

Abstract
The cobalamin or B12 cofactor supports sulfur and one-carbon metabolism and the catabolism of odd-chain fatty acids, branched-chain amino acids, and cholesterol. CblC is a B12-processing enzyme involved in an early cytoplasmic step in the cofactor-trafficking pathway. It catalyzes the glutathione (GSH)-dependent dealkylation of alkylcobalamins and the reductive decyanation of cyanocobalamin. CblC from Caenorhabditis elegans (ceCblC) also exhibits a robust thiol oxidase activity, converting reduced GSH to oxidized GSSG with concomitant scrubbing of ambient dissolved O2. The mechanism of thiol oxidation catalyzed by ceCblC is not known. In this study, we demonstrate that novel coordination chemistry accessible to ceCblC-bound cobalamin supports its thiol oxidase activity via a glutathionyl-cobalamin intermediate. Deglutathionylation of glutathionyl-cobalamin by a second molecule of GSH yields GSSG. The crystal structure of ceCblC provides insights into how architectural differences at the α- and β-faces of cobalamin promote the thiol oxidase activity of ceCblC but mute it in wild-type human CblC. The R161G and R161Q mutations in human CblC unmask its latent thiol oxidase activity and are correlated with increased cellular oxidative stress disease. In summary, we have uncovered key architectural features in the cobalamin-binding pocket that support unusual cob(II)alamin coordination chemistry and enable the thiol oxidase activity of ceCblC.
Keywords: adenosylcobalamin (AdoCbl), enzyme, oxidation-reduction (redox), spectroscopy, thiol, glutathione
Introduction
Cobalamin or B12 is an essential cofactor involved in homocysteine and one-carbon metabolism as well as in branched-chain amino acid and odd-chain fatty acid catabolism (1). Methylcobalamin (MeCbl)2 and adenosylcobalamin (AdoCbl) are the active cofactor derivatives found in mammals and are required by methionine synthase and methylmalonyl-CoA mutase, respectively (1). Mammals are unable to synthesize this essential cofactor and house an elaborate pathway for converting dietary cobalamins to the active cofactor forms and for delivering them to the target proteins (2–4). Defects in this pathway result in a functional B12 deficiency and lead to cobalamin disorders that are classified into 10 genetic complementation groups, cblA–G, cblJ, cblX, and mut (5–8).
CblC (also known as MMACHC for methylmalonic aciduria type C and homocystinuria) is a B12-processing protein that converts cobalamin derivatives entering the cytoplasm to a common intermediate that is subsequently partitioned for synthesis of the two biologically active alkylcobalamins, MeCbl and AdoCbl (3, 4). Mutations in human CblC (hCblC) represent the most common inborn error of cobalamin metabolism (9). CblC exhibits unusual chemical versatility with demonstrated glutathione transferase, reductive decyanase, and aquocobalamin (OH2Cbl) reductase activities (10–12). The Caenorhabdiitis elegans CblC (ceCblC) catalyzes essentially the same chemical reactions as hCblC but also harbors an additional oxygen-scrubbing and glutathione (GSH) oxidation activity, which was proposed to occur via Reactions 1–4 (13). The thiol oxidase activity associated with ceCblC is dependent on its dealkylation or decyanation activity and gives rise to an apparently futile redox cycle, wasting the GSH antioxidant pool and consuming oxygen. The thiol oxidase activity of hCblC is muted in contrast to that of ceCblC. Interestingly, two pathological mutants of hCblC, R161Q and R161G, exhibit elevated thiol oxidase activity, which, in turn, is correlated with increased oxidative stress that is associated with the CblC disorder (14).
In this study, a combination of kinetic, spectroscopic, computational, and crystallographic approaches has revealed the involvement of unusual cobalt coordination chemistry that is supported by the active-site architecture of ceCblC but not hCblC. These studies support a chemical mechanism of thiol oxidation in which glutathionyl-cobalamin (GSCbl) is formed as an intermediate and GSH-dependent deglutathionylation of GSCbl yields GSSG.
Results
Kinetics of thiol oxidase activity in the presence of cobalamin derivatives
The thiol oxidase activity of ceCblC was initially monitored during the dealkylation of AdoCbl by GSH by monitoring O2 consumption as well as GSSG and H2O2 generation (Fig. 1A). The corresponding kcat values for O2 consumption and GSSG and H2O2 production were estimated to be 17 ± 1, 19 ± 2, and 15.8 ± 0.4 min−1, respectively, at 20 °C. Neither GSSG nor H2O2 synthesis was supported by apo-ceCblC or ceCblC loaded with ethylphenylcobalamin (EtPhCbl), an analog that cannot be dealkylated (15) (Fig. 1B), indicating that the thiol oxidase activity requires dealkylation of the alkylcobalamin that is initially bound to CblC. Furthermore, the thiol oxidase activity is dependent on the presence of the initially formed cob(I)alamin or the subsequently oxidized cob(II)alamin state. The Km for GSH was estimated to be 181 ± 12 μm from the O2 consumption assay and 134 ± 7 μm from the GSSG production assay (Fig. 1C). By comparison, the Kact for GSH in the AdoCbl dealkylation assay was estimated to be 183 ± 11 μm (13).
Figure 1.
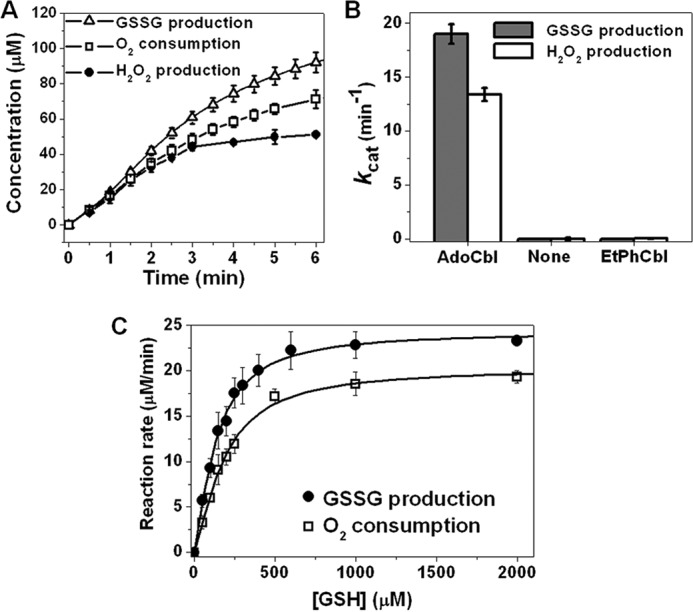
Thiol oxidase activity of ceCblC. A, time courses of GSSG production (triangles), O2 consumption (squares) and H2O2 production (circles) in reactions containing ceCblC (2.5 μm)-AdoCbl (1.25 μm) and 4 mm GSH. B, thiol oxidase activities of ceCblC (20 μm) in the presence of AdoCbl (10 μm), no cofactor, or EtPhCbl (10 μm), in the presence of 4 mm GSH. C, dependence of the rates of GSSG production (solid circles) and O2 consumption (empty squares) by ceCblC (2.5 μm)-AdoCbl (1.25 μm) on GSH concentration. The data represent the mean ± S.D. (error bars) of three independent experiments.
Next, we compared the thiol oxidase activity of ceCblC loaded with different cobalamin derivatives (supplemental Fig. S1). A pronounced lag phase was observed with some derivatives (e.g. AdoCbl and CNCbl). Comparing the rates of GSH-dependent cobalamin processing activities (i.e. dealkylation or reductive decyanation) and GSSG production, a correlation was observed between the rates of cobalamin processing and GSSG production (Table 1). When cofactor processing rates are high as with MeCbl and GSCbl, GSSG production rates are correspondingly high, reaching a maximal value of ∼100 min−1 under our experimental conditions. This result suggests that either cob(I)alamin (the initial product of dealkylation) or cob(II)alamin (product of reductive decyanation or formed by oxidation of cob(I)alamin) initiates the futile cycle.
Table 1.
Summary of ceCblC-catalyzed GSH-dependent cobalamin processing and GSSG production rates
Data represent the mean ± S.D. of at least three independent experiments.
| Cobalamin | kCbla | kGSSGb |
|---|---|---|
| min−1 | min−1 | |
| CNCbl | 0.046 ± 003c | 2.9 ± 0.1 |
| AdoCbl | 0.19 ± 0.03d | 19 ± 2 |
| OH2Cble | 0.46 ± 0.02 | 21 ± 2 |
| MeCbl | 13.5 ± 0.4d | 83 ± 9 |
| GSCbl | 108 ± 12 | 102 ± 9 |
a Cobalamin processing rates were determined by mixing ceCblC (40 μm)-cobalamin (20 μm) with 4 mm GSH in Buffer A under anaerobic conditions at 20 °C. The initial rates of the reactions were determined. With GSCbl, a single exponential fit was used to obtain the kobs.
b GSSG production rate was determined using ceCblC (1–10 μm)-cobalamin (0.5–5 μm) complex in the presence of 4 mm GSH under aerobic conditions.
c From Ref. 12.
d From Ref. 13.
e ceCblC-OH2Cbl complex was generated by oxidation of ceCblC-cob(II)alamin in the presence of the xanthine/xanthine oxidase system followed by rigorous washing to remove xanthine.
Redox cycling is not dependent on OH2Cbl reduction by GSH
Previously, we had proposed a mechanism of futile redox cycling involving OH2Cbl as an intermediate (Reactions 1–4) (12). Due to the technical challenge of loading ceCblC with “base-off” OH2Cbl (in which the endogenous dimethylbenzimadazole ligand is not coordinated to cobalt; see supplemental Fig. S2), OH2Cbl was generated in situ by oxidizing ceCblC-bound cob(II)alamin with O2˙̄. The resulting spectrum exhibited absorption maxima at 357 and 513 nm, in good agreement with the spectrum of ceCblC-aquocobinamide (OH2Cbi), which, lacking the dimethylbenzimadazole ligand, is naturally base-off (supplemental Fig. S2A).
