

Abstract
Manganese is an essential metal that becomes toxic at elevated levels. Loss-of-function mutations in SLC30A10, a cell-surface-localized manganese efflux transporter, cause a heritable manganese metabolism disorder resulting in elevated manganese levels and parkinsonian-like movement deficits. The underlying disease mechanisms are unclear; therefore, treatment is challenging. To understand the consequences of loss of SLC30A10 function at the organism level, we generated Slc30a10 knock-out mice. During early development, knock-outs were indistinguishable from controls. Surprisingly, however, after weaning and compared with controls, knock-out mice failed to gain weight, were smaller, and died prematurely (by ∼6–8 weeks of age). At 6 weeks, manganese levels in the brain, blood, and liver of the knock-outs were ∼20–60-fold higher than controls. Unexpectedly, histological analyses revealed that the brain and liver of the knock-outs were largely unaffected, but their thyroid exhibited extensive alterations. Because hypothyroidism leads to growth defects and premature death in mice, we assayed for changes in thyroid and pituitary hormones. At 6 weeks and compared with controls, the knock-outs had markedly reduced thyroxine levels (∼50–80%) and profoundly increased thyroid-stimulating hormone levels (∼800–1000-fold), indicating that Slc30a10 knock-out mice develop hypothyroidism. Importantly, a low-manganese diet produced lower tissue manganese levels in the knock-outs and rescued the phenotype, suggesting that manganese toxicity was the underlying cause. Our unanticipated discovery highlights the importance of determining the role of thyroid dysfunction in the onset and progression of manganese-induced disease and identifies Slc30a10 knock-out mice as a new model for studying thyroid biology.
Keywords: manganese, metal, thyroid, toxicity, transporter, SLC30, ZnT, cation diffusion facilitator, parkinsonism
Introduction
Manganese is an essential metal that becomes toxic at elevated levels (1). Historically, toxicity has been reported in occupational settings where workers are exposed to elevated manganese over prolonged periods (1). Recent studies suggest that toxicity may also develop after elevated exposure from environmental sources (2, 3). Additionally, manganese is primarily excreted in bile; therefore, patients with hepatic dysfunction fail to excrete manganese and may subsequently suffer from toxicity (4). The primary manifestation of overt manganese toxicity in occupationally exposed humans is the onset of a parkinsonian-like movement disorder (1, 5, 6). Epidemiological studies in children and adolescents show that elevated manganese exposure is associated with behavioral, cognitive, and motor defects (3, 7–15). The above neurological sequelae are attributed to direct neurotoxicity; manganese accumulates in the brain, particularly in the basal ganglia, and induces neuronal injury (1). That manganese, at elevated levels, is neurotoxic is well established; however, whether manganese induces damage to other organs in a manner that may, in turn, impact neurological function is unknown.
In 2012, homozygous mutations in SLC30A10 were reported to cause the first inherited disorder of manganese metabolism (16, 17). Patients exhibited large increases in blood manganese levels, had evidence of manganese deposition in the basal ganglia, and developed motor defects (16–19). Thus, the neurological sequelae were similar to that seen in individuals suffering from manganese toxicity caused by elevated exposure or liver disease (16, 17). Additionally, cirrhosis and polycythemia were reported (16, 17). Hepatic injury was likely a consequence of manganese deposition in the liver caused by decreased biliary excretion (16, 17) and could play a role in the neurotoxicity observed in these patients. Polycythemia was hypothesized to develop because of induction of erythropoietin gene by elevated manganese (16, 17, 20), but its contribution to observed neurological deficits is uncertain. Importantly, patients with SLC30A10 mutations had no history of exposure to elevated manganese, suggesting that homeostatic control of manganese was compromised (16, 17). Our recent studies provided insights into the molecular mechanisms. We discovered that SLC30A10 functioned as a cell-surface-localized manganese efflux transporter that protected cells and Caenorhabditis elegans against manganese toxicity (21, 22). Disease-causing mutations blocked the intracellular trafficking and manganese efflux activity of the transporter; consequently, cells and worms that expressed these mutants exhibited heightened sensitivity to manganese toxicity (21).
The SLC30 family of metal transporters has ten members, SLC30A1–A10, and is a constituent of the cation diffusion facilitator superfamily (23–25). Interestingly, although SLC30A10 mediates manganese efflux (21, 22, 26, 27), SLC30A1–A8 transport zinc (23–25). Our comparison of a structural prediction of SLC30A10 with the solved crystal structure of YiiP, a related bacterial zinc transporter (YiiP is the only cation diffusion facilitator that has a solved crystal structure (28, 29)), revealed that there were fundamental differences between a crucial zinc-binding site in the transmembrane domain of YiiP and the corresponding putative metal binding site of SLC30A10 (22). Such structural differences may contribute to the manganese transport specificity of SLC30A10.
Molecular studies are important in determining the mode of action of SLC30A10. However, to understand the process by which loss of function of SLC30A10 induces disease, studies in Slc30a10 knock-out mice are essential. Availability of genetically modified Slc30a10 strains will make it possible to determine whether a causal relationship exists between loss of SLC30A10 function and development of manganese-induced disease. Further, as described earlier, there is substantial overlap in the neurological presentation of patients who suffer from manganese toxicity caused by SLC30A10 mutations and those that develop toxicity caused by elevated manganese exposure or hepatic dysfunction (4–6, 16–19). Because of this overlap, Slc30a10 knock-out animals may become a useful model to study the mechanisms of manganese neurotoxicity in general. Finally, availability of these mice may aid efforts to develop small molecules for the management of manganese toxicity.
Here, we generated full-body, constitutive Slc30a10 knock-out mice and characterized their phenotype. We anticipated that the knock-out animals would develop motor defects. Instead, we made the surprising discovery that manganese toxicity induced severe hypothyroidism in these mice. Thus far, the relationship between thyroid function and manganese toxicity has received little attention, and whether human patients of manganese toxicity develop hypothyroidism is unknown. Importantly, however, thyroid hormone has profound effects on neuronal development and function (30), and if hypothyroidism is a feature of manganese toxicity, it may exacerbate the direct neurotoxic effects of manganese. Thus, an important implication of our findings is that it is now important to determine whether hypothyroidism contributes to the pathobiology of manganese toxicity in humans. Separately, our work identifies Slc30a10 knock-out mice to be a novel model to study thyroid biology.
Results
Generation of Slc30a10 knock-out mice
The mouse Slc30a10 gene has four exons and encodes a protein of 470 amino acids. Exon 1 encodes the first 205 amino acids, which encompasses a significant portion of the predicted transmembrane domain (amino acid 1–299). Because the transmembrane domain is required for metal transport (22), large deletions in this domain should inactivate the protein. In contrast, a partially active protein could be produced if downstream exons were deleted. Therefore, deletion of exon 1 provided the best approach to secure complete loss of function of Slc30a10; hence, we targeted exon 1.
We produced animals in which exon 1 of Slc30a10 was flanked by loxP sites, as described under “Experimental procedures.” Heterozygous and homozygous floxed mice appeared phenotypically normal. That is, unlike Slc30a10 knock-out mice, the body size and manganese levels of homozygous and heterozygous floxed animals were comparable with those of mice that were wild-type for the Slc30a10 gene (see Figs. 1 and 2 for phenotype of Slc30a10 knock-outs). Therefore, for this study, we considered homozygous and heterozygous floxed animals to be comparable with wild-types and designated these three genotypes as Slc30a10+/+.
Figure 1.

