

Abstract
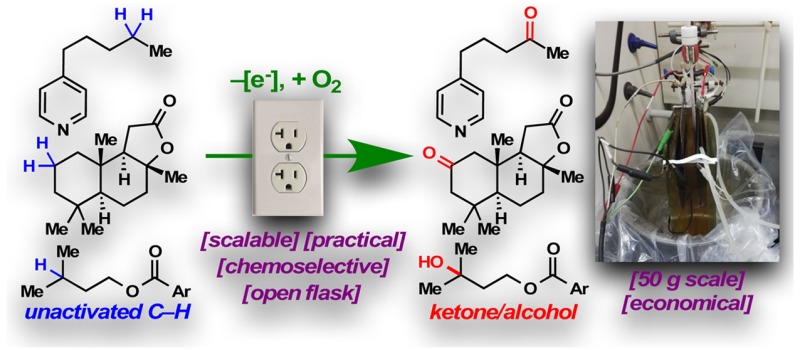
A practical electrochemical oxidation of unactivated C–H bonds is presented. This reaction utilizes a simple redox mediator, quinuclidine, with inexpensive carbon and nickel electrodes to selectively functionalize “deep-seated” methylene and methine moieties. The process exhibits a broad scope and good functional group compatibility. The scalability, as illustrated by a 50 g scale oxidation of sclareolide, bodes well for immediate and widespread adoption.
In 2016, we reported a scalable solution to allylic C–H oxidation using a simple small-molecule redox mediator via anodic oxidation.1 Unactivated methylene and methine systems, the most ubiquitous structural motifs in organic chemistry, have high redox potentials (oftentimes above 3.0 V vs SCE, see Figure 1A).2,3 Thus, under direct electrolysis, oxidative degradation of other functionalities and solvents is likely to occur prior to the desired C–H oxidation.4 Instead, chemists are generally beholden to strong oxidants, such as methyl(trifluoromethyl)dioxirane (TFDO),5,6 and metal complexes7,8 to effect such transformations. The complicated preparation of the former deters broad utilization, while the latter generally lacks economic viability owing to the high cost of catalyst systems. Consequently, a scalable means to oxidize unactivated C–H bonds remains elusive. As with prior studies, a mediated electrochemical oxidation was envisioned as a practical and sustainable solution to this lingering problem.9 A redox mediator can be oxidized anodically, generating a reactive species which could facilitate substrate oxidation through efficient homogeneous electron transfer. When an electron-rich mediator molecule is chosen, anodic oxidation can occur at relatively low potentials—chemoselective oxidation of unactivated C–H bonds can therefore be enabled. This Communication outlines the discovery of a simple redox mediator that, under electrochemical conditions, can achieve the chemoselective and scalable oxidation of unactivated C–H bonds previously only within the purview of strong chemical oxidants.
Figure 1.
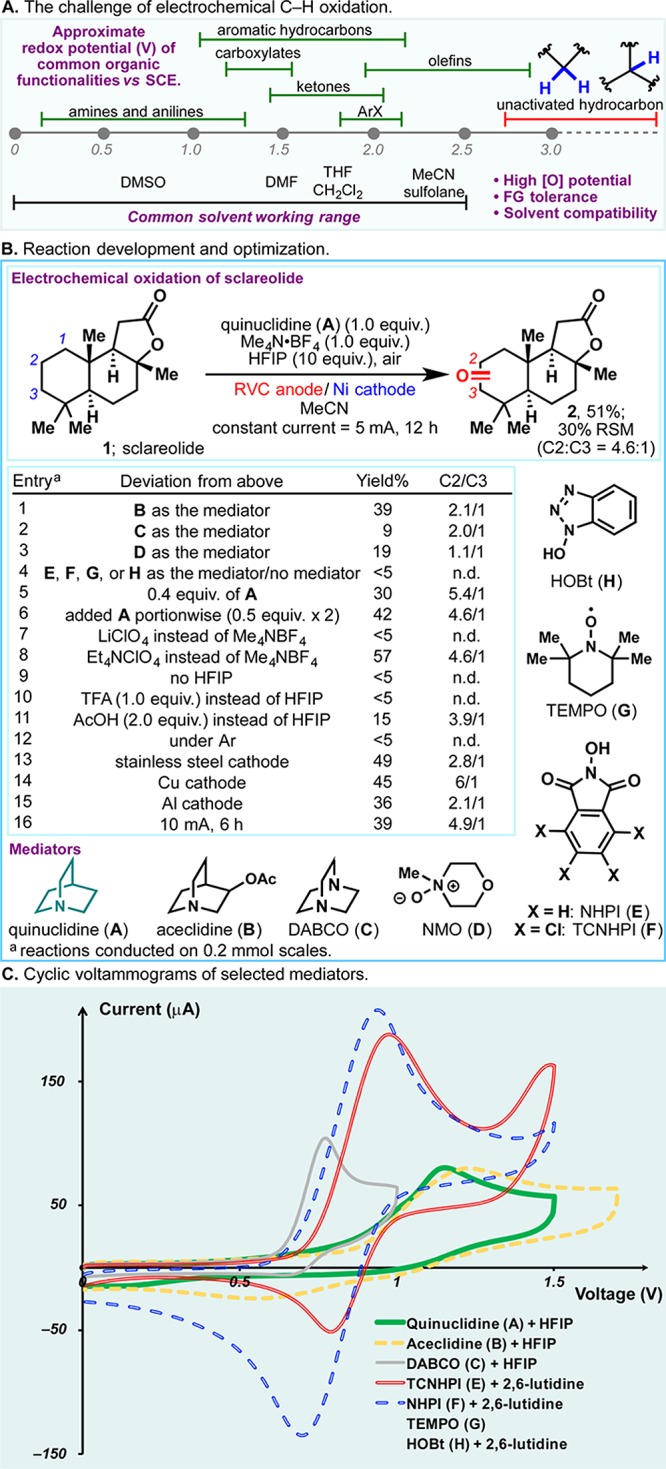
(A) The challenge of electrochemical C–H oxidation. (B) Reaction development and optimization. (C) Cyclic voltammograms of selected mediators.