Next, we tested the reactivity of ceCblC-OH2Cbl toward GSH under anaerobic conditions. As seen previously under aerobic conditions (12), cob(II)alamin (λmax = 473 nm) accumulation was observed (supplemental Fig. S3). Formation of GSCbl from OH2Cbl (by ligand exchange with GSH) was not observed, although GSCbl forms readily in solution. This result is consistent with ligand exchange being disfavored in the base-off state (16), which facilitates OH2Cbl reduction instead (17). The observed rate of cob(II)alamin formation from OH2Cbl and GSH was estimated to be 0.46 ± 0.02 min−1, which is significantly smaller than the rate of GSSG production under comparable conditions (ranging from ∼3 to 100 min−1 (Table 1). The discrepancy between the kobs for OH2Cbl reduction by GSH and the kobs for GSSG synthesis by ceCblC rules out OH2Cbl reduction as a kinetically competent step during redox cycling (Reaction 1).
An alternative mechanism for redox cycling
In light of the above results, we considered an alternative mechanism in which GSCbl is formed as a reaction intermediate and is deglutathionylated by GSH to form GSSG in a redox cycle in which O2 undergoes a net two-electron oxidation to H2O2 (Reactions 5–7). In this model, cob(I)alamin formed by dealkylation of MeCbl or AdoCbl enters the cycle via Reaction 6.
To test this mechanism, GSH-dependent deglutathionylation of GSCbl by ceCblC (Reaction 5) was monitored by stopped-flow spectrophotometry under anaerobic conditions. GSCbl binds to ceCblC in the base-off state and exhibits absorption maxima (417 and 506 nm) that are distinct from the base-on cofactor in solution (535 nm) and identical to ceCblC-bound glutathionylcobinamide (GSCbi) (supplemental Fig. S4). The addition of GSH initiates rapid deglutathionylation and yields cob(I)alamin (389 nm) with a kobs of 108 ± 12 min−1 (Fig. 2A, top and inset). An ∼0.2-s lag precedes formation of cob (I)alamin, during which time the spectrum of ceCblC-GSCbl shifts from 417 to 507 nm with isosbestic points at 453 and 532 nm (Fig. 2A, bottom). We tentatively assign the 507-nm species as the [ceCblC-GSCbl-GS−] intermediate.
Figure 2.

Formation of a common intermediate during GSH-dependent detglutathionylation of ceCblC-GSCbl or dealkylation of ceCblC-MeCbl. An intermediate with a λmax = 507 nm, assigned as [ceCblC-GSCbl-GS−], is observed in A–C. A, changes in the absorption spectrum of ceCblC (50 μm)-bound GSCbl (30 μm) mixed with 4 mm GSH under anaerobic conditions. Black trace, 0 s (λmax = 413 nm); red trace, 0.2 s (λmax = 507 nm); blue trace, 2.0 s (λmax = 389 nm); gray traces represent intermediate time points. The inset shows the time-dependent changes in absorbance at 389 nm (red) and 507 nm (black). B, spectral changes upon mixing ceCblC (50 μm)-bound GSCbl (30 μm) with 4 mm GSH under aerobic conditions. Black trace, 0 s (λmax = 413 nm); red trace, 0.2 s (λmax = 501–510 nm); blue trace, 7.0 s (λmax = 475 nm); orange trace, 20 s (λmax = 389 nm); gray traces represent intermediate time points. The inset shows the time-dependent changes in absorbance at 389 nm (red), 473 nm (blue), and 507 nm (black). C, change in the absorption spectra of ceCblC (50 μm)-bound MeCbl (30 μm) incubated with 4 mm GSH under aerobic conditions. Black trace, 0 s (λmax = 454 nm); red trace, 4.0 s (λmax = 487 nm); blue trace, 7.5 s (λmax = 479 nm); orange trace, 20 s (λmax = 389 nm); gray trace, intermediate time points. Inset, time-dependent changes in absorbance at 389 nm (red), 473 nm (blue), and 507 nm (black).
Next, we examined deglutathionylation of GSCbl under aerobic conditions. Due to its air sensitivity, cob(I)alamin accumulation was observed ∼7 s after initiation of the reaction (Fig. 2B, top), presumably when O2 in the reaction mixture had been scrubbed via redox cycling. The accumulation of cob(I)alamin under aerobic conditions is remarkable and was previously ascribed to thiol oxidase activity-mediated depletion of O2 during dealkylation of alklycobalamins (13). Before cob(I)alamin accumulation, formation of the 507-nm absorbing [ceCblC-GSCbl-GS−] intermediate was observed within 0.2 s (Fig. 2B, bottom). Between 0.2 and 7 s, a mixture of [ceCblC-GSCbl-GS−] and cob(II)alamin accumulated, as indicated by the absorption increase at 475 nm (Fig. 2B, middle). These observations are consistent with the intermediacy of GSCbl and cob(II)alamin (Reactions 7 and 6, respectively), during redox cycling. After 7 s, the absorbance associated with the 507-nm species decreased sharply and was accompanied by an increase in absorbance at 389 nm, consistent with cob(I)alamin accumulation. The final products of the reaction were a mixture of cob(I)alamin and cob(II)alamin.
GSCbl is an intermediate during dealkylation of MeCbl
Under anaerobic conditions, GSH-dependent dealkylation of MeCbl by ceCblC results in the full conversion to cob(I)alamin within 3 s (13). In contrast, under aerobic conditions, cob(I)alamin accumulation is observed only after 7.5 s (Fig. 2C, top). Between 0 and 4 s, an increase in absorbance between 500 and 550 nm is observed with a concomitant decrease in absorbance at 454 nm corresponding to base-off MeCbl (Fig. 2C, bottom).
To assess whether the spectral change in the 500–550 nm region was due to formation of the [ceCblC-GSCbl-GS−] intermediate, the spectral changes during 0–1 s were analyzed (supplemental Fig. S5). A reasonably good correspondence was seen between the experimental and predicted difference spectra for the conversion of ceCblC-MeCbl to [ceCblC-GSCbl-GS−], supporting the conclusion that GSCbl is also formed during aerobic dealkylation of ceCblC-MeCbl, where redox cycling occurs. Between 4–7.5 s, the spectral changes (Fig. 2C, middle) were similar to those observed during aerobic deglutathionylation of GSCbl (Fig. 2B, middle), consistent with the formation of both cob(II)alamin and GSCbl (Reactions 6 and 7).
Redox activity of cob(II)alamin bound to ceCblC
To further dissect the mechanism of redox cycling, we examined the mechanism of oxidation of cob(I)alamin and cob(II)alamin bound to ceCblC. Cob(I)alamin is the product of both the dealkylation and deglutathionylation reactions and is highly prone to oxidation. Single electron oxidation of cob(I)alamin by O2 generates cob(II)alamin and O2˙̄ (Reaction 6). The redox potential for the O2/O2˙̄ at pH 7.0 is −330 mV (18), and for base-off cob(II)alamin/cob(I)alamin, it is −500 mV (19); hence, cob(I)alamin oxidation by O2 is highly favorable. Although free cob(II)alamin is also readily oxidized in air (the redox potential of base-on OH2Cbl/cob(II)alamin is +190 mV) (19), cob(II)alamin bound to ceCblC is stabilized against oxidation (supplemental Fig. S6). The stabilization against air oxidation is consistent with OH2Cbl not being an intermediate in redox cycling (Reaction 4) and necessitates consideration of an alternative mechanism (Reaction 7).
ceCblC-cob(II)alamin exposed to O2˙̄ (generated by xanthine/xanthine oxidase) resulted in OH2Cbl formation (supplemental Fig. S7A and Fig. 3A). In the presence of superoxide dismutase, the rate of OH2Cbl formation was significantly diminished (supplemental Fig. S7B). The rates of O2˙̄ generation and ceCblC-cob(II)alamin oxidation were virtually identical (supplemental Fig. S7C), as reported previously with free cob(II)alamin (20). H2O2 (which could form via dismutation of O2˙̄) did not contribute to ceCblC-cob(II)alamin oxidation because catalase had no significant effect on the oxidation rate (supplemental Fig. S7, B and C).
Figure 3.
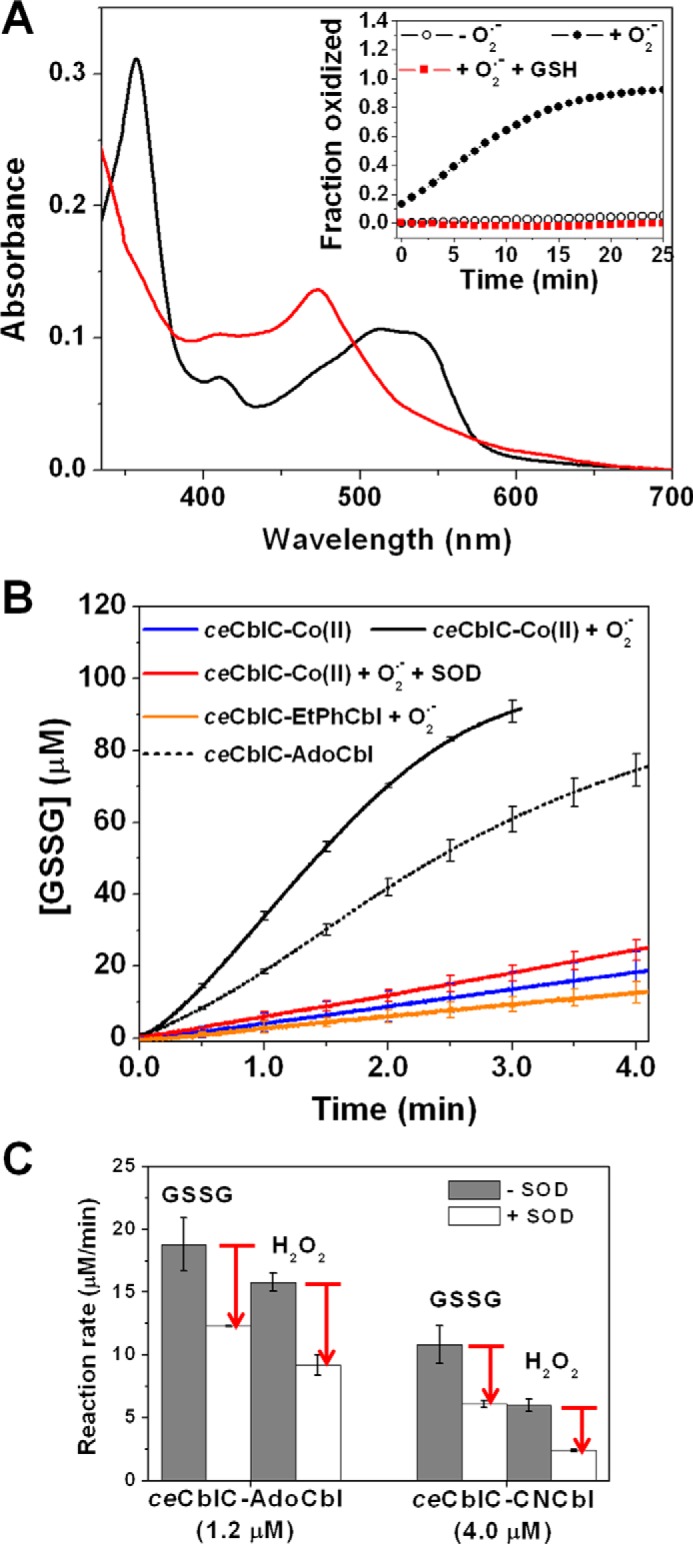
Superoxide is involved in futile redox cycling. A, absorption spectrum of ceCblC (40 μm)-bound cob(II)alamin (15 μm) after a 20-min incubation with O2˙̄ in the presence (red trace, λmax = 473 nm) and absence (black trace, λmax = 512 nm) of 4 mm GSH under aerobic conditions. Inset, time course of ceCblC-cob(II)alamin oxidation in the absence of O2˙̄ (open circles), presence of O2˙̄ (solid circles), or presence of 4 mm GSH and O2˙̄ (red squares). B, comparison of rates of GSSG production by ceCblC-cob(II)alamin, ceCblC-AdoCbl, and ceCblC-EtPhCbl, each with 1.2 μm cobalamin and 2.5 μm ceCblC, in the presence of 4 mm GSH. C, SOD (500 units/ml) partially inhibits redox cycling of ceCblC-AdoCbl and ceCblC-CNCbl in the presence of 4 mm GSH. The O2˙̄ flux was 1.2 μm/min in A and B. [SOD] = 500 units/ml in A–C. The data represent the mean ± S.D. (error bars) of three independent experiments.