Slc30a10 knock-out mice are smaller in size and weigh less than littermate controls. A, genomic DNA was extracted from tail snips, and PCR analyses were performed using conditions and primers described under “Experimental procedures.” B, Slc30a10+/+, Slc30a10+/−, and Slc30a10−/− mice were euthanized at 6 weeks of age, and RNA was extracted from liver and brain of each animal. RT-PCR analyses were then performed. M denotes male; F denotes female. C, photograph of a 6-week-old male Slc30a10−/− mouse and a sex-matched Slc30a10+/+ littermate control. D, body weights of male Slc30a10+/+, Slc30a10+/−, and Slc30a10−/− mice were measured at the indicated ages (n = 11 mice for +/+, 17 for +/−, and 13 for −/− genotypes; *, p < 0.05 for the difference between −/− and other genotypes at the indicated ages using two-way ANOVA and Sidak's post hoc test; weights of +/+ and +/− mice were comparable at all ages). E, weights of female Slc30a10+/+, Slc30a10+/−, and Slc30a10−/− mice were measured at the indicated ages (n = 9 mice for +/+, 14 for +/−, and 5 for −/− genotypes; *, p < 0.05 for the difference between −/− and other genotypes at the indicated ages using two-way ANOVA and Sidak's post hoc test; weights of +/+ and +/− mice were comparable at all ages). F, weights of Slc30a10−/− mice at day 27 from D and E above are expressed as percentages of corresponding Slc30a10+/+ controls (*, p < 0.05 using unpaired, two-tailed Student's t test).
Figure 2.

Manganese levels are elevated in 6-week-old Slc30a10 knock-out mice. A–D, Slc30a10+/+, Slc30a10+/−, and Slc30a10−/− mice were euthanized at 6 weeks of age. Samples of brain, liver, and blood were collected and used to measure absolute levels of manganese, zinc, copper, and iron (n = 8 males (M) and 4 females (F) for +/+, 7 males and 7 females for +/−, and 7 males and 5 females for −/− genotypes; the same animals were used to analyze all four metals; *, p < 0.05 for the difference between −/− and other genotypes for indicated measurements using two-way ANOVA and Sidak's post hoc test, irrespective of sex (main column effect); there were no differences between +/+ and +/− genotypes for any metal in any tissue; small, but statistically significant (p < 0.05 by two-way ANOVA and Sidak's post hoc test), sex-specific differences were seen in liver manganese, blood manganese, liver copper, and blood iron in Slc30a10−/− mice and in brain zinc and liver copper in Slc30a10+/− mice).
To delete exon 1, we bred floxed animals with transgenic Sox2Cre mice (see “Experimental procedures” for details about the Sox2Cre strain) and obtained animals in which exon 1 of one copy of Slc30a10 was deleted. These mice were designated as Slc30a10+/− and were heterozygous for the Slc30a10 knock-out. After another round of breeding, we obtained mice in which exon 1 of Slc30a10 was deleted in both chromosomes; these mice were the knock-out strain and designated Slc30a10−/−. Note that some Slc30a10+/− and Slc30a10−/− mice retained the Cre transgene; however, there was no effect of Cre expression on body size or manganese levels. Therefore, for these genotypes, we combined animals with or without Cre expression into one group.
To routinely genotype animals, we performed PCR from genomic DNA. We ran two separate reactions designed to either amplify a product from the wild-type/floxed allele or from the knock-out allele (Fig. 1A). In Slc30a10+/+ animals, a positive product was detected for the wild-type/floxed allele but not the knock-out allele; in Slc30a10+/−, positive products were detected for both reactions; and in Slc30a10−/−, a positive PCR product was detected for the knock-out, but not the wild-type/floxed allele (Fig. 1A). For selected animals, we verified the PCR results by performing reverse transcription-PCR assays, which confirmed that Cre-mediated recombination led to a loss of Slc30a10 mRNA in Slc30a10−/− mice (Fig. 1B). Despite extensive efforts, we were unable to validate loss of Slc30a10 gene product at the protein level in knock-outs using immunoblots because we could not identify any commercial antibody that specifically detected SLC30A10 in rodent tissue. To get around this hurdle, we generated an antibody against the C-terminal domain of human SLC30A10. Unfortunately, we discovered that although this antibody specifically detected SLC30A10 in human cell lines, it lost specificity when mouse tissue samples were used. Despite the lack of immunoblot data, the results obtained from the genomic DNA PCR were unequivocal and confirmed by reverse transcription-PCR. Therefore, we used PCR as an accurate means to genotype animals for further studies.
Slc30a10 knock-out mice are smaller in size and die prematurely
When we obtained the initial litters that contained knock-out mice, we observed that Slc30a10−/− animals were smaller than Slc30a10+/+ and Slc30a10+/− littermate controls. Further, knock-outs started dying by ∼6–8 weeks of age. This surprising finding led us to maintain heterozygous breeders and closely follow the growth and development of the knock-outs produced. Slc30a10−/− animals were born at expected Mendelian ratios and, until post-natal day 16–18, were indistinguishable from Slc30a10+/+ and Slc30a10+/− controls. However, after that, body weights of Slc30a10−/− mice failed to keep up with that of littermate controls (Fig. 1, C–E). The difference was exacerbated after weaning, and by 4 weeks of age, the weights of male and female Slc30a10−/− mice were ∼40 and ∼60% of corresponding littermate controls, respectively (Fig. 1, C–F). There was also a sex difference in the phenotype; the difference in weights between controls and knock-outs was greater and evident earlier in males (Fig. 1, D–F). Although we could not rigorously assay for the time of death in the knock-outs, because it would induce unnecessary suffering, observations in the first few litters suggested that male knock-outs died before females. There was no difference between weights of Slc30a10+/+ and Slc30a10+/− mice (Fig. 1, D and E). Thus, Slc30a10−/− animals are smaller in size than littermate controls and die prematurely, whereas Slc30a10+/− mice are unaffected.
Manganese levels are elevated in Slc30a10−/− mice
To determine whether the unexpected failure-to-thrive phenotype was related to alterations in manganese homeostasis, we assayed for the levels of manganese and other essential metals in blood, liver, and brain of knock-outs and littermate controls at 6 weeks of age. We discovered that compared with Slc30a10+/+ and Slc30a10+/− mice, in Slc30a10−/− animals, manganese levels were substantially elevated (∼20–60-fold) (Fig. 2A). There was no difference in tissue manganese levels between Slc30a10+/+ and Slc30a10+/− mice (Fig. 2A). Importantly, in the same animals, zinc levels were comparable across genotypes (Fig. 2B). In the Slc30a10−/− strain, there were minor increases in blood copper and iron and brain iron (Fig. 2, C and D), but the magnitude of these changes were far lesser than that seen with manganese (Fig. 2A), suggesting that changes in other metals were likely secondary to those in manganese. Overall, manganese levels are very high in Slc30a10−/− mice. These results suggest that loss of function of Slc30a10 in mice impacts manganese homeostasis in a manner similar to that seen in humans.
In Slc30a10−/− mice, there are extensive alterations in the thyroid
The high tissue manganese levels in Slc30a10−/− mice did not provide a direct clue to the underlying mechanism that led to their small body size and shortened life span. As the next step, we performed detailed pathological analyses of all organ systems at 6 weeks of age. Our expectation was that elevated manganese would induce neural and/or hepatic injury in the knock-outs. Remarkably, we did not find histological evidence of damage in the brain. Although the above finding did not imply that neural function of Slc30a10−/− animals was uncompromised, the lack of detectable tissue damage was surprising. Additionally, analyses of sections of the liver provided evidence of only mild degenerative changes (Fig. 3A). The expected diffuse glycogen vacuolation was evident in hepatocytes of Slc30a10+/+ and Slc30a10+/−, but not Slc30a10−/− animals, implying that glycogen levels were depleted in the knock-outs (Fig. 3A). Further, in the knock-outs, there was hepatocellular vacuolation centered on centrilobular areas (Fig. 3A), suggestive of mild degenerative change. It seemed unlikely that such limited hepatic injury could generate the severe phenotype observed. Therefore, we carefully analyzed other organs. Unexpectedly, we discovered that there were marked changes in the thyroid gland (Fig. 3B). Compared with Slc30a10+/+ and Slc30a10+/− genotypes, in Slc30a10−/− mice, there was a reduction in the amount of colloid within the thyroid follicles, and the follicular epithelial cells were hypertrophic (Fig. 3B). All other organ systems were unremarkable. Thus, the thyroid gland of Slc30a10−/− mice has extensive morphological alterations.