Figure 1B illustrates optimal conditions for this transformation alongside an abbreviated picture of reaction optimization on sclareolide (1). Identification of a viable redox mediator was the critical hurdle to overcome. To this end, numerous scaffolds were examined (e.g., A–H), and tertiary amines (e.g., A, B, and C) were singularly successful (entries 1–5).10
The use of quinuclidine (A) as a mediator allowed the selective electrochemical oxidation of C2 methylenes in 1 at ca. 1.8 V (with respect to Ag/AgCl reference electrode).11 Structurally similar aceclidine (B) and DABCO (C) gave inferior results, whereas TCNHPI (F), a highly effective mediator for allylic C–H oxidation, failed to afford ketone 2 in synthetically useful yields. NMO (D), TEMPO (G), and HOBT (H) have all proven ineffectual. Unsurprisingly, <5% oxidation product was observed in the absence of a mediator (entry 4). Attempts to reduce the quantities of A used led to lower yields (e.g., entry 5). Meanwhile, portion-wise addition of A had relatively little influence on yield and selectivity (entry 6). While it is conceivable that all mediators surveyed gave rise to radical or cationic intermediates capable of abstracting C–H bonds, the mixture of A and HFIP exhibited a high oxidation potential, rendering the corresponding radical cation sufficiently reactive to engage unactivated C–H bonds (Figure 1C). The mixtures of TCNHPI (F) or NHPI (E) with 2,6-lutidine, conversely, showcased a lower E1/2, making them more suited for the functionalization of weaker C–H bonds in allylic oxidation (Figure 1C).12 In the presence of HFIP, aceclidine (B) has E1/2 similar to that of A, and it is thus the second most effective mediator.
The choice of electrolyte also has a substantial impact on the reaction outcome. The use of lithium perchlorate led to virtually no product formation, presumably due to chelation of lithium cation to the quinuclidine mediator (entry 7). While Et4N·ClO4 and Me4N·BF4 were almost equally effective (entry 8), the latter was chosen owing to its superior safety profile and lower cost. After extensive experimentation, HFIP was identified as an essential additive; attempts to use other Brønsted acids such as TFA or acetic acid were unsuccessful (entries 9–11). Atmospheric oxygen ostensibly provides the terminal oxidant of this reaction, as no product was afforded under argon (entry 12).