In the presence of GSH, the absorption spectrum of ceCblC-cob(II)alamin was unaffected by exposure to O2˙̄ (Fig. 3A), although GSSG production, a hallmark of the thiol oxidase activity, was observed (Fig. 3B). GSSG production was diminished in the presence of SOD or when the dealkylation-inert EtPhCbl derivative was used. SOD inhibited GSSG formation when the thiol oxidase activity was monitored during dealkylation of AdoCbl or decyanation of CNCbl (Fig. 3C). These data are consistent with the involvement of O2˙̄ and cob(II)alamin in redox cycling (Reaction 7).
EPR and magnetic CD (MCD) analysis of ceCblC-cob(II)alamin
GSCbl formation during the redox cycle requires ligation of the GS− anion to the cobalt atom. However, ceCblC-OH2Cbl does not undergo ligand exchange to form GSCbl (supplemental Fig. S3), and 4-coordinate cob(I)alamin cannot form GSCbl directly. We therefore reasoned that cob(II)alamin, which is paramagnetic, is the source of GSCbl in the presence of O2˙̄ (Reaction 7). EPR analysis of ceCblC-cob(II)alamin revealed the presence of 5-coordinate H2O-ligated cob(II)alamin. Upon air exposure, a g = 2.0 signal corresponding to the O2˙̄-cob(III)alamin species was observed (Fig. 4, A and B) (21). Binding of GSH leads to expulsion of O2 and to the appearance of additional features in the EPR spectrum at g ∼2.3, which resembles the spectrum of cob(II)alamin bound to the methyltransferase, TsrM (22) (Fig. 4, C and D). The increase in g anisotropy and 59Co hyperfine coupling constants observed upon GSH binding to ceCblC-cob(II)alamin (Table 2) is consistent with a weakening of the axial Co(II)-ligand bonding interaction. To evaluate the possibility that GSH directly coordinates to the Co(II) ion in ceCblC-cob(II)alamin, density functional theory (DFT) geometry optimizations and EPR parameter calculations were performed for 5-coordinate cysteine-cob(II)inamide and H2O-cob(II)inamide (Table 2). Although DFT fails to accurately predict g values and hyperfine coupling constants for cob(II)alamin species, it reliably reproduces experimental trends in these parameters upon axial ligand perturbations (23). Thus, based on the computational prediction that replacement of the water ligand in H2O-cob(II)inamide by cysteine causes a substantial decrease in the g values and 59Co hyperfine coupling constants, direct coordination by GSH to cob(II)alamin was excluded.
Figure 4.
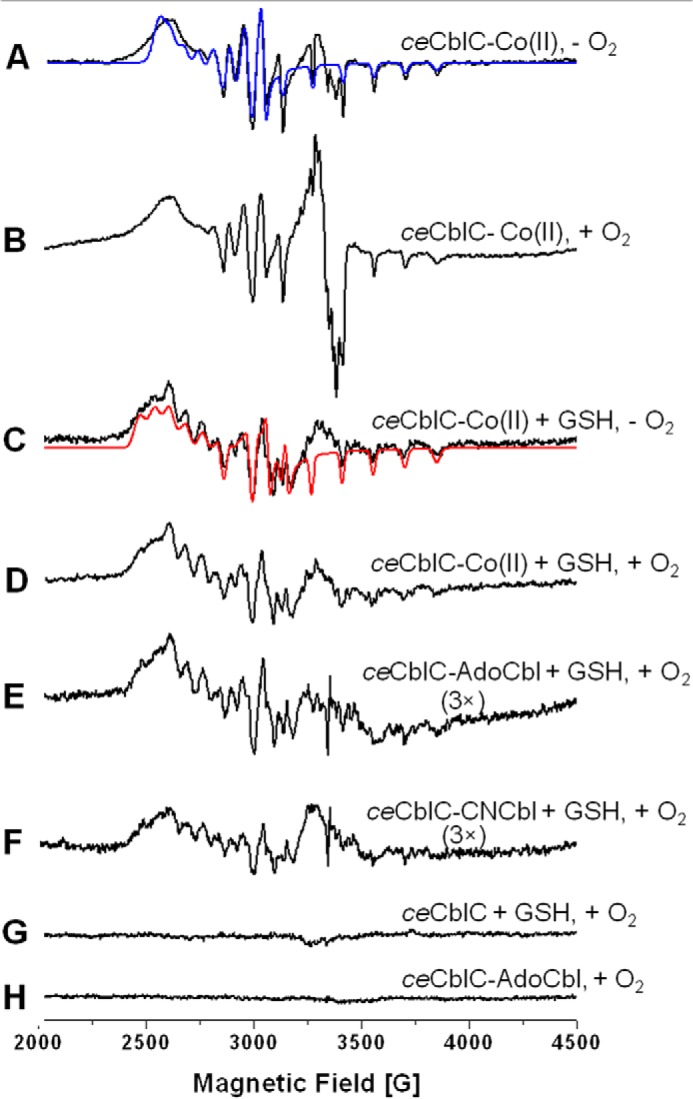
EPR spectra of ceCblC-cob(II)alamin. Cob(II)alamin was generated by photolysis of AdoCbl under anaerobic conditions, and ceCblC (100 μm) was mixed with cob(II)alamin (80 μm) anaerobically and frozen in EPR tubes without (A) or with (B) subsequent exposure to air. The g = 2.0 species in B corresponds to superoxo-cobalamin (Co(III)-O2˙̄) (55). Samples C and D were generated by adding 4 mm GSH to samples A and B, respectively, prior to freezing. The spectra in C and D are identical, and the absence of Co(III)-O2˙̄ in D indicates that GSH binding excludes oxygen. The blue trace in A and the red trace in C represent the simulated EPR spectra obtained using the fit parameters reported in Table 2. Sample E was prepared by incubating ceCblC (300 μm)-AdoCbl (200 μm) with 4 mm GSH for 2 min at room temperature under aerobic conditions before freezing. Sample F was made by incubating ceCblC (300 μm)-CNCbl (200 μm) with 4 mm GSH for 10 min at room temperature under aerobic conditions. Samples G and H were generated by mixing ceCblC (200 μm) with 4 mm GSH or 80 μm AdoCbl, respectively, under aerobic conditions. The data are representative of at least three experiments.
Table 2.
Summary of experimental and DFT-computed EPR g values and 59Co hyperfine coupling constants
The MCD spectrum of ceCblC-cob(II)alamin revealed the presence of an elongated Co-OH2 bond, as judged by the ∼320 cm−1 red shift in the lowest-energy, positively signed feature at 16,134 cm−1 compared with free cob(II)inamide (23) (Fig. 5A). GSH caused an additional red shift by ∼520 cm−1 (Fig. 5B), indicating a further weakening of the Co–OH2 bond. We speculate that lengthening of the Co–O bond facilitates ligand exchange, promoting formation of a transient GS−-cob(II)alamin intermediate. EPR and MCD spectra of samples frozen during redox cycling (Figs. 4 (E and F) and 5C) are similar to those of ceCblC-cob(II)alamin plus GSH, consistent with the presence of the same species in both sets of samples.
Figure 5.
