., Xu L., and Tall G. G. (2015) Adhesion G protein-coupled receptors are activated by exposure of a cryptic tethered agonist. Proc. Natl. Acad. Sci. U.S.A. 112, 6194–6199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Demberg L. M., Rothemund S., Schöneberg T., and Liebscher I. (2015) Identification of the tethered peptide agonist of the adhesion G protein-coupled receptor GPR64/ADGRG2. Biochem. Biophys. Res. Commun. 464, 743–747 [DOI] [PubMed] [Google Scholar]
- 12. Peeters M. C., Fokkelman M., Boogaard B., Egerod K. L., van de Water B., IJzerman A. P., and Schwartz T. W. (2015) The adhesion G protein-coupled receptor G2 (ADGRG2/GPR64) constitutively activates SRE and NFκB and is involved in cell adhesion and migration. Cell. Signal. 27, 2579–2588 [DOI] [PubMed] [Google Scholar]
- 13. Piao X., Hill R. S., Bodell A., Chang B. S., Basel-Vanagaite L., Straussberg R., Dobyns W. B., Qasrawi B., Winter R. M., Innes A. M., Voit T., Ross M. E., Michaud J. L., Déscarie J. C., Barkovich A. J., et al. (2004) G protein-coupled receptor-dependent development of human frontal cortex. Science 303, 2033–2036 [DOI] [PubMed] [Google Scholar]
- 14. Jeong S. J., Luo R., Singer K., Giera S., Kreidberg J., Kiyozumi D., Shimono C., Sekiguchi K., and Piao X. (2013) GPR56 functions together with α3β1 integrin in regulating cerebral cortical development. PLoS One 8, e68781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Koirala S., Jin Z., Piao X., and Corfas G. (2009) GPR56-regulated granule cell adhesion is essential for rostral cerebellar development. J. Neurosci. 29, 7439–7449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Iguchi T., Sakata K., Yoshizaki K., Tago K., Mizuno N., and Itoh H. (2008) Orphan G protein-coupled receptor GPR56 regulates neural progenitor cell migration via a Gα12/13 and Rho pathway. J. Biol. Chem. 283, 14469–14478 [DOI] [PubMed] [Google Scholar]
- 17. Giera S., Deng Y., Luo R., Ackerman S. D., Mogha A., Monk K. R., Ying Y., Jeong S. J., Makinodan M., Bialas A. R., Chang B. S., Stevens B., Corfas G., and Piao X. (2015) The adhesion G protein-coupled receptor GPR56 is a cell-autonomous regulator of oligodendrocyte development. Nat. Commun. 6, 6121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Chiang N. Y., Peng Y. M., Juang H. H., Chen T. C., Pan H. L., Chang G. W., and Lin H. H. (2017) GPR56/ADGRG1 activation promotes melanoma cell migration via NTF dissociation and CTF-mediated Gα12/13/RhoA signaling. J. Invest. Dermatol. 137, 727–736 [DOI] [PubMed] [Google Scholar]
- 19. Yang L., Chen G., Mohanty S., Scott G., Fazal F., Rahman A., Begum S., Hynes R. O., and Xu L. (2011) GPR56 regulates VEGF production and angiogenesis during melanoma progression. Cancer Res. 71, 5558–5568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Shashidhar S., Lorente G., Nagavarapu U., Nelson A., Kuo J., Cummins J., Nikolich K., Urfer R., and Foehr E. D. (2005) GPR56 is a GPCR that is overexpressed in gliomas and functions in tumor cell adhesion. Oncogene 24, 1673–1682 [DOI] [PubMed] [Google Scholar]
- 21. Xu L., Begum S., Hearn J. D., and Hynes R. O. (2006) GPR56, an atypical G protein-coupled receptor, binds tissue transglutaminase, TG2, and inhibits melanoma tumor growth and metastasis. Proc. Natl. Acad. Sci. U.S.A. 103, 9023–9028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Dunér P., Al-Amily I. M., Soni A., Asplund O., Safi F., Storm P., Groop L., Amisten S., and Salehi A. (2016) Adhesion G protein-coupled receptor G1 (ADGRG1/GPR56) and pancreatic β-cell function. J. Clin. Endocrinol. Metab. 101, 4637–4645 [DOI] [PubMed] [Google Scholar]