The use of expensive electrode materials (e.g., platinum electrode) was consciously avoided at the outset of this study. Reticulated vitreous carbon (RVC) electrode (ca. $3 per electrode) was used as the anode, while nickel, copper, and even stainless steel were all found to be viable materials for the cathode (entries 13–15). The reaction proceeds at room temperature in a simple undivided cell under constant current conditions (25 mA/mmol).
With the optimized conditions in hand, the scope of this electrochemical C–H oxidation method was probed next (Scheme 1). Methylene groups in linear, cyclic, bicyclic, heteroarene-containing, and natural-product-derived substrates could all be selectively transformed into ketones. The yields and selectivities were comparable to those of TFDO oxidation13,14 or Barton-Gif-type (iron-based) processes.8
Scheme 1. Scope of the Quinuclidine-Mediated Electrochemical C–H Oxidation.

Oxidation of linear systems occurred preferentially, as illustrated by 3–5, at the methylene unit distal to electron-withdrawing functional groups.15 Whereas TFDO oxidation delivered 4 as a 1.9:1 mixture with the β-hydroxy derivative, higher selectivity of δ-methylene oxidation was obtained with this electrochemical protocol (3.2:1). This δ selectivity was also observed in cycloheptyl (6, 11) and bicyclic (7) scaffolds. Conversely, γ-methylene units were oxidized preferentially in 6-membered rings (8–10), presumably due to the higher statistical probability of γ-oxidation and strain-release considerations.5c
The quinuclidine mediator allowed electrochemical oxidation to occur at a relatively low potential. As a result, a host of functionalities are tolerated under the reaction conditions. For example, esters, arenes, ketones, amides, and silyl ethers were left untouched during the reaction. While TFDO oxidation failed to produce 9 in meaningful yields, this electrochemical protocol exhibited compatibility with the free hydroxyl group. In 11, not only was the phthalimide tolerated under the reaction conditions, but the α-C–H bond was also preserved—no noticeable α-oxidation (Shono-type) took place.1612 was obtained with complete retention of stereochemical integrity, despite the presence of two epimerizable centers. Selective distal methylene oxidation was observed in 13 wherein the pyridine motif and the benzylic methylene group both remained intact. TFDO oxidation, in contrast, led exclusively to N-oxide formation and no methylene oxidation.17 This compatibility with azine motifs bodes well for applications in the pharmaceutical industry. The capacity of this method to selectively modify complex natural scaffolds was illustrated through the oxidation of isosteviol ethyl ester, which proceeded with exquisite selectivity. As shown earlier (Figure 1B), electrochemical C–H oxidation of sclareolide occurred selectively at C2. Intriguingly, TFDO elicited mainly C3 methylene oxidation.
Activated methylenes, such as benzylic (15) and α-alkoxy (16) C–H bonds, could also be oxidized effectively. While this quinuclidine-based redox mediator system furnished 17 in high yields through selective allylic C–H oxidation, the TCNHPI-based protocol still proved superior on densely functionalized substrates, particularly those bearing multiple olefin motifs. The TCNHPI system delivered 18 in high yields; attempted anodic oxidation of valencene with quinuclidine mediator failed to afford the same product. This quinuclidine-mediated protocol could also be used to oxidize tertiary C–H bonds, affording the corresponding alcohols as products (19–21).
The scalability of this technology is readily evident from the efficient preparations of 6 and 12 in gram and 500 mg scales, respectively. This prompted us to examine its applicability in a process setting: A 50 g scale oxidation of sclareolide was carried out using inexpensive RVC anode and stainless steel cathode in a beaker that was left open to air (Figure 2A, inset photograph). 2 was afforded in a yield (47%) and selectivity (5.6:1) similar to those found in mmol-scale experiments. The remaining mass balance could largely be accounted for by the recovered starting material (26%), further attesting to the mildness of the reaction conditions. Reagents/solvents (including quinuclidine, HFIP, Me4N·BF4, MeCN) and electricity used in this 50 g scale process cost ca. $2517 in total, while the analogous TFDO reaction or Fe(PDP)-mediated oxidation would have been evidently more expensive, as the trifluoroacetone (TFDO precursor) or Fe(PDP) catalyst alone would cost $9120 and $37 229, respectively, on this scale.18
Figure 2.