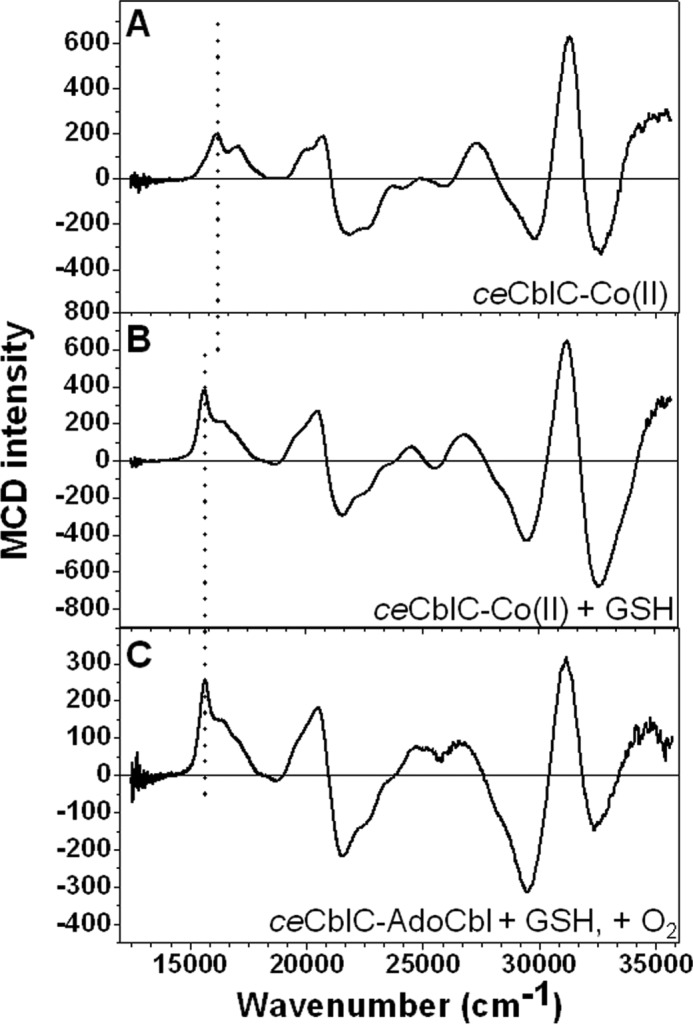
MCD spectra of ceCblC-cob(II)alamin at 4 K. A, ceCblC (250 μm)-cob(II)alamin (200 μm); B, ceCblC (250 μm)-cob(II)alamin (200 μm) + 4 mm GSH; C, ceCblC (500 μm)-AdoCbl (400 μm) + 4 mm GSH, incubated for 2 min at room temperature. The position of the lowest-energy feature, which correlates with the Co–OH2 bond length, is highlighted by the vertical dotted line. The peaks are at 16,134, 15,659, and 15,649 cm−1 for A, B, and C, respectively. Upon the binding of GSH, the Co–OH2 bond is significantly weakened in the ceCblC active site, as indicated by the red shift of this peak.
Crystal structure of ceCblC
The structure of ceCblC(ΔC8) with MeCbl bound was solved at 1.35 Å resolution (Table 3). The overall fold of ceCblC is identical to that of hCblC, with which it shares 35% sequence identity (Fig. 6A) (24, 25). Based on structural superposition, the root mean square deviations for Cα atoms and for all atoms were 0.78 and 1.04 Å, respectively. Clear electron density for MeCbl and tartrate (present in the crystallization buffer) was observed in the active site. The ceCblC form used for crystallization had 6 additional residues (ENLYFQ) that remained following cleavage of the C-terminal His6 tag. Surprisingly, the electron density for this C-terminal extension was observed with the tripeptide YFQ, protruding into the active site of an adjacent monomer (Fig. 6B and supplemental Fig. S8A). Using the interactions observed between ceCblC, tartrate, and Gln-270 from the tripeptide, GSH was modeled in the active site, which placed its sulfur atom at a 3.1-Å distance from the methyl group of MeCbl, oriented for methyl group transfer (Fig. 6C).
Table 3.
X-ray crystallography data collection and refinement statistics for ceCblC
Values in parentheses are for the highest-resolution shell.
| Parameter | Value |
|---|---|
| Data collection | |
| Space group | P212121 |
| Cell dimensions | |
| a, b, c (Å) | 53.0, 66.1, 92.9 |
| α, β, γ (degrees) | 90, 90, 90 |
| Wavelength (Å) | 1.033 |
| Resolution (Å) | 46.0–1.35 (1.37–1.35) |
| Rmeas (%) | 5.8 (59.1) |
| I/σI | 15 (1.9) |
| CC½ | 0.997 (0.618) |
| Completeness (%) | 92.9 (58.7) |
| Redundancy | 3.8 (2.3) |
| Refinement | |
| Resolution (Å) | 53.8–1.35 |
| No. of reflections | 63,476 |
| Rwork/Rfree | 0.132/0.163 |
| No. of atoms | |
| Protein | 4445 |
| Water | 273 |
| Methylcobalamin | 179 |
| B-factors | |
| Protein | 17.5 |
| Water | 28.1 |
| Methylcobalamin | 11.1 |
| Root mean square deviations | |
| Bond lengths (Å) | 0.015 |
| Bond angles (degrees) | 2.013 |
| Ramachandran plot (%) | |
| Favored/allowed/outliers | 98.92/1.08/0.00 |
| MolProbity score | 1.01 (99th percentile) |
| Protein Data Bank code | 5UJC |
Figure 6.
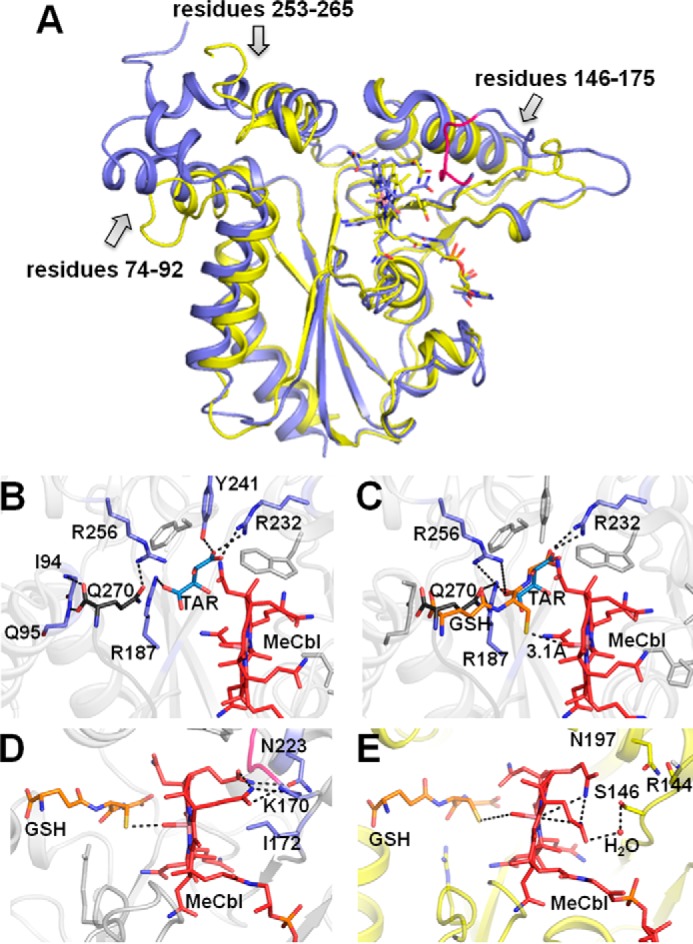
Crystal structure of ceCblC. A, comparison of structures of ceCblC-MeCbl (blue) (PDB entry 5UJC) and hCblC-MeCbl (yellow) (PDB entry 3SC0). Base-off MeCbl is shown in a stick representation. Three regions exhibiting some structural differences between ceCblC and hCblC are indicated with arrows. The portion of the β-hairpin loop in ceCblC that adopts the “closed” conformation is shown in pink. B, close-up of the active site of ceCblC. Tartarate (TAR shown in blue) forms hydrogen bonds with Arg-187, Tyr-241, and Arg-232. The C-terminal extension from the adjacent monomer (shown in gray) protrudes into the active site. The carboxyl group of the C-terminal residue, Gln-270, forms hydrogen bonds with the backbone of Ile-94 and Gln-95, and its side chain interacts with Arg-256. C, modeled structure of GSH (orange) in ceCblC, which was generated by aligning the glutamate and glycine moieties in GSH with Gln-270 and tartrate, respectively. The distance between the sulfur atom of the modeled GSH and the methyl group of MeCbl is 3.1 Å. The predicted hydrogen bonds between GSH and Arg-187, Arg-232, and Arg-256 are shown. Selected residues are shown in a stick representation. D, due to the extended β-hairpin loop (pink) in ceCblC being in the closed conformation, Lys-170 is pushed in toward MeCbl and forms electrostatic interactions with a propionamide chain. The hydrophobic side chain of Ile-172 is directly under the cobalt. The position of GSH (orange) is modeled as in C. E, the propionamide side chains in hCblC (yellow) curve toward the corrin ring and do not interact with Arg-144. In contrast to ceCblC, the α-face of MeCbl is hydrophilic and solvent-accessible, as evidenced by the presence of a water molecule, bridging between a propionamide and Ser-146. The position of GSH (orange) in hCblC·MeCbl was predicted by overlaying the structure of hCblC·MeCbl (PDB entry 3SC0) and hCblC·AntiB12·GSH (PDB entry 5UOS).3
The position of the modeled GSH is supported by the newly obtained structure of hCblC with GSH and a cobalamin analog bound.3 The modeled GSH interacts with Arg-187, Arg-232, and Arg-256, which form an “arginine-rich” pocket, which was previously implicated in GSH binding (25). The active site is commodious, and in fact, a second molecule of GSH can be accommodated in it (supplemental Fig. S8, B and C), supporting the plausibility of the postulated [ceCblC-GSCbl-GS−] intermediate.
Two major structural differences exist between ceCblC and hCblC, which could be important for differential tuning of their thiol oxidase activity (Fig. 6, D and E). The first is the conformation of a β-hairpin loop (residues 146–175) that is folded over the cofactor pocket in ceCblC but is flayed away from it in hCblC (Fig. 6A). In this “closed” conformation in ceCblC, electrostatic interactions between the backbone amido and carbonyl groups of Lys-170 and two propionamide chains from the corrin ring push the cofactor closer toward GSH (Fig. 6D). In hCblC, the two propionamide chains bend in toward the corrin ring and avoid contact with Arg-144 (Fig. 6E). The second feature is the presence in ceCblC of the hydrophobic and bulky Ile-172 directly beneath the cobalt on the α-face of the corin ring. In hCblC, the α-face is hydrophilic and has an ordered water molecule that bridges a propionamide side chain with Ser-146.