- 23. Chang G. W., Hsiao C. C., Peng Y. M., Vieira Braga F. A., Kragten N. A., Remmerswaal E. B., van de Garde M. D., Straussberg R., König G. M., Kostenis E., Knäuper V., Meyaard L., van Lier R. A., van Gisbergen K. P., Lin H. H., et al. (2016) The adhesion G protein-coupled receptor GPR56/ADGRG1 is an inhibitory receptor on human NK cells. Cell Rep. 15, 1757–1770 [DOI] [PubMed] [Google Scholar]
- 24. Peng Y. M., van de Garde M. D., Cheng K. F., Baars P. A., Remmerswaal E. B., van Lier R. A., Mackay C. R., Lin H. H., and Hamann J. (2011) Specific expression of GPR56 by human cytotoxic lymphocytes. J. Leukoc. Biol. 90, 735–740 [DOI] [PubMed] [Google Scholar]
- 25. White J. P., Wrann C. D., Rao R. R., Nair S. K., Jedrychowski M. P., You J. S., Martínez-Redondo V., Gygi S. P., Ruas J. L., Hornberger T. A., Wu Z., Glass D. J., Piao X., and Spiegelman B. M. (2014) G protein-coupled receptor 56 regulates mechanical overload-induced muscle hypertrophy. Proc. Natl. Acad. Sci. U.S.A. 111, 15756–15761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Wu M. P., Doyle J. R., Barry B., Beauvais A., Rozkalne A., Piao X., Lawlor M. W., Kopin A. S., Walsh C. A., and Gussoni E. (2013) G-protein coupled receptor 56 promotes myoblast fusion through serum response factor- and nuclear factor of activated T-cell-mediated signalling but is not essential for muscle development in vivo. FEBS J. 280, 6097–6113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Saito Y., Kaneda K., Suekane A., Ichihara E., Nakahata S., Yamakawa N., Nagai K., Mizuno N., Kogawa K., Miura I., Itoh H., and Morishita K. (2013) Maintenance of the hematopoietic stem cell pool in bone marrow niches by EVI1-regulated GPR56. Leukemia 27, 1637–1649 [DOI] [PubMed] [Google Scholar]
- 28. Santos-Silva R., Passas A., Rocha C., Figueiredo R., Mendes-Ribeiro J., Fernandes S., Biskup S., and Leão M. (2015) Bilateral frontoparietal polymicrogyria: a novel GPR56 mutation and an unusual phenotype. Neuropediatrics 46, 134–138 [DOI] [PubMed] [Google Scholar]
- 29. Bahi-Buisson N., Poirier K., Boddaert N., Fallet-Bianco C., Specchio N., Bertini E., Caglayan O., Lascelles K., Elie C., Rambaud J., Baulac M., An I., Dias P., des Portes V., Moutard M. L., et al. (2010) GPR56-related bilateral frontoparietal polymicrogyria: further evidence for an overlap with the cobblestone complex. Brain 133, 3194–3209 [DOI] [PubMed] [Google Scholar]
- 30. Luo R., Jeong S. J., Yang A., Wen M., Saslowsky D. E., Lencer W. I., Araç D., and Piao X. (2014) Mechanism for adhesion G protein-coupled receptor GPR56-mediated RhoA activation induced by collagen III stimulation. PLoS One 9, e100043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kishore A., and Hall R. A. (2016) Versatile signaling activity of adhesion GPCRs. Handb. Exp. Pharmacol. 234, 127–146 [DOI] [PubMed] [Google Scholar]
- 32. Liebscher I., Ackley B., Araç D., Ariestanti D. M., Aust G., Bae B. I., Bista B. R., Bridges J. P., Duman J. G., Engel F. B., Giera S., Goffinet A. M., Hall R. A., Hamann J., Hartmann N., et al. (2014) New functions and signaling mechanisms for the class of adhesion G protein-coupled receptors. Ann. N.Y. Acad. Sci. 1333, 43–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Langenhan T., Aust G., and Hamann J. (2013) Sticky signaling—adhesion class G protein-coupled receptors take the stage. Sci. Signal. 6, re3. [DOI] [PubMed] [Google Scholar]
- 34. Wilde C., Fischer L., Lede V., Kirchberger J., Rothemund S., Schöneberg T., and Liebscher I. (2016) The constitutive activity of the adhesion GPCR GPR114/ADGRG5 is mediated by its tethered agonist. FASEB J. 30, 666–673 [DOI] [PubMed] [Google Scholar]