(A) 50 g scale C–H oxidation of sclareolide enabled the synthesis of 2-oxo-yahazunone. (B) Putative mechanism for the quinuclidine-mediated electrochemical C–H oxidation.
Earlier studies from our laboratory have revealed sclareolide as a convenient starting material for the divergent synthesis of meroterpenoids.19 To this end, the ability to procure ample amounts of 2-oxo-sclareolide (2, Figure 2A, inset photograph) via electrochemical C–H oxidation affords the unique opportunity to examine the 2-oxo analogues of these bioactive terpenoids. In our previous strategy toward meroterpenoids, sclareolide was converted into its borono analogue, which served as a “terpenyl radical” progenitor. It was envisioned that, in a complementary approach, decarboxylative Negishi coupling of redox-active ester 22 and arylzinc species could afford various meroterpenoid analogues.20
Indeed, 22 was synthesized from 2 in three steps, whereupon nickel-catalyzed cross coupling afforded 23 in 57% yield. Removal of the protecting groups with a combination of cerium ammonium nitrate and 24 furnished (+)-2-oxo-yahazunone (25).21
Mechanistically, the electrochemical C–H oxidation likely involves a quinuclidine radical cation generated in anodic oxidation. This high-energy species could homolytically cleave an unactivated C–H bond; reaction between the ensuing carbon-centered radical (Figure 2B) and molecular oxygen then affords the oxidation product. HFIP is believed to serve as the electron acceptor to generate H2 in the cathodic process.22
In summary, the oxidation of unactivated C–H bonds, heretofore commonly achieved with highly reactive, exotic, and expensive reagents, has been enabled in an exceedingly simple fashion through an electrochemical process. This practical method employs quinuclidine as the mediator in an unsophisticated electrochemical setup using inexpensive carbon and nickel electrodes. A diverse range of functional groups were shown to be compatible with the chemoselective process. The scalability of the method, as evident through the successful oxidation of sclareolide on a 50 g scale, has the potential to facilitate the synthesis of complex materials.
Acknowledgments
Financial support for this work was provided by NIH (GM-118176), Pfizer, and Asymchem. We thank Prof. A. L. Rheingold, Dr. C. E. Moore, and Dr. M. Gembicky (UCSD) for X-ray crystallographic analysis, and Drs. D.-H. Huang and L. Pasternack (TSRI) for NMR analysis. We are grateful to Dr. E. Horn for efforts during the early stage of the investigation. We thank Mr. M. Collins (Pfizer Inc., La Jolla, CA) for providing substrates in this study. We thank Dr. E. Zhang (Asymchem) for helpful discussions. We acknowledge Drs. G. Che and Q. Hou (Asymchem) for technical assistance.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.7b03539.
The authors declare no competing financial interest.