Discussion
The thiol oxidase activity involves unusual cobalamin chemistry, and differences in accessible coordination chemistry are correlated with promoting this reactivity in ceCblC but suppressing it in hCblC. Both cob(I)alamin generated from dealkylation and cob(II)alamin formed from GSH-dependent reduction of CNCbl or OH2Cbl can enter the redox cycle (Fig. 7). Formation of GSCbl, a key intermediate in the cycle, is supported by the spectroscopic detection of a [ceCblC-GSCbl-GS−] species (Fig. 2) and by kinetic studies, which established that GSCbl is deglutathionylated (kobs = 108 ± 12 min−1) at a rate that is comparable with GSSG production (kGSSG = 102 ± 9 min−1). Thus, deglutathionylation of GSCbl is rate-limiting (step 6) within the redox cycle. Because GSCbl does not form via ligand exchange of ceCblC-OH2Cbl (supplemental Fig. S4), we propose that ligand exchange occurs at the cob(II)alamin oxidation level instead (step 3).
Figure 7.

Proposed mechanism of the ceCblC-catalyzed thiol oxidase activity. Dealkylation of MeCbl or AdoCbl by GSH generates cob(I)alamin and the corresponding thioether (GSR) (step 1). Oxidation of cob(I)alamin to cob(II)alamin by O2 generates O2˙̄ (step 2). Ligand substitution of O2˙̄ by GSH or by H2O generates GS−-cob(II)alamin or H2O-cob(II)alamin (steps 3 and 7), respectively. GS−-cob(II)alamin is oxidized by O2˙̄, forming GSCbl and H2O2 (step 4). Binding of a second equivalent of GSH results in the formation of the [ceCblC-GSCbl-GS−] intermediate (step 5) and promotes the deglutathionylation of GSCbl to form GSSG and cob(I)alamin (step 6). Alternatively, cob(II)alamin formed either during reductive decyanation of CNCbl (step 10) or by GSH-dependent reduction of OH2Cbl (step 9) can participate in redox cycling. GS−-cob(II)alamin is postulated to form from H2O-cob(II)alamin via ligand exchange (step 8). The glutathionyl radical (GS•) could react with a second equivalent of GSH and O2 to give GSSG and O2˙̄ (Reactions 2 and 3) (26). For clarity, the overall charge of the cobalamin derivatives is not shown.
Why is the thiol oxidase chemistry muted in hCblC? The crystal structure of ceCblC reveals a more compact active site than in hCblC (Fig. 6, D and E) and suggests that GSH might be positioned closer to cobalamin, favoring Co–S bond formation. Additionally, we posit that in hCblC, the equilibrium between O2˙̄-cob(II)alamin and O2˙̄-cob(III)alamin favors the latter via α-face H2O ligation, stabilizing the preferred 6-coordinate geometry and taking it out of the redox cycle (step 11). Alternatively, in hCblC, H2O coordination at the α-face of cob(II)alamin, which prefers 5-coordinate geometry, would stabilize it against further chemistry on the β-face and curtail redox cycling.
DFT geometry optimizations performed for the 5-coordinate cysteine-cob(II)inamide model furnished a 2.51-Å bond distance for Co-S(cysteine) and support the feasibility of a base-off GS−-cob(II)alamin intermediate (Table 2 and supplemental Table S1). However, the GS−-cob(II)alamin intermediate was not observed during the redox cycle. EPR and MCD analyses revealed that although the H2O ligand is retained in ceCblC-cob(II)alamin in the presence of GSH, the Co–O bond is elongated, indicating bond weakening. It is possible that ligand exchange (step 8) is kinetically coupled to GS−-cob(II)alamin oxidation (step 4), leading to the lack of GS−-cob(II)alamin accumulation.
Entering the redox cycle from OH2Cbl or CNCbl (steps 9 and 10) poses the following challenge. The redox potential for GS•/GSH is +920 mV at pH 7.4 (27), which is more positive than the potentials for base-off OH2Cbl/cob(II)alamin (+510 mV) or CNCbl/cob(II)alamin (-110 mV) (28). Based on the crystal structure of ceCblC, the α-side of cobalamin faces a hydrophobic environment, which disfavors binding of water and, therefore, formation of 6-coordinate OH2Cbl. Consequently, the redox potential of the OH2Cbl/cob(II)alamin or CNCbl/cob(II)alamin couples in the ceCblC active site could be higher, rendering reduction more favorable. In addition, kinetic coupling of the unfavorable B12 reduction step to the highly favorable quenching of the thiyl radical (GS•) could provide the driving force (Reactions 2 and 3).
Thiol oxidation by free cobalamins and cobinamides has been reported previously (29). Cobinamides, lacking the lower axial base, are ∼103 times more active than the corresponding free cobalamins in oxidizing thiols. CblC binds base-off cobalamins, which not only facilitates reduction but also has the potential to promote adventitious redox cycling, as observed with ceCblC. Although futile redox cycling is suppressed in wild-type hCblC, it is unleashed by two disease-causing mutations (R161Q and R161G) (14). We posit that architectural constraints lead to a larger separation between the cobalt and thiolate (of GSH) ions on the β-face in hCblC than in ceCblC, inhibiting ligand exchange, and that loss of the interaction between Arg-161 and GSH allows greater proximity and promotes thiol oxidation. The increased thiol oxidase activity of the Arg-161 mutants is correlated with elevated reactive oxygen species in cell lines harboring CblC mutations (30).
In summary, key architectural differences in the cobalamin-binding pocket between the C. elegans and human CblCs support a role for stereoelectronic and coordination chemistry control of the thiol oxidase activity. Pathogenic mutations that corrupt the precise positioning of GSH in hCblC on the β-face and/or water coordination on the α-face can potentially unmask its latent thiol oxidase activity.
Experimental procedures
Materials
Cerium nitrate hexahydrate, AdoCbl, MeCbl, CNCbl, OHCbl, GSH, GSSG, NADPH (reduced), bovine superoxide dismutase, and bovine liver catalase were purchased from Sigma-Aldrich. Ammonium hydroxide, methanol, and acetonitrile were purchased from Fisher. EtPhCbl was synthesized as described previously (15).
Expression and purification of ceCblC
ceCblC was expressed and purified as described previously (12) and was dialyzed into Buffer A containing 100 mm HEPES, pH 7.0, 150 mm KCl, and 10% glycerol. All assays were performed in Buffer A unless otherwise specified.
Synthesis of OH2Cbi
OH2Cbi was prepared by cereous hydroxide hydrolysis of OH2Cbl by modification of the published procedure (31). In brief, NaOH (110 mg, 2.75 mmol) was dissolved in 13 ml of H2O followed by the addition of cerium nitrate hexahydrate (585 mg, 1.35 mmol). A solution of OHCbl·HCl (50 mg, 36.1 μmol in 2 ml of H2O was added to the suspension, and the reaction mixture was heated to 100 °C with vigorous stirring. After 1.5 h, the reaction was cooled to room temperature, and the pH was adjusted to 8.5 with concentrated ammonium hydroxide. The mixture was centrifuged for 15 min at 2236 × g. The supernatant was decanted, and the residue was washed twice with 10 ml of water. The supernatants were pooled, and the solution was desalted using an RP-18 cartridge (Waters). The crude OH2Cbi product was purified using a carboxymethyl cellulose (2.5 × 10-cm) column (Sigma-Aldrich). Unreacted OH2Cbl was removed by washing the column with 100 ml of 10 mm phosphate buffer, pH 8.5. OH2Cbi was eluted with 50 ml of 0.1 m NaCl in 10 mm phosphate buffer, pH 8.5, and desalted using an RP-18 cartridge. The cartridge was washed with 10 ml of water, 1 ml of 0.1 m NaCl followed by 10 ml of water. The product was eluted with 50% aqueous MeOH and lyophilized to obtain 9.9 mg of OH2Cbi·Cl (28% yield) as a red powder. Further purification was achieved by preparative HPLC on an RP-18 column (Luna C-18 250 × 10 mm; Phenomenex) with the following solvent system: A: 0.1% acetic acid, pH 3.0; B: 0.1% acetic acid in acetonitrile; 0–2 min, 2% B isocratic; 2–20 min, 2–40% B; 20–25 min, 40–60% B; 25–30 min, 60% B isocratic; 30–32 min, 60 to 2% B; 32–35 min, 2% B isocratic.
Synthesis of GSCbl and GSCbi
GSCbl was prepared as previously described (32). GSCbi was prepared in an analogous way as described for N-acetylcysteinylcobalamin (32). OH2Cbi·Cl (3.8 mg, 3.7 μmol) was dissolved in 250 μl of 100 mm MES buffer, pH 6.0, in a black 1.5-ml sample tube. The solution was cooled to 0 °C, and GSH (65 μl of a 100 mm stock solution in MES buffer, pH 6.0) was added dropwise. The reaction mixture was stirred for 1.5 h, after which time only one major peak was observed by analytical HPLC. The reaction mixture was then loaded onto an RP-18 cartridge (Waters). The column was washed sequentially with 10 ml of water, 1 ml of 0.1 m NaCl, and 5 ml of water, and the product was eluted with 10 ml of 50% aqueous MeOH. Following lyophilization, an orange product was obtained, which was further purified by preparative HPLC on an RP-18 column (Phenomenex Luna C-18 250 × 10 mm) with the following solvent system: solvent A: 10 mm phosphate buffer, pH 6.5; solvent B: acetonitrile; 0–2 min, 2% B isocratic; 2–12 min, 2–15% B; 12–25 min, 15–18% B; 25–30 min, 18–40% B; 30–33 min, 40–60% B; 33–40 min, 60% B isocratic; 40–43 min, 60–2% B; 43–47 min, 2% B. Fractions containing GSCbi were pooled and desalted using an RP-18 cartridge. The product was eluted with 50% aqueous MeOH and dried by lyophilization to obtain 1.6 mg of GSCbi (1.2 μmol, 32% yield).