Figure 3.
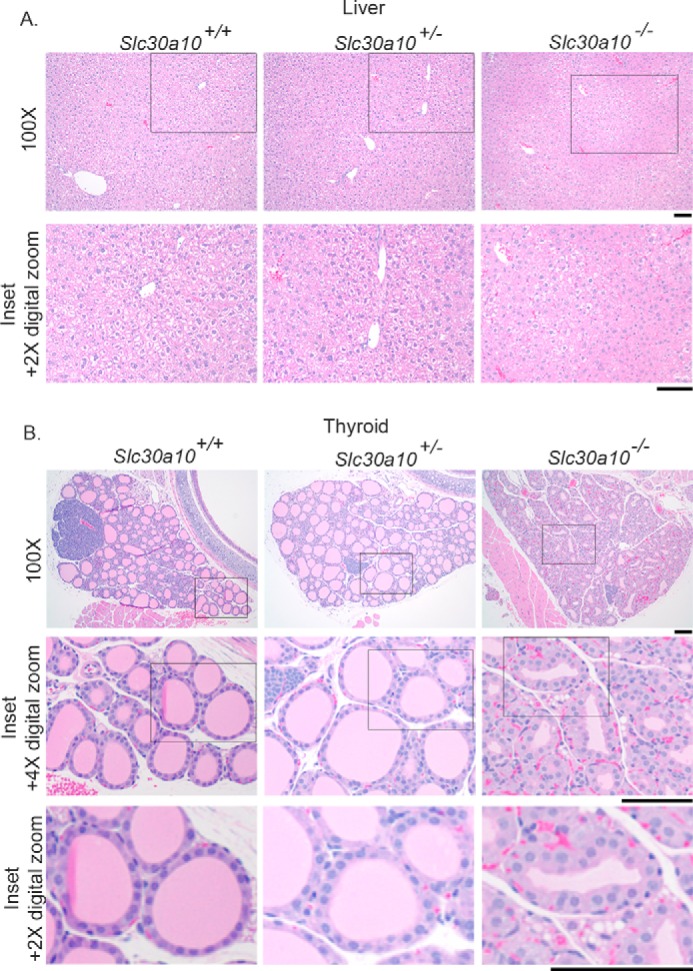
Alterations in the thyroid gland of Slc30a10 knock-out mice. A and B, sections of liver (A) and thyroid (B) were generated from 6-week-old, male Slc30a10+/+, Slc30a10+/−, and Slc30a10−/− mice, stained with hematoxylin-eosin, and imaged. Scale bars, 100 μm.
Slc30a10 knock-out mice suffer from hypothyroidism
The pathological changes observed raised the possibility that Slc30a10−/− mice were suffering from hypothyroidism. The decrease in colloid could be indicative of a reduction in thyroid hormone production, and hypertrophy of the follicular epithelial cells could be a compensatory response to enhance production. Moreover, small body size and premature death are evident in other knock-out strains in which animals develop severe hypothyroidism (as examples, in Pax8 or thyroid-stimulating hormone receptor knock-out (31, 32)). Therefore, we assayed for serum levels of the thyroid hormone thyroxine. Because functionality of the thyroid gland is closely regulated by thyroid-stimulating hormone produced by the anterior pituitary, we assayed for serum levels of this hormone as well. In 6-week-old knock-out mice, compared with littermate controls, there was an enormous increase in levels of thyroid-stimulating hormone (∼800–1,000-fold), whereas thyroxine levels were substantially reduced (∼50–80%) (Fig. 4, A and B). These changes were consistent with the onset of primary hypothyroidism in which a defect in the thyroid gland decreases thyroxine levels leading to a compensatory increase in thyroid-stimulating hormone caused by loss of negative feedback regulation. To rule out the possibility that the increase in thyroid-stimulating hormone was due to an alteration in anterior pituitary function, we measured serum levels of prolactin, growth hormone, follicle-stimulating hormone, and adrenocorticotrophic hormone in 6-week-old animals. We observed that in Slc30a10−/− mice, compared with littermate controls, there was a modest reduction in prolactin levels, and no change in levels of the other hormones assayed (Fig. 5), suggesting that overall functionality of the anterior pituitary was not compromised. We also noted that in Slc30a10−/− mice, thyroxine levels of male knock-outs were lower and thyroid-stimulating hormone levels were greater than females (Fig. 4), consistent with the lower body weight of males (Fig. 1F). The results of the hormone measurement and pathology assays put together indicate that by 6 weeks of age, Slc30a10−/− animals develop severe hypothyroidism and that male knock-outs are more sensitive. These results further suggest that the hypothyroidism may be due to a primary defect in the thyroid gland and not due to alterations in anterior pituitary function.
Figure 4.

Thyroid-stimulating hormone levels are elevated, whereas thyroxine levels are reduced, in 6-week-old Slc30a10 knock-out mice. A and B, Slc30a10−/− mice and littermate controls were euthanized at 6 weeks of age. Serum samples were prepared and used to measure thyroid-stimulating hormone (TSH; A) or thyroxine (T4; B). For each sex, animals of the +/+ and +/− genotypes were combined into a control group (n = 14 males (M) and 9 females (F) for control, and 6 males and 7 females for −/− (denoted as Knockout) groups; *, p < 0.05 for the difference between control and −/− groups using two-way ANOVA and Sidak's post hoc test; the nature of the data (two genotypes and two sexes) necessitated that comparisons be performed in a sex-specific manner across genotypes; also, there was a sex-specific difference in levels of both hormones in Slc30a10−/− mice (p < 0.05 by two-way ANOVA and Sidak's post hoc test)).
Figure 5.

Measurements of additional anterior pituitary hormones in 6-week-old Slc30a10 knock-out mice and littermate controls. A–D, levels of indicated anterior pituitary hormones were measured in serum samples obtained from 6-week-old Slc30a10−/− mice and littermate controls (n = 14 males (M) and 9 females (F) for control, and 7 males and 7 females for −/− (denoted as Knockout) groups; the animals used were the same as those used in Fig. 4 with the exception that samples from one additional male knock-out were analyzed; *, p < 0.05 for the difference between control and −/− groups using two-way ANOVA and Sidak's post hoc test; there were sex-specific differences in levels of follicle-stimulating hormone (FSH) in control and −/− groups and in adrenocorticotrophic hormone (ACTH) in the control group (p < 0.05 using two-way ANOVA and Sidak's post hoc test)). GH, growth hormone.
As part of these studies, we felt it was also important to determine whether Slc30a10 knock-out mice were hypothyroid at a time when the failure-to-thrive phenotype was not yet evident. For this, we performed assays in 18-day-old animals. As described earlier, at this age, body weights of knock-out animals were comparable with those of controls (Fig. 1, D and E). Importantly, at 18 days, there were no differences in serum levels of thyroxine or thyroid-stimulating hormone between controls and knock-outs (Fig. 6, A and B), although by this age, blood manganese levels of knock-outs were already ∼10-fold greater than controls (Fig. 6C).
Figure 6.