- 35. Kishore A., Purcell R. H., Nassiri-Toosi Z., and Hall R. A. (2016) Stalk-dependent and stalk-independent signaling by the adhesion G protein-coupled receptors GPR56 (ADGRG1) and BAI1 (ADGRB1). J. Biol. Chem. 291, 3385–3394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Fromm C., Coso O. A., Montaner S., Xu N., and Gutkind J. S. (1997) The small GTP-binding protein Rho links G protein-coupled receptors and Gα12 to the serum response element and to cellular transformation. Proc. Natl. Acad. Sci. U.S.A. 94, 10098–10103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Tao Y. X., and Conn P. M. (2014) Chaperoning G protein-coupled receptors: from cell biology to therapeutics. Endocr. Rev. 35, 602–647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Reiter E., and Lefkowitz R. J. (2006) GRKs and β-arrestins: roles in receptor silencing, trafficking and signaling. Trends Endocrinol. Metab. 17, 159–165 [DOI] [PubMed] [Google Scholar]
- 39. Tohgo A., Pierce K. L., Choy E. W., Lefkowitz R. J., and Luttrell L. M. (2002) β-Arrestin scaffolding of the ERK cascade enhances cytosolic ERK activity but inhibits ERK-mediated transcription following angiotensin AT1a receptor stimulation. J. Biol. Chem. 277, 9429–9436 [DOI] [PubMed] [Google Scholar]
- 40. Tian X., Kang D. S., and Benovic J. L. (2014) β-Arrestins and G protein-coupled receptor trafficking. Handb. Exp. Pharmacol. 219, 173–186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Xue Y., Ren J., Gao X., Jin C., Wen L., and Yao X. (2008) GPS 2.0, a tool to predict kinase-specific phosphorylation sites in hierarchy. Mol. Cell. Proteomics 7, 1598–1608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Goswami T., Li X., Smith A. M., Luderowski E. M., Vincent J. J., Rush J., and Ballif B. A. (2012) Comparative phosphoproteomic analysis of neonatal and adult murine brain. Proteomics 12, 2185–2189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Singh A., Hildebrand M. E., Garcia E., and Snutch T. P. (2010) The transient receptor potential channel antagonist SKF96365 is a potent blocker of low-voltage-activated T-type calcium channels. Br. J. Pharmacol. 160, 1464–1475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Merritt J. E., Armstrong W. P., Benham C. D., Hallam T. J., Jacob R., Jaxa-Chamiec A., Leigh B. K., McCarthy S. A., Moores K. E., and Rink T. J. (1990) SK&F 96365, a novel inhibitor of receptor-mediated calcium entry. Biochem. J. 271, 515–522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Prömel S., Langenhan T., and Araç D. (2013) Matching structure with function: the GAIN domain of adhesion-GPCR and PKD1-like proteins. Trends Pharmacol. Sci. 34, 470–478 [DOI] [PubMed] [Google Scholar]
- 46. Scholz N., Gehring J., Guan C., Ljaschenko D., Fischer R., Lakshmanan V., Kittel R. J., and Langenhan T. (2015) The adhesion GPCR latrophilin/CIRL shapes mechanosensation. Cell Rep. 11, 866–874 [DOI] [PubMed] [Google Scholar]
- 47. Jin Z., Tietjen I., Bu L., Liu-Yesucevitz L., Gaur S. K., Walsh C. A., and Piao X. (2007) Disease-associated mutations affect GPR56 protein trafficking and cell surface expression. Hum. Mol. Genet. 16, 1972–1985 [DOI] [PubMed] [Google Scholar]
- 48. Chiang N. Y., Hsiao C. C., Huang Y. S., Chen H. Y., Hsieh I. J., Chang G. W., and Lin H. H. (2011) Disease-associated GPR56 mutations cause bilateral frontoparietal polymicrogyria via multiple mechanisms. J. Biol. Chem. 286, 14215–14225 [DOI] [PMC free article] [PubMed] [Google Scholar]