GSCbi was characterized by UV-visible and NMR spectroscopy and by mass spectrometry. UV-visible spectroscopy: (H2O): λmax (ϵ) = 497 (7.88), 428 (7.30), 331 (8.95), 269 (8.85) nm (mm−1 cm−1); 1H NMR: (400 MHz, 298 K, D2O, c = 2.9 mm,) δ [ppm] = 0.56 (dd, 1H J = 7.0, 11.5 Hz, H2C-Cys-β), 0.82 (dd, 1H J = 7.0, 11.5 Hz, H2C-Cys-β), 1.02–1.12 (m, 11H, H3C-12B, H3C-17B, H3C-177 H2C-81), 1.43 (s, 3H, H3C-1A), 1.55 (s, 3H, H3C-2A), 1.60 (s, 3H, H3C-12A), 1.70–1.97 (m, 8H, H2C-81, H2C-171, H2C-172, H2C-Glu-γ), superimposed by 1.79 (s, 3H, H3C-7A), 2.00–2.68 (m, 24H, H2C-21, H2C-31, H2C-71, H2C-81, H2C-82, H2C-32, H2C-131, H2C-132, superimposed by 2.21 (s, 3H, H3C-51), 2.36 (s, 3H, H3C-151)), 2.82–2.88 (m, 2H, H2C-181), 3.00–3.25 (m, 5H, H2C-175, HC-18), 3.35–3.75 (m, 5H, HC-8, HC-13, HC-Glu-α, H2C-Gly), 3.78–3.92 (m, 2H, HC176, HC-Cys-α), 4.05 (d, 1H, HC-3), 4.68 (d, J = 8.6 Hz, 1H, HC-19), 6.86 (s, 1H, HC-10); MS: electrospray ionization positive ion, m/z = 1047.5 [M − GSH + HO + Na]+ 1024.5 [M − GSH + H2O + H]+, 989.5 [M − GSH − H2O]+, 648.2 [M + 2H]2+.
GSSG quantification by a coupled glutathione reductase assay
Dealkylation of ceCblC (10–50 μm)-bound AdoCbl (5–25 μm) in the presence of 4 mm GSH was carried out in Buffer A under aerobic conditions. The reactions were stopped at the desired time points by precipitating the protein with an equal volume of metaphosphoric acid solution (16.8 mg/ml metaphosphoric acid, 2 mg/ml EDTA, and 9 mg/ml NaCl). The supernatant (40–80 μl) was mixed with 0.7 units/ml glutathione reductase (from bakers' yeast; Sigma-Aldrich) and 0.2 mm NADPH in 50 mm Tris-HCl, pH 7.5, to a total volume of 400 μl and incubated for 20 min at room temperature to allow for the complete reduction of GSSG and stoichiometric oxidation of NADPH. The amount of GSSG produced in the dealkylation reaction was quantified spectrophotometrically using a Δϵ340 nm = 6.2 mm−1 cm−1 for the oxidation of NADPH. Alternatively, GSSG production rates were measured in a continuous assay in reactions containing ceCblC (1–4 μm)-cobalamin (0.5–2 μm) (MeCbl, AdoCbl, CNCbl, GSCbl, OH2Cbl, or OH2Cbi), 4 mm GSH, 7 units/ml glutathione reductase, and 0.2 mm NADPH in 200 μl of Buffer A. The change in absorbance at 340 nm was monitored. To determine the Km for GSH, ceCblC (2.4 μm)-AdoCbl (1.2 μm) was mixed with 0.05–2 mm GSH. The initial rate of the reaction was plotted against GSH concentration and fitted to the Michaelis-Menten equation.
| (Eq. 1) |
H2O2 quantification using xylenol orange
Dealkylation of ceCblC (2–20 μm)-AdoCbl (1–10 μm) in the presence of 4 mm GSH was carried out in Buffer A under aerobic conditions. The reactions were stopped at the desired time points by mixing 10–30 μl of the reaction mixture with 400 μl of the H2O2 detection reagent containing 125 μm xylenol orange, 0.1 m sorbitol, 0.25 mm ammonium ferrous sulfate, and 25 mm sulfuric acid (PierceTM quantitative peroxide assay kits). After a 30-min incubation at room temperature, the amount of H2O2 was quantified by the change in absorbance at 560 nm. A standard curve was generated by mixing 10 μl of H2O2 (0–400 μm) with 400 μl of the H2O2 detection reagent.
Oxygen consumption assay
The reactions were performed with ceCblC (2–20 μm)-AdoCbl (1–10 μm) complex in Buffer A in a total volume of 1.6 ml in a Gilson type chamber equipped with a Clark oxygen electrode at room temperature. The reactions were started by injection of GSH, and oxygen consumption was recorded on a Kipp and Zonen BD single channel chart recorder. To determine the Km for GSH, ceCblC (2.4 μm)-AdoCbl (1.2 μm) was mixed with 0.05–2 mm GSH. The initial rate of the reaction was plotted against GSH concentration and fitted to the Michaelis-Menten equation.
Oxidation of ceCblC-cob(II)alamin by superoxide
Cob(II)alamin was generated by photolysis of 60 μm AdoCbl in Buffer A and mixed with 100 μm ceCblC under anaerobic conditions. Subsequently, ceCblC-cob(II)alamin was mixed with aerobic Buffer A in a 1:2 (v/v) ratio. A superoxide flux was generated by the addition of 1.4 mm xanthine and varying concentrations (0–40 μg/ml) of bovine milk xanthine oxidase (Sigma-Aldrich) to the ceCblC-cob(II)alamin mixture. The superoxide flux generated at a given concentration of xanthine oxidase was estimated by cytochrome c oxidation (Δϵ550 nm = 21 mm−1 cm−1) separately, as described previously (20). Oxidation of cob(II)alamin to OH2Cbl was monitored by UV-visible spectroscopy and quantified by the change in absorption at 525 nm (Δϵ525 nm = 5.5 mm−1 cm−1).
Estimation of the binding affinity of ceCblC for cobalamins
Cobalamins (20 μm) and ceCblC (0–200 μm) were mixed in a total volume of 200 μl of Buffer A. Unbound cobalamin was separated from protein by filtration using Nanosep® centrifugal devices (cutoff molecular weight = 10 kDa; Pall Life Sciences) at 4 °C to estimate the free cofactor concentration. The ratio of bound cofactor to total cofactor was estimated using the following equation, where E0 and L0 are the total concentration of ceCblC and cofactor, respectively.
| (Eq. 2) |
Binding of OH2Cbi was followed by the change in absorbance at 550 nm (ΔA550 nm), and the fractional change in absorbance (ΔA550 nm/ΔA550 nm max) was estimated using the following equation.
| (Eq. 3) |
Deglutathionylation of GSCbl by ceCblC under anaerobic conditions
The reaction mixtures contained 50 μm ceCblC and 30 μm GSCbl in Buffer A. The reactions were initiated by the addition of 4 mm GSH and carried out at 20 °C under anaerobic conditions. Formation of cob(I)alamin was followed for 5 s by the increase in absorbance at 389 nm (Δϵ = 19 mm−1 cm−1) using stopped-flow spectrophotometry and fitted to the single exponential equation below to obtain kobs.
| (Eq. 4) |
Deglutathionylation of GSCbl and dealkylation of MeCbl by ceCblC under aerobic conditions
The reaction mixtures contained 50 μm ceCblC and 30 μm GSCbl or MeCbl in Buffer A. The reactions were initiated by the addition of 4 mm GSH and carried out at 20 °C under aerobic conditions. The reactions were followed for 28 s using stopped-flow spectrophotometry.
EPR Spectroscopy
One set of samples was anaerobic and contained 100 μm ceCblC and 80 μm cob(II)alamin ± 4 mm GSH in Buffer A. A second set of samples was prepared to monitor the EPR spectrum of ceCblC-cob(II)alamin under aerobic conditions. For this, anaerobic ceCblC (300 μm reconstituted with 240 μm cob(II)alamin) was mixed with aerobic Buffer A (1:2, v/v) with or without 4 mm GSH and incubated on ice for 20 min before freezing in liquid nitrogen. A third set of samples was prepared to test whether cob(II)alamin is formed during redox cycling. For this, ceCblC (300 μm), AdoCbl or CNCbl (200 μm), and 4 mm GSH were mixed aerobically and incubated in the dark for 2–10 min at 15 °C before freezing in liquid nitrogen. The settings for the EPR spectrometer were as follows: temperature, 70 K; microwave power, 20 milliwatts; microwave frequency, 9.32 GHz; receiver gain, 0.5 × 105; modulation amplitude, 10.0 G; modulation frequency, 100 kHz. EPR spectral simulations were performed using Dr. Mark Nilges' SIMPOW program (33).
DFT calculations
Models of H2O- and Cys-cob(II)inamide were derived from the X-ray crystal structure of cob(II)alamin (34). In each case, the entire nucleotide loop and all other corrin ring substituents were replaced by hydrogen atoms separated by 1.08 Å from the neighboring carbon atoms. Full geometry optimizations were performed in ORCA version 3.0 (35), using DFT in conjunction with the BP86 functional (36, 37). The SV(P) (Ahlrichs polarized split valence) basis (38) in conjunction with the SV/C auxiliary basis (39) were used for all atoms except for the cobalt ion and the atoms directly bound to it, for which the TZVP (Ahlrichs polarized triple-ζ valence) basis (40) was used instead.
Molecular g values and 59Co hyperfine coupling parameters were also calculated with ORCA version 3.0 using DFT by solving the coupled-perturbed self-consistent field (SCF) equations (41) and employing the B3LYP hybrid functional (42, 43). The “core properties” with extended polarization (CP(PPP)) basis (44) and Kutzelnigg's NMR/EPR (IGLOIII) basis (45) were used to treat the cobalt ion and all ligating atoms (nitrogen and oxygen or sulfur), respectively, whereas the same basis sets described above were used for all other atoms. A high-resolution radial grid with an integration accuracy of 7 was used for the cobalt ion, and spin-orbit coupling contributions were included in the calculation of the 59Co hyperfine tensor (46).
MCD spectroscopy
Samples containing 250 μm ceCblC and 200 μm cob(II)alamin with or without 4 mm GSH were prepared anaerobically in buffer containing 0.1 m HEPES, pH 7.0, 150 mm KCl, and 55% glycerol. A third sample was prepared by mixing 500 μm ceCblC, 400 μm AdoCbl, and 4 mm GSH aerobically in the same buffer and incubating in the dark for 2 min at room temperature before freezing in liquid nitrogen. MCD and low-temperature absorption spectra were collected on a Jasco J-715 spectropolarimeter in conjunction with an Oxford Instruments SM-4000 8T magnetocryostat. All MCD spectra presented herein were obtained by taking the difference between spectra collected with the magnetic field oriented parallel and antiparallel to the light propagation axis to remove contributions from the natural CD and glass strain.