Measurements of thyroid-stimulating hormone, thyroxine, and blood manganese in preweanling mice. A and B, Slc30a10−/− mice and littermate controls were euthanized at 18 days of age. Serum levels of thyroid-stimulating hormone (TSH; A) or thyroxine (T4; B) were then measured. For each sex, animals of the +/+ and +/− genotypes were combined into a control group (n = 3 males (M) and 3 females (F) for control and −/− (denoted as Knockout) groups; there were no genotype- or sex-specific differences between groups (p > 0.05 using two-way ANOVA and Sidak's post hoc test)). C, blood manganese levels were measured in 15-day-old Slc30a10−/− mice and littermate controls. Animals of the +/+ and +/− genotypes were combined into a control group. Additionally, the sexes were combined for analyses (n = 10 for control (6 males and 4 females), and 5 for −/− (denoted as Knockout; 3 males and 2 females) groups; *, p < 0.05 using unpaired, two-tailed Student's t test).
A low-manganese diet rescues the phenotype of Slc30a10−/− mice
To determine whether manganese toxicity was the underlying cause of the phenotype, we sought to reduce manganese levels and test whether this intervention could rescue. Manganese levels are very high in commercial rodent chow (∼84 μg/g). We purchased a commercially available low-manganese diet that had ∼11 μg manganese/g chow. For the experiment, we changed the diet of animals at 10 days of age, meaning that when litters were 10 days old, but pups were still suckling, the diet of the mother was swapped from regular rodent chow to a low-manganese diet. We decided to switch the diet at this early stage because pups often have access to solid food before they are weaned as small particles of food pellets may fall into the cage, and by ∼2 weeks of age, pups are large enough to reach pellets placed on the top of the cage. After weaning, pups continued to receive the modified low-manganese diet for the duration of the experiment. To assess the effectiveness of the new diet, we determined metal levels in brain, liver, and blood at 6 weeks of age. Tissue manganese levels of Slc30a10−/− mice were still greater than those of littermate controls (Fig. 7A). Importantly, however, manganese levels in the brain and liver of knock-out mice fed the low-manganese diet were significantly lower than those of knock-outs fed regular rodent chow (Fig. 7B). For reasons that are unclear, the low-manganese diet only minimally reduced blood manganese in female knock-outs and had no effect in males (Fig. 7B). Despite the lack of effect in the blood, the above data showed that the low-manganese diet was effective, at least partially, in reducing the body burden of manganese in Slc30a10−/− mice. We note two additional points about the specificity of the modified diet. First, there was no difference in manganese levels of control animals fed regular rodent chow or the low-manganese diet (Fig. 7B), implying that the diet, by itself, did not change the manganese status of animals that did not carry mutations in Slc30a10. Second, measurements of other metals revealed that in animals fed the low-manganese diet, between Slc30a10−/− and control mice, there were only modest changes in tissue zinc, copper, and iron levels (Fig. 7, C–E), indicating that the diet did not unexpectedly change tissue levels of another metal. Based on the results of these metal measurement assays, we moved forward with the rescue experiment. Importantly, at the 6-week time point, in Slc30a10−/− mice fed the low-manganese diet, levels of thyroid-stimulating hormone and thyroxine were comparable with controls (Fig. 8, A and B). Additionally, at this time point, body weights of knock-out mice were also comparable with controls (Fig. 8, C–E). Thus, although manganese levels in Slc30a10−/− animals on the modified diet were more than controls, the reduction achieved over the regular diet was sufficient to rescue the phenotype. These results indicate that manganese toxicity induces the failure-to-thrive and hypothyroidism phenotype seen in Slc30a10−/− mice.
Figure 7.

The modified low-manganese diet produces lower manganese levels in Slc30a10 knock-out mice. A, Slc30a10−/− mice and littermate controls were fed the modified low-manganese diet and euthanized at 6 weeks of age. Samples of brain, liver, and blood were collected and processed for measurements of absolute amounts of manganese. For each sex, Slc30a10+/+ and Slc30a10+/− animals were combined into a control group (n = 6 males (M) and 8 females (F) for control and 5 males and 5 females for −/− (denoted as Knockout) groups; *, p < 0.05 for the difference between control and −/− groups using two-way ANOVA and Sidak's post hoc test; there were small, but statistically significant, sex-specific differences in brain and liver manganese in Slc30a10−/− mice (p < 0.05 using two-way ANOVA and Sidak's post hoc test)). B, Slc30a10+/+ and Slc30a10+/− mice used to measure manganese levels in Fig. 2A were combined into a control group named “control animals on regular diet” (sexes were kept separate). Mean manganese levels in the brain, liver, and blood of these mice were calculated separately for both sexes. After this, the fold difference in manganese levels between this group and Slc30a10−/− mice fed regular rodent chow (from Fig. 2A; denoted as Knockout animals on regular diet), control mice fed the modified diet (from Fig. 7A above; denoted as Control animals on low Mn diet), or Slc30a10−/− mice fed the modified diet (also from Fig. 7A above; denoted as Knockout animals on low Mn diet) was calculated in a sex-specific manner (*, p < 0.05 for indicated comparisons using two-way ANOVA and Sidak's post hoc test; for blood, between Knockout animals on regular diet and Knockout animals on low Mn diet, there was a modest, but statistically significant, difference in females (F), but no difference in males (M); there were no differences between the Control animals on regular diet and Control animals on low Mn diet groups in any of the comparisons). C–E, absolute levels of zinc, copper, and iron were measured in the samples used to assay for manganese in Fig. 7A (*, p < 0.05 for the difference between control and −/− (denoted as Knockout) groups using two-way ANOVA and Sidak's post hoc test; small, but statistically significant, sex-specific differences were evident in liver copper and iron in control animals (p < 0.05 using two-way ANOVA and Sidak's post hoc test)).
Figure 8.

The modified low-manganese diet rescues the phenotype of Slc30a10 knock-out mice. A and B, Slc30a10−/− mice and littermate controls were fed the modified low-manganese diet and euthanized at 6 weeks of age. Slc30a10+/+ and Slc30a10+/− animals were combined into a control group. Serum samples were used to assay for levels of thyroid-stimulating hormone (TSH; A) or thyroxine (T4; B) (n = 5 males (M) and 5 females (F) for control, and 3 males and 3 females for −/− (denoted as Knockout) groups; for both panels, hormone levels were comparable across genotypes (p > 0.05 using two-way ANOVA and Sidak's post hoc test); there was a sex-specific difference in levels of thyroid-stimulating hormone in the control group (p < 0.05 using two-way ANOVA and Sidak's post hoc test)). C, photograph of a 6-week-old male Slc30a10−/− mouse and a sex-matched Slc30a10+/+ littermate control. Both animals were fed the modified low-manganese diet. D, male Slc30a10−/− mice and littermate controls were fed the modified low-manganese diet and weighed at indicated ages. Slc30a10+/+ and Slc30a10+/− animals were combined into a control group (n = 7 mice for control and 6 for −/− (denoted as Knockout) groups; *, p < 0.05 for the difference between control and −/− groups at the indicated ages using two-way ANOVA and Sidak's post hoc test; by the end of the assay, on day 41, the weights of mice in control and −/− groups were comparable (p > 0.05)). E, female Slc30a10−/− mice and littermate controls on the low-manganese diet were weighed at indicated ages. Slc30a10+/+ and Slc30a10+/− animals were combined into a control group (n = 8 mice for control and 5 for −/− (denoted as Knockout) groups; *, p < 0.05 for the difference between control and −/− groups at the indicated ages using two-way ANOVA and Sidak's post hoc test; by the end of the assay, on day 41, the weights of mice in control and −/− groups were comparable (p > 0.05)).