Supplementary Material
References
- Horn E. J.; Rosen B. R.; Chen Y.; Tang J.; Chen K.; Eastgate M. D.; Baran P. S. Nature 2016, 533, 77. 10.1038/nature17431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eberson L.; Nyberg K. Tetrahedron 1976, 32, 2185. 10.1016/0040-4020(76)85132-0. [DOI] [Google Scholar]
- a Luca O. R.; Gustafson J. L.; Maddox S. M.; Fenwick A. Q.; Smith D. C. Org. Chem. Front. 2015, 2, 823. 10.1039/C5QO00075K. [DOI] [Google Scholar]; b Roth H. G.; Romero N. A.; Nicewicz D. A. Synlett 2016, 27, 714. 10.1055/s-0035-1561297. [DOI] [Google Scholar]
- For examples of direct anodic oxidations of unactivated sp3 C–H bonds, see:; a Koch V. R.; Miller L. L. J. Am. Chem. Soc. 1973, 95, 8631. 10.1021/ja00807a022. [DOI] [Google Scholar]; b Fritz H. P.; Würminghausen T. J. Chem. Soc., Perkin Trans. 1 1976, 610. 10.1039/P19760000610. [DOI] [Google Scholar]; c Hembrock A.; Schäfer H. J.; Zimmermann G. Angew. Chem., Int. Ed. Engl. 1985, 24, 1055. 10.1002/anie.198510551. [DOI] [Google Scholar]
- a Curci R.; D’Accolti L.; Fusco C. Acc. Chem. Res. 2006, 39, 1. 10.1021/ar050163y. [DOI] [PubMed] [Google Scholar]; b Newhouse T. R.; Baran P. S. Angew. Chem., Int. Ed. 2011, 50, 3362. 10.1002/anie.201006368. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Chen K.; Eschenmoser A.; Baran P. S. Angew. Chem., Int. Ed. 2009, 48, 9705. 10.1002/anie.200904474. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Crandall J. K.; Curci R.; D’Accolti L.; Fusco C.; Fusco C.; D’Accolti L.; Annese C.. Methyl(trifluoromethyl)dioxirane. In Encyclopedia of Reagents for Organic Synthesis; Fuchs P., Bode J. W., Charette A. B., Rovis T., Eds.; Wiley: New York, 2016. [Google Scholar]
- For selected examples of C–H oxidation with oxaziridine species, see:; a Litvinas N. D.; Brodsky B. H.; Du Bois J. Angew. Chem., Int. Ed. 2009, 48, 4513. 10.1002/anie.200901353. [DOI] [PubMed] [Google Scholar]; b DesMarteau D. D.; Donadelli A.; Montanari V.; Petrov V. A.; Resnati G. J. Am. Chem. Soc. 1993, 115, 4897. 10.1021/ja00064a063. [DOI] [Google Scholar]
- For some reviews on nondirected metal-catalyzed oxidation of unactivated C–H bonds, see:; a Huang X.; Groves J. T. JBIC, J. Biol. Inorg. Chem. 2017, 22, 185. 10.1007/s00775-016-1414-3. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Shilov A. E.; Shul’pin G. B. Chem. Rev. 1997, 97, 2879. 10.1021/cr9411886. [DOI] [PubMed] [Google Scholar]; c Olivo G.; Cussó O.; Costas M. Chem. - Asian J. 2016, 11, 3148. 10.1002/asia.201601170. [DOI] [PubMed] [Google Scholar]; d Que L. Jr.; Tolman W. B. Nature 2008, 455, 333. 10.1038/nature07371. [DOI] [PubMed] [Google Scholar]
- For selected examples of nondirected metal-catalyzed oxidation of unactivated C–H bonds, see:; a Barton D. H. R.; Doller D. Acc. Chem. Res. 1992, 25, 504. 10.1021/ar00023a004. [DOI] [Google Scholar]; b Kim C.; Chen K.; Kim J.; Que L. Jr. J. Am. Chem. Soc. 1997, 119, 5964. 10.1021/ja9642572. [DOI] [Google Scholar]; c Roelfes G.; Lubben M.; Hage R.; Que L. Jr.; Feringa B. L. Chem. - Eur. J. 2000, 6, 2152.. [DOI] [PubMed] [Google Scholar]; d Howell J. M.; Feng K.; Clark J. R.; Trzepkowski L. J.; White M. C. J. Am. Chem. Soc. 2015, 137, 14590. 10.1021/jacs.5b10299. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Chen M. S.; White M. C. Science 2010, 327, 566. 10.1126/science.1183602. [DOI] [PubMed] [Google Scholar]; f Das S.; Incarvito C. D.; Crabtree R. H.; Brudvig G. W. Science 2006, 312, 1941. 10.1126/science.1127899. [DOI] [PubMed] [Google Scholar]