Expression and purification of ceCblC for crystallization
A construct missing the last eight C-terminal residues (ceCblC(ΔC8)) and fused to a C-terminal cleavable His6 tag was generated for crystallization. The deletion of the last 8 residues was needed to circumvent the insolubility of the full-length construct. ceCblC(ΔC8)-His6 was expressed and purified as described previously (12). The His6 tag was removed following purification on a nickel-nitrilotriacetic acid-agarose column using tobacco etch virus protease and dialyzed overnight against 1 liter of dialysis buffer containing 50 mm Tris-HCl, pH 8.0, 150 mm KCl, 1 mm DTT, and 10% glycerol at 4 °C. The dialyzed protein was passed through a second nickel-nitrilotriacetic acid-agarose column (2.5 × 2 cm), and unbound protein containing ceCblC(ΔC8) was collected, concentrated, and loaded on to a Superdex S200 column (120 ml) pre-equilibrated with buffer containing 50 mm Tris-HCl, pH 8.0. ceCblC(ΔC8) eluted as a monomer and was reconstituted with 1.2 eq of MeCbl in the presence of 0.5 mm tris(2-carboxyethyl)phosphine and was concentrated to at least 30 mg/ml for crystallization.
Crystallization of ceCblC(ΔC8)
A stock solution (12 mg/ml) of MeCbl-loaded ceCblC in 50 mm Tris-HCl, pH 8.0, and 0.5 mm tris(2-carboxyethyl)phosphine was used for crystallization. ceCblC crystals were obtained by the sitting-drop vapor-diffusion method at 4 °C by mixing the protein with 21.5% PEG 3350, 40 mm spermidine-HCl, 0.2 m ammonium tartrate. Harvested crystals were cryoprotected in a solution containing 18.3% PEG 3350, 34 mm spermidine-HCl, 0.17 m ammonium tartrate, 42 mm Tris, pH 7.5, and 15% glycerol. Crystals of ceCblC were of space group P 21 21 21 (a = 53.0 Å, b = 66.1 Å, c = 92.9 Å). One molecule of ceCblC was present per asymmetric unit (Matthews coefficient Vm = 2.61/Da for one molecule/asymmetric unit, 52.9% solvent content).
Data collection and structure determination
Diffraction data were collected at GM/CA-CAT 23-IDB (Advanced Photon Source, Argonne National Laboratory) on a Mar 300 detector and processed with XDS (47) to 1.35 Å resolution. Initial phases were obtained by the molecular replacement method with Phaser (48) using the apo-hCblC structure (PDB entry 3SBZ) as a search model (24). The resulting Phaser solution was used for refinement with PHENIX (49), including rigid body refinement of the individual domains followed by simulated annealing in torsional and Cartesian space, coordination minimization, and restrained individual B-factor adjustment with maximum-likelihood targets. Refmac5 (50) in the CCP4i suite (51) was subsequently employed for restrained refinement by using a mixture of anisotropic individual B-factors for protein and cofactor atoms and isotropic individual B-factors for water molecules with maximum-likelihood targets and also using the Babinet model for bulk solvent scaling. Refinement was followed by model building and modification with Coot (52). Several iterative rounds of refinement followed by model building/modifications were performed before the cobalamin cofactor was modeled in the electron density and further refined with Refmac5. All residues corresponding to the ceCblC(ΔC8) construct with the exception of residues 1 and 2 were traced and accounted for in the electron density of the final model, including residues (ENLYFQ) left from the cleavage of the C-terminal His tag. Crystallographic information as well as refinement statistics are provided in Table 3. The geometric quality of the model and its agreement with the structure factors were assessed with MolProbity (53). For ceCblC, MolProbity reported a clash and a molprobity score of 2.35 (99th percentile) and 1.01 (99th percentile), respectively, whereas 98.92% of the residues were in the favored Ramachandran plot regions with no residues in outlier regions. Crystal structure figures were generated with PyMOL (54).
Author contributions
Z. L. designed, performed, and analyzed the experiments and wrote the manuscript. A. S., K. Y., and M. K. solved the crystal structure of ceCblC. M. R. synthesized cobalamin derivatives and together with B. K. contributed to the proposed reaction scheme. N. A. L. expressed and purified recombinant ceCblC for crystallography. T. C. B. performed the MCD analysis, EPR simulations, and DFT calculation. R. B. helped conceive the experiments, analyzed the data, and co-wrote the manuscript. All authors approved the final version of the manuscript.
Supplementary Material
Acknowledgments
GM/CA at the Advanced Photon Source is funded by NCI, National Institutes of Health (NIH), Grant ACB-12002 and NIGMS, NIH, Grant AGM-12006. This research utilized resources at the Advanced Photon Source, operated for the United States Department of Energy Office of Science by Argonne National Laboratory under contract DE-AC02-06CH11357.
This work was supported in part by National Institutes of Health Grant DK45776 (to R. B.) and by American Heart Association Grant 13SDG14560056 (to M. K.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
The atomic coordinates and structure factors (code 5UJC) have been deposited in the Protein Data Bank (http://wwpdb.org/).
This article contains supplemental Table S1 and Figs. S1–S8.
Ruetz, M., Shanmuganathan, A., Gherasim, C., Karasik, A., Salchner, R., Kieninger, C., Wurst, K., Banerjee, R., Koutmos, M., and Kräutler, B. (2017) Antivitamin B12 inhibition of the human B12-processing enzyme CblC: Crystal structure of an inactive ternary complex with glutatione as the cosubstrate. Angew. Chem. Int. Ed. Engl. 10.1002/anie.201701583.
- MeCbl
- methylcobalamin
- AdoCbl
- 5′-deoxyadenosylcobalamin
- CNCbl
- cyanocobalamin
- GSCbl
- glutathionylcobalamin
- GSCbi
- glutathionylcobinamide
- OH2Cbl
- aquocobalamin
- OH2Cbi
- aquocobinamide
- EtPhCbl
- ethylphenylcobalamin
- hCblC
- human CblC
- ceCblC
- C. elegans CblC
- GS−-cob(II)alamin
- glutathionyl-cob(II)alamin
- MCD
- magnetic circular dichroism
- SOD
- superoxide dismutase
- DFT
- density functional theory
- PDB
- Protein Data Bank.
References
- 1. Banerjee R., and Ragsdale S. W. (2003) The many faces of vitamin B12: catalysis by cobalamin-dependent enzymes. Annu. Rev. Biochem. 72, 209–247 [DOI] [PubMed] [Google Scholar]
- 2. Banerjee R. (2006) B12 trafficking in mammals: A for coenzyme escort service. ACS Chem. Biol. 1, 149–159 [DOI] [PubMed] [Google Scholar]
- 3. Banerjee R., Gherasim C., and Padovani D. (2009) The tinker, tailor, soldier in intracellular B12 trafficking. Curr. Opin. Chem. Biol. 13, 484–491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Gherasim C., Lofgren M., and Banerjee R. (2013) Navigating the B12 road: assimilation, delivery and disorders of cobalamin. J. Biol. Chem. 288, 13186–13193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Chen H. P., and Marsh E. N. (1997) Adenosylcobalamin-dependent glutamate mutase: examination of substrate and coenzyme binding in an engineered fusion protein possessing simplified subunit structure and kinetic properties. Biochemistry 36, 14939–14945 [DOI] [PubMed] [Google Scholar]
- 6. Gulati S., Baker P., Li Y. N., Fowler B., Kruger W., Brody L. C., and Banerjee R. (1996) Defects in human methionine synthase in cblG patients. Hum. Mol. Genet. 5, 1859–1865 [DOI] [PubMed] [Google Scholar]
- 7. Leclerc D., Campeau E., Goyette P., Adjalla C. E., Christensen B., Ross M., Eydoux P., Rosenblatt D. S., Rozen R., and Gravel R. A. (1996) Human methionine synthase: cDNA cloning and identification of mutations in patients of the cblG complementation group of folate/cobalamin disorders. Hum. Mol. Genet. 5, 1867–1874 [DOI] [PubMed] [Google Scholar]
- 8. Ledley F. D., Lumetta M. R., Zoghbi H. Y., VanTuinen P., Ledbetter S. A., and Ledbetter D. H. (1988) Mapping of human methylmalonyl CoA mutase (MUT) locus on chromosome 6. Am. J. Hum. Genet. 42, 839–846 [PMC free article] [PubMed] [Google Scholar]
- 9. Lerner-Ellis J. P., Tirone J. C., Pawelek P. D., Doré C., Atkinson J. L., Watkins D., Morel C. F., Fujiwara T. M., Moras E., Hosack A. R., Dunbar G. V., Antonicka H., Forgetta V., Dobson C. M., Leclerc D., et al. (2006) Identification of the gene responsible for methylmalonic aciduria and homocystinuria, cblC type. Nat. Genet. 38, 93–100 [DOI] [PubMed] [Google Scholar]
- 10. Kim J., Gherasim C., and Banerjee R. (2008) Decyanation of vitamin B12 by a trafficking chaperone. Proc. Natl. Acad. Sci. U.S.A. 105, 14551–14554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kim J., Hannibal L., Gherasim C., Jacobsen D. W., and Banerjee R. (2009) A human vitamin B12 trafficking protein uses glutathione transferase activity for processing alkylcobalamins. J. Biol. Chem. 284, 33418–33424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Li Z., Gherasim C., Lesniak N. A., and Banerjee R. (2014) Glutathione-dependent one-electron transfer reactions catalyzed by a B12 trafficking protein. J. Biol. Chem. 289, 16487–16497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Li Z., Lesniak N. A., and Banerjee R. (2014) Unusual aerobic stabilization of Cob(I)alamin by a B12-trafficking protein allows chemoenzymatic synthesis of organocobalamins. J. Am. Chem. Soc. 136, 16108–16111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Gherasim C., Ruetz M., Li Z., Hudolin S., and Banerjee R. (2015) Pathogenic mutations differentially affect the catalytic activities of the human B12-processing chaperone CblC and increase futile redox cycling. J. Biol. Chem. 290, 11393–11402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ruetz M., Gherasim C., Gruber K., Fedosov S., Banerjee R., and Kräutler B. (2013) Access to organometallic arylcobaltcorrins through radical synthesis: 4-ethylphenylcobalamin, a potential “antivitamin B12”. Angew. Chem. Int. Ed. Engl. 52, 2606–2610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Krautler B. (1987) Thermodynamic trans-effects of the nucleotide base in the B-12 coenzymes. Helv. Chim. Acta 70, 1268–1278 [Google Scholar]