Manganese levels are elevated in the thyroid gland of Slc30a10−/− mice
As the final step of the current work, we assayed for metal levels in the thyroid. In 1-month-old knock-out mice, thyroid manganese levels were ∼10-fold higher than controls; zinc, copper, and iron were not enhanced (Fig. 9, A–D). Thus, manganese levels are substantially elevated in the thyroid gland of knock-out mice. It was tempting to attribute the observed hypothyroidism to direct toxicity induced by manganese in the thyroid. To test the feasibility of this idea, we compared manganese levels in thyroid, pituitary, and brain of 1-month-old animals. We chose the pituitary and brain for comparison because thyroid-stimulating hormone, produced by the pituitary, and thyrotropin-releasing hormone, produced by the hypothalamus in the brain, control thyroxine production by the thyroid (30). Interestingly, in Slc30a10−/− animals, the magnitude of increase in thyroid manganese levels (∼10-fold) was significantly greater than in the pituitary (only ∼2-fold) but significantly lesser than the ∼20-fold increase observed in the brain (Fig. 9, E–G). These results suggest that elevated thyroid manganese may not be the only driving mechanism inducing hypothyroidism in Slc30a10−/− mice, and they underscore the importance of generating tissue-specific (brain or thyroid) Slc30a10 knock-out strains for further assays.
Figure 9.

Manganese levels are elevated in the thyroid gland of Slc30a10−/− mice. A–D, metal levels were measured in samples of the thyroid gland prepared from 1-month-old Slc30a10−/− mice and littermate controls. Animals of the +/+ and +/− genotypes were combined into a control group (n = 4 males (M) and 3 females (F) for control and −/− (denoted as Knockout) groups; the same animals were used to analyze all four metals; *, p < 0.05 for indicated differences using two-way ANOVA and Sidak's post hoc test). E and F, manganese levels were measured in samples of the brain (E) or pituitary (F) obtained from 1-month-old Slc30a10−/− mice and littermate controls. Animals of the +/+ and +/− genotypes were combined into a control group (n = 10 males (M) and 13 females (F) for control and 7 males and 4 females for −/− (denoted as Knockout) groups; brain and pituitary samples were obtained from the same animals; *, p < 0.05 for indicated differences using two-way ANOVA and Sidak's post hoc test; there was a sex-specific difference in brain manganese levels in Slc30a10−/− mice). G, fold differences in manganese levels between knock-out and control animals from A, E, and F were calculated after combining sexes (*, p < 0.05 for indicated differences using one-way ANOVA and Tukey Kramer post hoc test).
Discussion
The discovery that Slc30a10−/− mice develop hypothyroidism raises the possibility that thyroid dysfunction may be a feature of manganese toxicity in humans. Thyroid hormone plays an essential role in neurological development and function, and hypothyroidism induces severe neurological defects (30). Indeed, hypothyroidism during fetal or neonatal life or during early childhood may lead to the onset of cretinism; in this syndrome, affected children may exhibit severe growth defects, suffer from mental retardation, have wide-ranging neurological deficits, such as compromised motor skills, and develop hearing loss (30, 33). Hypothyroidism in early life may also induce defects in learning, memory, and attention abilities (33). Interestingly, in prior rodent studies of manganese toxicity, animals exposed to manganese during early life (post-natal days 1–21) developed behavioral, learning, attentional, and fine motor deficits (34–36). Findings from these animal assays were consistent with epidemiological reports linking manganese exposure in children and adolescents to defects in cognitive, behavioral, and fine motor function (3, 7–15). In our model, knock-out mice started developing growth deficits at approximately post-natal day 21 and were severely hypothyroid by post-natal day 42. These ages correspond to a human age of ∼2–18 y (37). Thus, our current results likely have more relevance to manganese exposure and hypothyroidism in early life. A direct implication of our work is that it is now important to determine the thyroid status of manganese-exposed children. Decreased thyroid hormone levels, if present in such children, may cause, contribute to, or exacerbate the neurotoxic effects of manganese. An important corollary is that thyroxine supplementation may provide a straightforward means to mitigate some of the observed neurological deficits.
Thus far, only a handful of studies have investigated the relationship between thyroid function and manganese toxicity; however, a few prior findings provide historical precedent for our discovery (38). As examples, in 1977, Buthieau and Autissier (39) showed that, in rats 24 h after injection of MnCl2 (2 mg/100 g), there was a reduction in the uptake of radioactive iodine in the thyroid and a decrease in serum thyroxine. In 1983, the same authors (40) demonstrated that rats treated with manganese for a longer period (1 mg MnSO4·H2O/100 g daily for 5 weeks) had decreases in body weight, and serum thyroxine and thyroid-stimulating hormone levels. In 1985, Kawada et al. (41) reported that ingestion of 200 mg/liter MnCl2·4H2O for 7 weeks induced goiter in female mice. In that study, manganese-treated animals also exhibited a mild reduction in serum thyroxine levels (41). Finally, a study by the National Toxicology Program demonstrated that mice fed very high manganese (15,000 μg manganese/g) for 2 years developed thyroid follicular dilatation and hyperplasia (42).
At this time, we can only speculate about the molecular mechanisms leading to hypothyroidism in Slc30a10−/− mice. Thyroid manganese levels were elevated in Slc30a10−/− animals, and increased thyroid manganese may interfere with one or more steps of thyroxine production. As one example, elaborate trafficking pathways mediate transport of thyroglobulin between the cytoplasm of thyroid follicular epithelial cells and the lumen (30), and intracellular protein trafficking is altered in manganese-treated cells and animals (43–45). Additionally, as described above, studies in rodents showed that manganese blocked uptake of iodine into thyroid (39). An effect on iodine homeostasis, if present in the knock-outs, may contribute to the development of hypothyroidism. It is also noteworthy that, in Slc30a10−/− mice, accumulation of manganese in the brain exceeded that in the thyroid. Because thyroid function is regulated by thyrotropin-releasing hormone produced by the hypothalamus (30), it remains possible that yet-to-be-discovered manganese-induced functional alterations in the hypothalamus or other brain regions initiate or contribute to the thyroid dysfunction.
It was previously reported that CD-1 mice expressed greater levels of Slc30a10 gene product in the pituitary compared with the thyroid (46). In our studies in Slc30a10−/− mice, accumulation of manganese in the thyroid was greater than that in the pituitary. The reasons for this are unclear; it may be reflective of strain-specific differences or of the capability of another transporter to compensate for the loss of Slc30a10 in the pituitary but not the thyroid.
There was a clear sex-specific difference in the phenotype of Slc30a10−/− mice; thyroxine levels and normalized body weights were lower in males. Sex-specific differences in the effects of manganese on thyroid function were also described in the report by Kawada et al. (41). Unlike our study, in that of Kawada et al. (41), female mice were sensitive. The difference between our work and that of Kawada et al. with regards to which sex is more sensitive may relate to differences in manganese levels in the mice used in these separate studies. Interestingly, Kawada et al. (41) showed that castration of male mice made them sensitive to developing manganese-induced goiters, and this effect was rescued by testosterone treatment. Separately, estrogen exerted a protective effect on manganese toxicity in primary neuronal and astrogial cultures and in mice (47–49). Considering that there are sex differences in thyroid disease in humans, that gonadal steroid hormones modulate effects of thyroid hormones, and that a euthyroid state is necessary for normal reproductive function (50), manganese exposure may change how androgens and estrogens influence thyroid function. Future castration and ovariectomy experiments in Slc30a10−/− mice, together with hormone replacement studies, may provide insight into the mechanisms leading to the sex-specific phenotypic differences we observed.