- For a review on the use of mediators in electrochemistry, see:Francke R.; Little R. D. Chem. Soc. Rev. 2014, 43, 2492. 10.1039/c3cs60464k. [DOI] [PubMed] [Google Scholar]
- For some examples of tertiary aminium radical-mediated functionalizations of unactivated C–H bonds, see:; a Michaudel Q.; Thevenet D.; Baran P. S. J. Am. Chem. Soc. 2012, 134, 2547. 10.1021/ja212020b. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Bloom S.; Pitts C. R.; Miller D. C.; Haselton N.; Holl M. G.; Urheim E.; Lectka T. Angew. Chem., Int. Ed. 2012, 51, 10580. 10.1002/anie.201203642. [DOI] [PubMed] [Google Scholar]; c Pitts C. R.; Bloom S.; Woltornist R.; Auvenshine D. J.; Ryzhkov L. R.; Siegler M. A.; Lectka T. J. Am. Chem. Soc. 2014, 136, 9780. 10.1021/ja505136j. [DOI] [PubMed] [Google Scholar]
- For the direct C2–H chlorination and bromination of sclareolide through an intermolecular HLF reaction, see:; a Quinn R. K.; Könst Z. A.; Michalak S. E.; Schmidt Y.; Szklarski A. R.; Flores A. R.; Nam S.; Horne D. A.; Vanderwal C. D.; Alexanian E. J. J. Am. Chem. Soc. 2016, 138, 696. 10.1021/jacs.5b12308. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Schmidt V. A.; Quinn R. K.; Brusoe A. T.; Alexanian E. J. J. Am. Chem. Soc. 2014, 136, 14389. 10.1021/ja508469u. [DOI] [PubMed] [Google Scholar]
- All cyclic voltammograms were recorded in MeCN using Me4N·BF4 as the electrolyte. For details, see the SI.
- For the preparations of compound 3, 6, and 7 via TFDO oxidation, see:Asensio G.; Castellano G.; Mello R.; González Núñez M. E. J. Org. Chem. 1996, 61, 5564. 10.1021/jo9604189. [DOI] [Google Scholar]
- Yields and selectivity of compounds 4, 8, 9, 10, and 2 through TFDO oxidation were determined experimentally. See SI for detailed information.
- Minisci F.; Galli R.; Galli A.; Bernardi R. Tetrahedron Lett. 1967, 8, 2207. 10.1016/S0040-4039(00)90798-6. [DOI] [Google Scholar]
- For reviews on Shono oxidation, see:; a Shono T. Tetrahedron 1984, 40, 811. 10.1016/S0040-4020(01)91472-3. [DOI] [Google Scholar]; b Jones A. M.; Banks C. E. Beilstein J. Org. Chem. 2014, 10, 3056. 10.3762/bjoc.10.323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- This was observed experimentally. See SI for details.
- Calculations based on prices of commercially available quinuclidine, Me4N·BF4, MeCN, HFIP (for electrochemical protocol), trifluoroacetone (for TFDO oxidation), and Fe(PDP) (for Fe-mediated process) from Sigma Aldrich.
- Dixon D. D.; Lockner J. W.; Zhou Q.; Baran P. S. J. Am. Chem. Soc. 2012, 134, 8432. 10.1021/ja303937y. [DOI] [PubMed] [Google Scholar]
- a Cornella J.; Edwards J. T.; Qin T.; Kawamura S.; Wang J.; Pan C.-M.; Gianatassio R.; Schmidt M.; Eastgate M. D.; Baran P. S. J. Am. Chem. Soc. 2016, 138, 2174. 10.1021/jacs.6b00250. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Qin T.; Cornella J.; Li C.; Malins L. R.; Edwards J. T.; Kawamura S.; Maxwell B. D.; Eastgate M. D.; Baran P. S. Science 2016, 352, 801. 10.1126/science.aaf6123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gansäuer A.; Justicia J.; Rosales A.; Worgull D.; Rinker B.; Cuerva J. M.; Oltra J. E. Eur. J. Org. Chem. 2006, 2006, 4115. 10.1002/ejoc.200600389. [DOI] [Google Scholar]
- This is evidenced by the observation of effervescence (putatively H2 evolution) during some experiments. Nevertheless, the potential of the cathode during the course of the reaction was found to be −1.1 V (vs Ag/AgCl reference electrode); oxygen reduction at the cathode may also be operative.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.