- 17. Lexa D., and Saveant J. M. (1976) Electrochemistry of vitamin B12. I. Role of the base-on/base-off reaction in the oxidoreduction mechanism of the B12r-B12s system. J. Am. Chem. Soc. 98, 2652–2658 [DOI] [PubMed] [Google Scholar]
- 18. Wood P. M. (1988) The potential diagram for oxygen at pH 7. Biochem. J. 253, 287–289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Lexa D., and Saveant J. M. (1983) The Electrochemistry of vitamin B12. Acc. Chem. Res. 16, 235–243 [Google Scholar]
- 20. Suarez-Moreira E., Yun J., Birch C. S., Williams J. H., McCaddon A., and Brasch N. E. (2009) Vitamin B12 and redox homeostasis: cob(II)alamin reacts with superoxide at rates approaching superoxide dismutase (SOD). J. Am. Chem. Soc. 131, 15078–15079 [DOI] [PubMed] [Google Scholar]
- 21. Pilbrow J. R. (1982) EPR of B12-dependent enzyme reactions and related systems. In B12 (Dolphin D., ed) pp. 431–462, Wiley-Interscience, New York [Google Scholar]
- 22. Blaszczyk A. J., Silakov A., Zhang B., Maiocco S. J., Lanz N. D., Kelly W. L., Elliott S. J., Krebs C., and Booker S. J. (2016) Spectroscopic and electrochemical characterization of the iron-sulfur and cobalamin cofactors of TsrM, an unusual radical S-adenosylmethionine methylase. J. Am. Chem. Soc. 138, 3416–3426 [DOI] [PubMed] [Google Scholar]
- 23. Stich T. A., Buan N. R., Escalante-Semerena J. C., and Brunold T. C. (2005) Spectroscopic and computational studies of the ATP:corrinoid adenosyltransferase (CobA) from Salmonella enterica: insights into the mechanism of adenosylcobalamin biosynthesis. J. Am. Chem. Soc. 127, 8710–8719 [DOI] [PubMed] [Google Scholar]
- 24. Koutmos M., Gherasim C., Smith J. L., and Banerjee R. (2011) Structural basis of multifunctionality in a vitamin B12-processing enzyme. J. Biol. Chem. 286, 29780–29787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Froese D. S., Krojer T., Wu X., Shrestha R., Kiyani W., von Delft F., Gravel R. A., Oppermann U., and Yue W. W. (2012) Structure of MMACHC reveals an arginine-rich pocket and a domain-swapped dimer for its B12 processing function. Biochemistry 51, 5083–5090 [DOI] [PubMed] [Google Scholar]
- 26. Hoffman M. Z., and Hayon E. (1973) Pulse-radiolysis study of sulfhydryl compounds in aqueous-solution. J. Phys. Chem. 77, 990–996 [Google Scholar]
- 27. Madej E., and Wardman P. (2007) The oxidizing power of the glutathione thiyl radical as measured by its electrode potential at physiological pH. Arch. Biochem. Biophys. 462, 94–102 [DOI] [PubMed] [Google Scholar]
- 28. Zheng D., and Lu T. (1997) Electrochemical reactions of cyanocobalamin in acidic media. J. Electroanal. Chem. 429, 61–65 [Google Scholar]
- 29. Jacobsen D. W., Troxell L. S., and Brown K. L. (1984) Catalysis of thiol oxidation by cobalamins and cobinamides: reaction-products and kinetics. Biochemistry 23, 2017–2025 [Google Scholar]
- 30. Richard E., Jorge-Finnigan A., Garcia-Villoria J., Merinero B., Desviat L. R., Gort L., Briones P., Leal F., Pérez-Cerdá C., Ribes A., Ugarte M., and Pérez B., and MMACHC Working Group (2009) Genetic and cellular studies of oxidative stress in methylmalonic aciduria (MMA) cobalamin deficiency type C (cblC) with homocystinuria (MMACHC). Hum. Mutat. 30, 1558–1566 [DOI] [PubMed] [Google Scholar]
- 31. Ishida A., Ichikawa M., Kobayashi K., Hitomi T., Kojima S., and Toraya T. (1993) Importance of the nucleotide loop moiety coordinated to the cobalt atom of adenosylcobalamin for coenzymic function in the diol dehydrase reaction. J. Nutr. Sci. Vitaminol. 39, 115–125 [DOI] [PubMed] [Google Scholar]
- 32. Suarez-Moreira E., Hannibal L., Smith C. A., Chavez R. A., Jacobsen D. W., and Brasch N. E. (2006) A simple, convenient method to synthesize cobalamins: synthesis of homocysteinylcobalamin, N-acetylcysteinylcobalamin, 2-N-acetylamino-2-carbomethoxyethanethiolatocobalamin, sulfitocobalamin and nitrocobalamin. Dalton Trans. 10.1039/b610158e [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Nilges M. J. (1979) Electron paramagnetic resonance studies of low symmetry nickel(I) and molybdenum(V) complexes. Ph.D. thesis, University of Illinois at Urbana-Champaign, Urbana, IL [Google Scholar]
- 34. Kräutler B., Keller W., and Kratky C. (1989) Coenzyme B12 chemistry: the crystal and molecular structure of Cob(II)alamin. J. Am. Chem. Soc. 111, 8936–8938 [Google Scholar]
- 35. Neese F. (2014) ORCA: An ab Initio, Density Functional, and Semiempirical Program Package, Version 3.0.3, Max Planck Institute for Chemical Energy Conversion, Mülheim, Germany [Google Scholar]
- 36. Becke A. D. (1988) Density functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A 38, 3098–3100 [DOI] [PubMed] [Google Scholar]
- 37. Perdew J. P. (1986) Density functional approximation for the correlation energy of the inhomogeneous electron gas. Phys. Rev. B Condens. Matter 33, 8822–8824 [DOI] [PubMed] [Google Scholar]
- 38. Schäfer A., Horn H., and Ahlrichs R. (1992) Fully optimized contracted Gaussian-basis sets for atoms Li to Kr. J. Chem. Phys. 97, 2571–2577 [Google Scholar]
- 39. Weigend F., and Ahlrichs R. (2005) Balanced basis sets of split valence, triple ζ valence and quadruple ζ valence quality for H to Rn: design and assessment of accuracy. Phys. Chem. Chem. Phys. 7, 3297–3305 [DOI] [PubMed] [Google Scholar]
- 40. Schäfer A., Huber C., and Ahlrichs R. (1994) Fully optimized contracted Gaussian basis sets of triple ζ valence quality for atoms Li to Kr. J. Chem. Phys. 100, 5829–5835 [Google Scholar]
- 41. Neese F. (2001) Prediction of electron paramagnetic resonance g values using coupled perturbed Hartree-Fock and Kohn-Sham theory. J. Chem. Phys. 115, 11080–11096 [Google Scholar]
- 42. Becke A. D. (1993) Density-functional thermochemistry. 3. The role of exact exchange. J. Chem. Phys. 98, 5648–5652 [Google Scholar]
- 43. Lee C., Yang W., and Parr R. G. (1988) Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B Condens. Matter 37, 785–789 [DOI] [PubMed] [Google Scholar]
- 44. Neese F. (2002) Prediction and interpretation of the Fe-57 isomer shift in Mossbauer spectra by density functional theory. Inorg. Chem. Acta 337, 181–192 [Google Scholar]
- 45. Kutzelnigg W., Fleischer U., and Schindler M. (eds) (1990) The IGLO Method: Ab Initio Calculation and Interpretation of NMR Chemical Shifts and Magnetic Susceptibilities, Vol. 23, Springer-Verlag, Heidelberg, Germany [Google Scholar]
- 46. Neese F. (2003) Metal and ligand hyperfine couplings in transition metal complexes: the effect of spin-orbit coupling as studied by coupled perturbed Kohn-Sham theory. J. Chem. Phys. 118, 3939–3948 [Google Scholar]
- 47. Kabsch W. (2010) XDS. Acta Crystallogr. D 66, 125–132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. McCoy A. J., Grosse-Kunstleve R. W., Adams P. D., Winn M. D., Storoni L. C., and Read R. J. (2007) Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Adams P. D., Grosse-Kunstleve R. W., Hung L. W., Ioerger T. R., McCoy A. J., Moriarty N. W., Read R. J., Sacchettini J. C., Sauter N. K., and Terwilliger T. C. (2002) PHENIX: building new software for automated crystallographic structure determination. Acta Crystallogr. D Biol. Crystallogr. 58, 1948–1954 [DOI] [PubMed] [Google Scholar]
- 50. Murshudov G. N., Skubák P., Lebedev A. A., Pannu N. S., Steiner R. A., Nicholls R. A., Winn M. D., Long F., and Vagin A. A. (2011) REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr. D Biol. Crystallogr. 67, 355–367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Potterton E., Briggs P., Turkenburg M., and Dodson E. (2003) A graphical user interface to the CCP4 program suite. Acta Crystallogr. D Biol. Crystallogr. 59, 1131–1137 [DOI] [PubMed] [Google Scholar]
- 52. Emsley P., Lohkamp B., Scott W. G., and Cowtan K. (2010) Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Davis I. W., Leaver-Fay A., Chen V. B., Block J. N., Kapral G. J., Wang X., Murray L. W., Arendall W. B. 3rd, Snoeyink J., Richardson J. S., and Richardson D. C. (2007) MolProbity: all-atom contacts and structure validation for proteins and nucleic acids. Nucleic Acids Res. 35, W375–W383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. DeLano W. L. (2010) The PyMOL Molecular Graphics System, Version 1.3 rl, Schroedinger, LLC, New York [Google Scholar]
- 55. Frasca V., Banerjee R. V., Dunham W. R., Sands R. H., and Matthews R. G. (1988) Cobalamin-dependent methionine synthase from Escherichia coli B: electron paramagnetic resonance spectra of the inactive form and the active methylated form of the enzyme. Biochemistry 27, 8458–8465 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.