In summary, Slc30a10 knock-out mice develop severe hypothyroidism because of manganese toxicity, raising the possibility that thyroid dysfunction may play a role in the pathobiology of manganese-induced disease in humans.
Experimental procedures
Animal experiments
All experiments with mice were approved by the Institutional Animal Care and Use Committee of the University of Texas at Austin. The conditional gene targeting vector with loxP sites flanking exon 1 of Slc30a10 was constructed by Cyagen Biosciences (Santa Clara, CA). We electroporated the targeting vector into V6.5 embryonic stem cells (C57BL/6 × 129/Sv) (51). Electroporation, PCR screening, Southern analysis of embryonic stem cell clones, and blastocyst injection experiments were performed by the University of Texas at Austin Mouse Genetic Engineering Facility following standard methods (52). Germ-line chimeric males were bred with wild-type C57BL/6J mice, obtained from The Jackson Laboratory (Bar Harbor, ME), to produce mice heterozygous for the floxed allele. These heterozygous floxed mice were intercrossed to produce homozygous floxed animals. Because there was no detectable phenotype in heterozygous and homozygous floxed mice, we considered homozygous and heterozygous floxed animals to be comparable with wild-types and designated these three genotypes as Slc30a10+/+.
To delete exon 1 of Slc30a10, we bred floxed animals with Sox2Cre transgenic mice. In this transgenic strain, Cre gene product induces recombination in all cells that arise from the epiblast by embryonic day 6.5 (53). Because of recombination at this early stage of life, the mutation is introduced into male and female gametes and is transmitted to future generations without further need for Cre expression. Through this breeding strategy, we obtained animals in which exon 1 of Slc30a10 was deleted in one chromosome. These animals were heterozygous for the Slc30a10 knock-out (designated Slc30a10+/−). A further round of breeding produced animals in which exon 1 was deleted in both chromosomes; these mice constituted the knock-out strain (designated Slc30a10−/−). Some Slc30a10+/− and Slc30a10−/− mice retained the Cre transgene. However, because there was no effect of Cre expression on body size or manganese levels, for these genotypes, we combined animals with or without Cre expression into one group. The Sox2Cre strain was obtained from The Jackson Laboratory.
The mice were housed in the specific pathogen-free facility of the University of Texas at Austin in a room maintained at ∼21 °C with a 12-h light-dark cycle (lights on between 7 p.m. and 7 a.m.). After weaning, 3–4 littermates of the same sex were kept per cage. Animals had free access to food and water. The regular diet was PicoLab Rodent Diet 20, which contains ∼84 μg manganese/g chow. The low-manganese diet was AIN-93G, which contains ∼11 μg manganese/g chow.
PCR and reverse transcription-PCR
For PCR, genomic DNA was extracted from tail snips using the Extracta DNA Prep for PCR tissue kit (Quanta Biosciences, Beverly, MA). PCR was performed using the AccuStart II PCR Supermix reagent (Quanta). The primers used were as follows: (i) to amplify the wild-type/floxed Slc30a10 allele: forward, 5′-CGG ACT GGG AGC ACT TTG TT-3′, and reverse, 5′-ATA CTG CCA CAG GAG AGG GG-3′; (ii) to amplify the recombined/knock-out Slc30a10 allele: forward 5′-CAC CTT TAA GGG CAC TAG GC-3′, and the reverse primer used for the wild-type/floxed allele above; and (iii) to amplify Cre: forward, 5′-GGA CAT GTT CAG GGA TCG CCA GGC-3′, and reverse, 5′-CGA CGA TGA AGC ATG TTT AGC TG-3′. PCR conditions for all three reactions were as follows: 95 °C for 3 min; followed by 30 cycles of 95 °C for 30 s, 60 °C for 30 s, and 72 °C for 35 s; and a final step of 72 °C for 1 min. All three reactions generated a single PCR product ∼250 base pairs in size. For reverse transcription-PCR, the mice were euthanized by decapitation after isoflurane (Piramal Enterprises, Mumbai, India) anesthesia, and samples of liver and brain were flash-frozen in liquid nitrogen. RNA was extracted and reverse-transcribed into cDNA as described by us previously (21). After this, PCR was performed using the AccuStart II reagent described above. Primers used were as follows: (i) for Slc30a10, forward, 5′-GTA GCA GGT GAT TCC CTG AAC-3′, and reverse, 5′-GTG ATG ACC ACA ACC ACG GAC-3′, and (ii) for 18S, forward, 5′-CAT TAA GGG CGT GGG GCG G-3′ and reverse, 5′-GTC GTG GGT TCT GCA TGA TG-3′. PCR conditions were as follows: 95 °C for 3 min; followed by 30 cycles of 95 °C for 30 s, 50 °C for 30 s, and 72 °C for 35 s; and a final step of 72 °C for 1 min. Both PCR products were <200 base pairs in size.
Metal measurements
For brain, liver, and blood samples, the animals were anesthetized with isoflurane and euthanized by decapitation. Midbrain and liver samples were collected in microcentrifuge tubes that had been previously washed with 10% nitric acid to remove metal contamination, as described by us previously (21), flash-frozen in liquid nitrogen, and stored at −80 °C. Blood samples were collected in heparinized tubes (BD Vacutainer tubes; Becton Dickinson) and stored at −80 °C. Subsequently, the metal levels were analyzed by inductively coupled plasma mass spectrometry, as described by us previously (34). For thyroid samples, the animals were euthanized using carbon dioxide. Thyroid was then dissected. Further processing was as described for brain and liver.
Pathology
Pathology analyses were performed at the M. D. Anderson Cancer Center by a board-certified anatomic veterinary pathologist. The mice were exsanguinated under deep isoflurane anesthesia. Immediately following exsanguination, thoracotomy was performed, and a complete necropsy was done. Organs were dissected, fixed in formalin, and embedded in paraffin. 3–5-μm-thick sections were cut and stained using hematoxylin and eosin. Images were captured using an Olympus BX43 microscope at 100× magnification using a Diagnostic Instrument camera.
Hormone assays
The animals were euthanized by decapitation after isoflurane anesthesia. To ensure that levels did not reflect diurnal variations, all animals were euthanized at approximately the same time of day (between 2 and 4 p.m.). To obtain serum samples, blood was collected in microcentrifuge tubes and allowed to clot for 30 min at room temperature. After this, tubes were spun at 1000 rpm for 10 min at room temperature. The supernatant, which was the serum, was transferred to new microcentrifuge tubes and frozen at −80 °C until use. Anterior pituitary hormones were measured with the Milliplex MAP mouse pituitary magnetic bead panel (EMD Millipore, Billerica, MA) using recommendations made by the manufacturer (54). Thyroxine was measured using the AccuDiag T4 ELISA kit (Diagnostic Automation/Cortex Diagnostics, Inc., Woodland Hills, CA); the manufacturer's recommendations were followed.
Chemicals
Unless specified, chemicals and reagents were from Thermo Fisher Scientific or Sigma-Aldrich.
Statistical analyses
The animal numbers are provided in the figure legends. Comparisons between multiple groups were performed using one-way or two-way ANOVA.2 Comparisons between two groups were performed using Student's t test. All analyses were performed using the Prism 6 software (GraphPad, La Jolla, CA). p < 0.05 was considered to be significant. Asterisks in graphs, where present, denote statistically significant differences.
Author contributions
S. H., C. L., T. J., W. S., and B. K. C. performed experiments; W. Y. provided essential technical expertise; S. M. conceived the project and designed studies with assistance from D. R. S., M. A., and A. C. G.; S. M. and D. R. S. analyzed data; S. M. wrote the manuscript with critical contributions from D. R. S., M. A., and A. C. G. All authors reviewed and approved the final version of the manuscript.
Acknowledgments
We thank Dr. Steven Vokes (University of Texas at Austin) for advice about knock-out animal generation; Dr. Anthony Hollenberg and Dr. Kristen Vella (Harvard Medical School) for technical assistance with thyroxine measurement assays; and Dr. Edward Mills (University of Texas at Austin) and Dr. Adam Linstedt (Carnegie Mellon University) for critical comments on the manuscript.
This work was supported by NIEHS, National Institutes of Health Grants R01-ES024812 and R00-ES020844 (to S. M.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
- ANOVA
- analysis of variance.
References
- 1. Aschner M., Erikson K. M., Herrero Hernández E., and Tjalkens R. (2009) Manganese and its role in Parkinson's disease: from transport to neuropathology. Neuromolecular Med. 11, 252–266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Lucchini R. G., Guazzetti S., Zoni S., Benedetti C., Fedrighi C., Peli M., Donna F., Bontempi E., Borgese L., Micheletti S., Ferri R., Marchetti S., and Smith D. R. (2014) Neurofunctional dopaminergic impairment in elderly after lifetime exposure to manganese. Neurotoxicology 45, 309–317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Lucchini R. G., Guazzetti S., Zoni S., Donna F., Peter S., Zacco A., Salmistraro M., Bontempi E., Zimmerman N. J., and Smith D. R. (2012) Tremor, olfactory and motor changes in Italian adolescents exposed to historical ferro-manganese emission. Neurotoxicology 33, 687–696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Butterworth R. F. (2013) Parkinsonism in cirrhosis: pathogenesis and current therapeutic options. Metab. Brain Dis. 28, 261–267 [DOI] [PubMed] [Google Scholar]
- 5. Olanow C. W. (2004) Manganese-induced parkinsonism and Parkinson's disease. Ann. N.Y. Acad. Sci. 1012, 209–223 [DOI] [PubMed] [Google Scholar]
- 6. Perl D. P., and Olanow C. W. (2007) The neuropathology of manganese-induced Parkinsonism. J. Neuropathol. Exp. Neurol 66, 675–682 [DOI] [PubMed] [Google Scholar]
- 7. Bhang S. Y., Cho S. C., Kim J. W., Hong Y. C., Shin M. S., Yoo H. J., Cho I. H., Kim Y., and Kim B. N. (2013) Relationship between blood manganese levels and children's attention, cognition, behavior, and academic performance: a nationwide cross-sectional study. Environ. Res. 126, 9–16 [DOI] [PubMed] [Google Scholar]
- 8. Bouchard M., Laforest F., Vandelac L., Bellinger D., and Mergler D. (2007) Hair manganese and hyperactive behaviors: pilot study of school-age children exposed through tap water. Environ. Health Perspect. 115, 122–127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Bouchard M. F., Sauvé S., Barbeau B., Legrand M., Brodeur M. È., Bouffard T., Limoges E., Bellinger D. C., and Mergler D. (2011) Intellectual impairment in school-age children exposed to manganese from drinking water. Environ. Health Perspect. 119, 138–143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Claus Henn B., Ettinger A. S., Schwartz J., Téllez-Rojo M. M., Lamadrid-Figueroa H., Hernández-Avila M., Schnaas L., Amarasiriwardena C., Bellinger D. C., Hu H., and Wright R. O. (2010) Early postnatal blood manganese levels and children's neurodevelopment. Epidemiology 21, 433–439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Khan K., Factor-Litvak P., Wasserman G. A., Liu X., Ahmed E., Parvez F., Slavkovich V., Levy D., Mey J., van Geen A., and Graziano J. H. (2011) Manganese exposure from drinking water and children's classroom behavior in Bangladesh. Environ. Health Perspect. 119, 1501–1506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Khan K., Wasserman G. A., Liu X., Ahmed E., Parvez F., Slavkovich V., Levy D., Mey J., van Geen A., Graziano J. H., and Factor-Litvak P. (2012) Manganese exposure from drinking water and children's academic achievement. Neurotoxicology 33, 91–97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Oulhote Y., Mergler D., Barbeau B., Bellinger D. C., Bouffard T., Brodeur M. È., Saint-Amour D., Legrand M., Sauvé S., and Bouchard M. F. (2014) Neurobehavioral function in school-age children exposed to manganese in drinking water. Environ. Health Perspect. 122, 1343–1350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Riojas-Rodríguez H., Solís-Vivanco R., Schilmann A., Montes S., Rodríguez S., Ríos C., and Rodríguez-Agudelo Y. (2010) Intellectual function in Mexican children living in a mining area and environmentally exposed to manganese. Environ. Health Perspect. 118, 1465–1470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Wasserman G. A., Liu X., Parvez F., Ahsan H., Levy D., Factor-Litvak P., Kline J., van Geen A., Slavkovich V., LoIacono N. J., Cheng Z., Zheng Y., and Graziano J. H. (2006) Water manganese exposure and children's intellectual function in Araihazar, Bangladesh. Environ. Health Perspect. 114, 124–129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Quadri M., Federico A., Zhao T., Breedveld G. J., Battisti C., Delnooz C., Severijnen L. A., Di Toro Mammarella L., Mignarri A., Monti L., Sanna A., Lu P., Punzo F., Cossu G., Willemsen R., et al. (2012) Mutations in SLC30A10 cause parkinsonism and dystonia with hypermanganesemia, polycythemia, and chronic liver disease. Am. J. Hum. Genet. 90, 467–477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Tuschl K., Clayton P. T., Gospe S. M. Jr., Gulab S., Ibrahim S., Singhi P., Aulakh R., Ribeiro R. T., Barsottini O. G., Zaki M. S., Del Rosario M. L., Dyack S., Price V., Rideout A., Gordon K., et al. (2012) Syndrome of hepatic cirrhosis, dystonia, polycythemia, and hypermanganesemia caused by mutations in SLC30A10, a manganese transporter in man. Am. J. Hum. Genet. 90, 457–466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Lechpammer M., Clegg M. S., Muzar Z., Huebner P. A., Jin L. W., and Gospe S. M. Jr. (2014) Pathology of inherited manganese transporter deficiency. Ann. Neurol. 75, 608–612 [DOI] [PubMed] [Google Scholar]
- 19. Tuschl K., Mills P. B., Parsons H., Malone M., Fowler D., Bitner-Glindzicz M., and Clayton P. T. (2008) Hepatic cirrhosis, dystonia, polycythaemia and hypermanganesaemia: a new metabolic disorder. J. Inherit. Metab. Dis. 31, 151–163 [DOI] [PubMed] [Google Scholar]
- 20. Ebert B. L., and Bunn H. F. (1999) Regulation of the erythropoietin gene. Blood 94, 1864–1877 [PubMed] [Google Scholar]
- 21. Leyva-Illades D., Chen P., Zogzas C. E., Hutchens S., Mercado J. M., Swaim C. D., Morrisett R. A., Bowman A. B., Aschner M., and Mukhopadhyay S. (2014) SLC30A10 is a cell surface-localized manganese efflux transporter, and Parkinsonism-causing mutations block its intracellular trafficking and efflux activity. J. Neurosci. 34, 14079–14095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Zogzas C. E., Aschner M., and Mukhopadhyay S. (2016) Structural elements in the transmembrane and cytoplasmic domains of the metal transporter SLC30A10 are required for its manganese efflux activity. J. Biol. Chem. 291, 15940–15957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Huang L., and Tepaamorndech S. (2013) The SLC30 family of zinc transporters: a review of current understanding of their biological and pathophysiological roles. Mol. Aspects Med. 34, 548–560 [DOI] [PubMed] [Google Scholar]
- 24. Kambe T., Tsuji T., Hashimoto A., and Itsumura N. (2015) The physiological, biochemical, and molecular roles of zinc transporters in zinc homeostasis and metabolism. Physiol. Rev. 95, 749–784 [DOI] [PubMed] [Google Scholar]
- 25. Kolaj-Robin O., Russell D., Hayes K. A., Pembroke J. T., and Soulimane T. (2015) Cation diffusion facilitator family: structure and function. FEBS Lett. 589, 1283–1295 [DOI] [PubMed] [Google Scholar]
- 26. Chen P., Bowman A. B., Mukhopadhyay S., and Aschner M. (2015) SLC30A10: a novel manganese transporter. Worm 4, e1042648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Nishito Y., Tsuji N., Fujishiro H., Takeda T. A., Yamazaki T., Teranishi F., Okazaki F., Matsunaga A., Tuschl K., Rao R., Kono S., Miyajima H., Narita H., Himeno S., and Kambe T. (2016) Direct comparison of manganese detoxification/efflux proteins and molecular characterization of ZnT10 protein as a manganese transporter. J. Biol. Chem. 291, 14773–14787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lu M., Chai J., and Fu D. (2009) Structural basis for autoregulation of the zinc transporter YiiP. Nat. Struct. Mol. Biol. 16, 1063–1067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Lu M., and Fu D. (2007) Structure of the zinc transporter YiiP. Science 317, 1746–1748 [DOI] [PubMed] [Google Scholar]
- 30. Hall J. E. (2016) Thyroid metabolic hormones. In Textbook of Medical Physiology, pp. 951–963, Elsevier, Philadelphia, PA [Google Scholar]
- 31. Mansouri A., Chowdhury K., and Gruss P. (1998) Follicular cells of the thyroid gland require Pax8 gene function. Nat. Genet. 19, 87–90 [DOI] [PubMed] [Google Scholar]
- 32. Marians R. C., Ng L., Blair H. C., Unger P., Graves P. N., and Davies T. F. (2002) Defining thyrotropin-dependent and -independent steps of thyroid hormone synthesis by using thyrotropin receptor-null mice. Proc. Natl. Acad. Sci. U.S.A. 99, 15776–15781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Zoeller R. T., Tan S. W., and Tyl R. W. (2007) General background on the hypothalamic-pituitary-thyroid (HPT) axis. Crit. Rev. Toxicol. 37, 11–53 [DOI] [PubMed] [Google Scholar]
- 34. Beaudin S. A., Nisam S., and Smith D. R. (2013) Early life versus lifelong oral manganese exposure differently impairs skilled forelimb performance in adult rats. Neurotoxicol. Teratol. 38, 36–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Beaudin S. A., Strupp B. J., Strawderman M., and Smith D. R. (2017) Early postnatal manganese exposure causes lasting impairment of selective and focused attention and arousal regulation in adult rats. Environ. Health Perspect. 125, 230–237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kern C. H., Stanwood G. D., and Smith D. R. (2010) Preweaning manganese exposure causes hyperactivity, disinhibition, and spatial learning and memory deficits associated with altered dopamine receptor and transporter levels. Synapse 64, 363–378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Semple B. D., Blomgren K., Gimlin K., Ferriero D. M., and Noble-Haeusslein L. J. (2013) Brain development in rodents and humans: identifying benchmarks of maturation and vulnerability to injury across species. Prog. Neurobiol. 106–107, 1–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Soldin O. P., and Aschner M. (2007) Effects of manganese on thyroid hormone homeostasis: potential links. Neurotoxicology 28, 951–956 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Buthieau A. M., and Autissier N. (1977) [The effect of Mn2+ on thyroid iodine metabolism in rats]. C. R. Seances Soc. Biol. Fil. 171, 1024–1028 [PubMed] [Google Scholar]
- 40. Buthieau A. M., and Autissier N. (1983) Effects of manganese ions on thyroid function in rat. Arch. Toxicol. 54, 243–246 [DOI] [PubMed] [Google Scholar]
- 41. Kawada J., Nishida M., Yoshimura Y., and Yamashita K. (1985) Manganese ion as a goitrogen in the female mouse. Endocrinol. Jpn. 32, 635–643 [DOI] [PubMed] [Google Scholar]
- 42. National Toxicology Program (1993) NTP toxicology and carcinogenesis studies of manganese (II) sulfate monohydrate (CAS no. 10034–96-5) in F344/N rats and B6C3F1 mice (feed studies). Natl. Toxicol. Program Tech. Rep. Ser. 428, 1–275 [PubMed] [Google Scholar]
- 43. Mukhopadhyay S., Bachert C., Smith D. R., and Linstedt A. D. (2010) Manganese-induced trafficking and turnover of the cis-Golgi glycoprotein GPP130. Mol. Biol. Cell 21, 1282–1292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Mukhopadhyay S., and Linstedt A. D. (2011) Identification of a gain-of-function mutation in a Golgi P-type ATPase that enhances Mn2+ efflux and protects against toxicity. Proc. Natl. Acad. Sci. U.S.A. 108, 858–863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Mukhopadhyay S., and Linstedt A. D. (2012) Manganese blocks intracellular trafficking of Shiga toxin and protects against Shiga toxicosis. Science 335, 332–335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Zhong M. L., Chi Z. H., Shan Z. Y., Teng W. P., and Wang Z. Y. (2012) Widespread expression of zinc transporter ZnT (SLC30) family members in mouse endocrine cells. Histochem. Cell Biol. 138, 605–616 [DOI] [PubMed] [Google Scholar]
- 47. Lee E. S., Sidoryk M., Jiang H., Yin Z., and Aschner M. (2009) Estrogen and tamoxifen reverse manganese-induced glutamate transporter impairment in astrocytes. J. Neurochem. 110, 530–544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Lee E. S., Yin Z., Milatovic D., Jiang H., and Aschner M. (2009) Estrogen and tamoxifen protect against Mn-induced toxicity in rat cortical primary cultures of neurons and astrocytes. Toxicol. Sci. 110, 156–167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Moreno J. A., Streifel K. M., Sullivan K. A., Hanneman W. H., and Tjalkens R. B. (2011) Manganese-induced NF-κB activation and nitrosative stress is decreased by estrogen in juvenile mice. Toxicol. Sci. 122, 121–133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Morshed S. A., Latif R., and Davies T. F. (2012) Delineating the autoimmune mechanisms in Graves' disease. Immunol. Res. 54, 191–203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Eggan K., Akutsu H., Loring J., Jackson-Grusby L., Klemm M., Rideout W. M. 3rd, Yanagimachi R., and Jaenisch R. (2001) Hybrid vigor, fetal overgrowth, and viability of mice derived by nuclear cloning and tetraploid embryo complementation. Proc. Natl. Acad. Sci. U.S.A. 98, 6209–6214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Behringer R., Gertsenstein M., Vintersten Nagy K., and Nagy A. (2014) Manipulating the Mouse Embryo: A Laboratory Manual, 4th ed., Cold Spring Harbor Laboratory, Cold Spring Harbor, NY [Google Scholar]
- 53. Hayashi S., Lewis P., Pevny L., and McMahon A. P. (2002) Efficient gene modulation in mouse epiblast using a Sox2Cre transgenic mouse strain. Mech. Dev. 119, S97–S101 [DOI] [PubMed] [Google Scholar]
- 54. Yin W., Maguire S. M., Pham B., Garcia A. N., Dang N. V., Liang J., Wolfe A., Hofmann H. A., and Gore A. C. (2015) Testing the critical window hypothesis of timing and duration of estradiol treatment on hypothalamic gene networks in reproductively mature and aging female rats. Endocrinology 156, 2918–2933 [DOI] [PMC free article] [PubMed] [Google Scholar]